
Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Sample Treatment
2.3. Data Analysis
3. Results
3.1. Physicochemical Properties of Rhizosphere and Non-Rhizosphere Soils
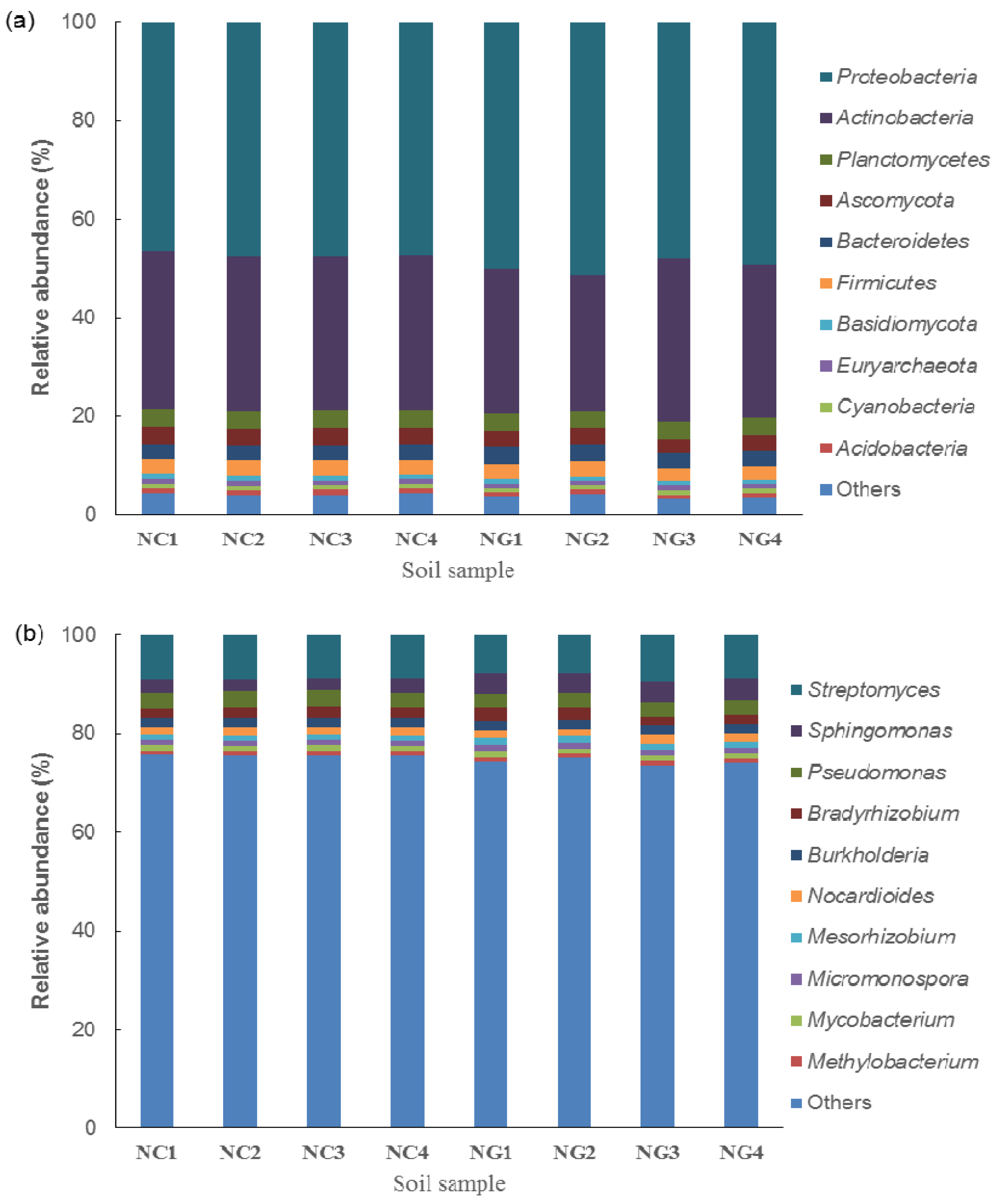
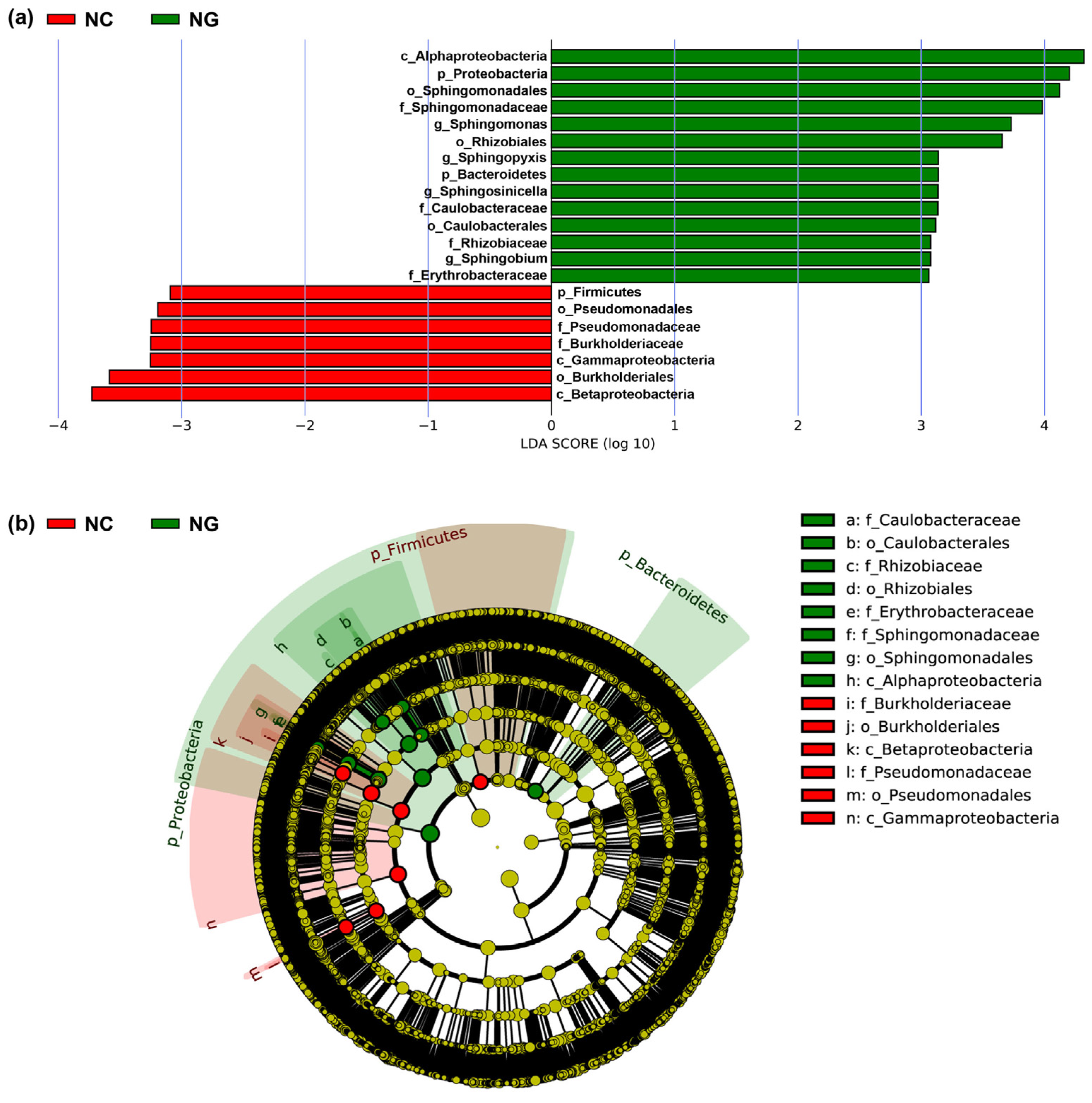
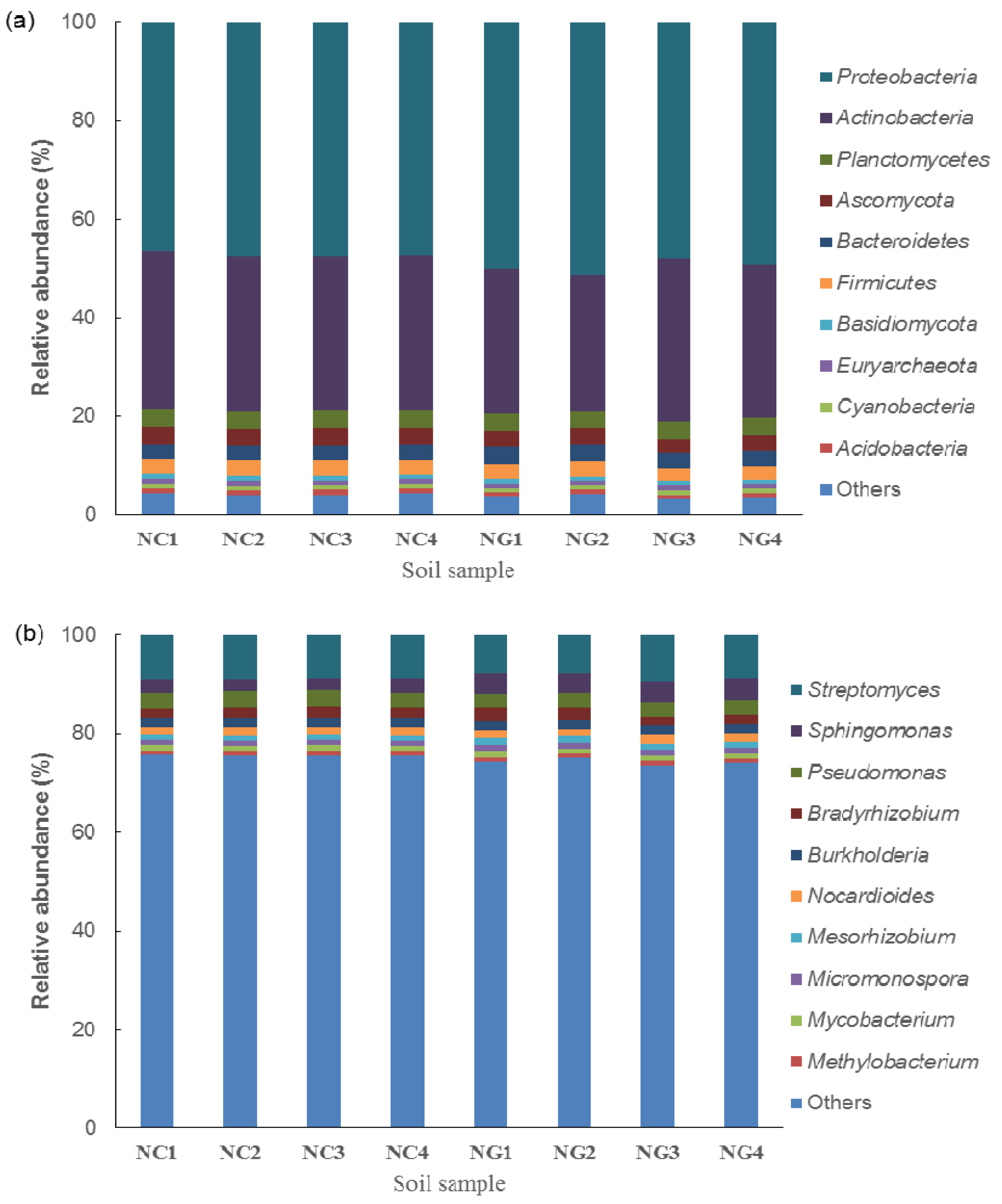
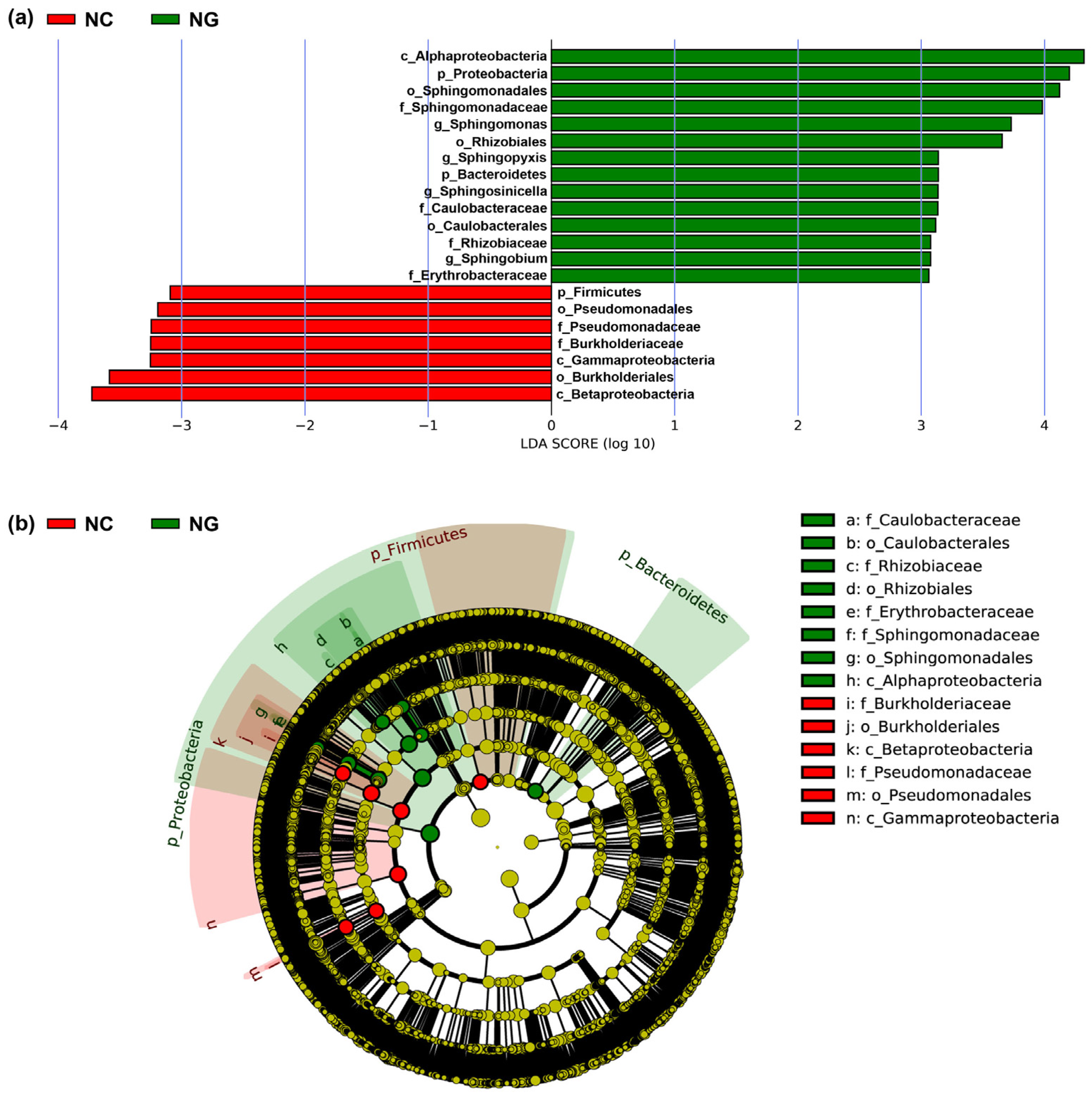
3.2. Microbial Composition of Poa alpigena L. Soils on Bird Island
3.3. Microbial Diversity of Poa alpigena L. Soils on Bird Island
3.3.1. Microbial α-Diversity
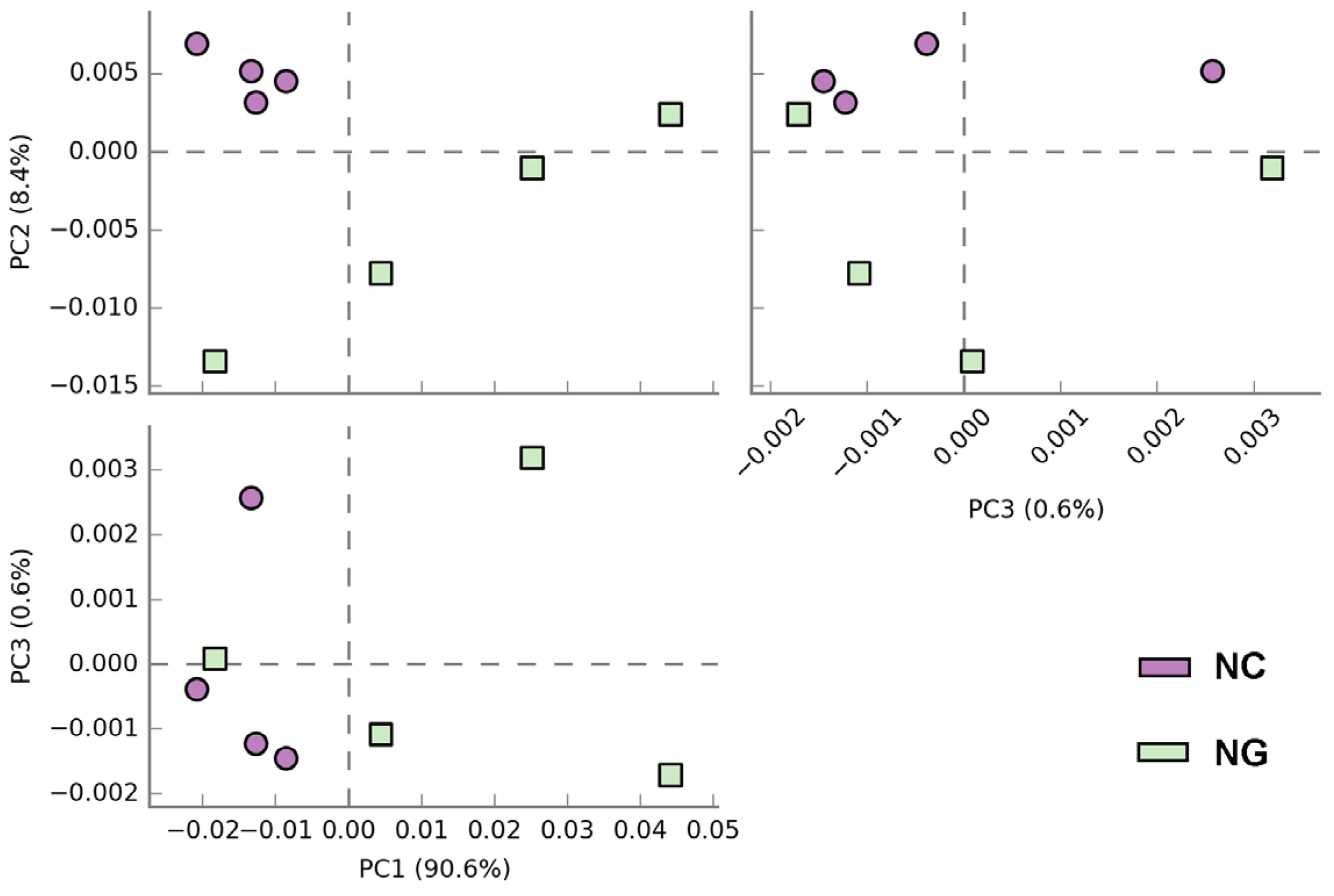
3.3.2. Microbial β-Diversity
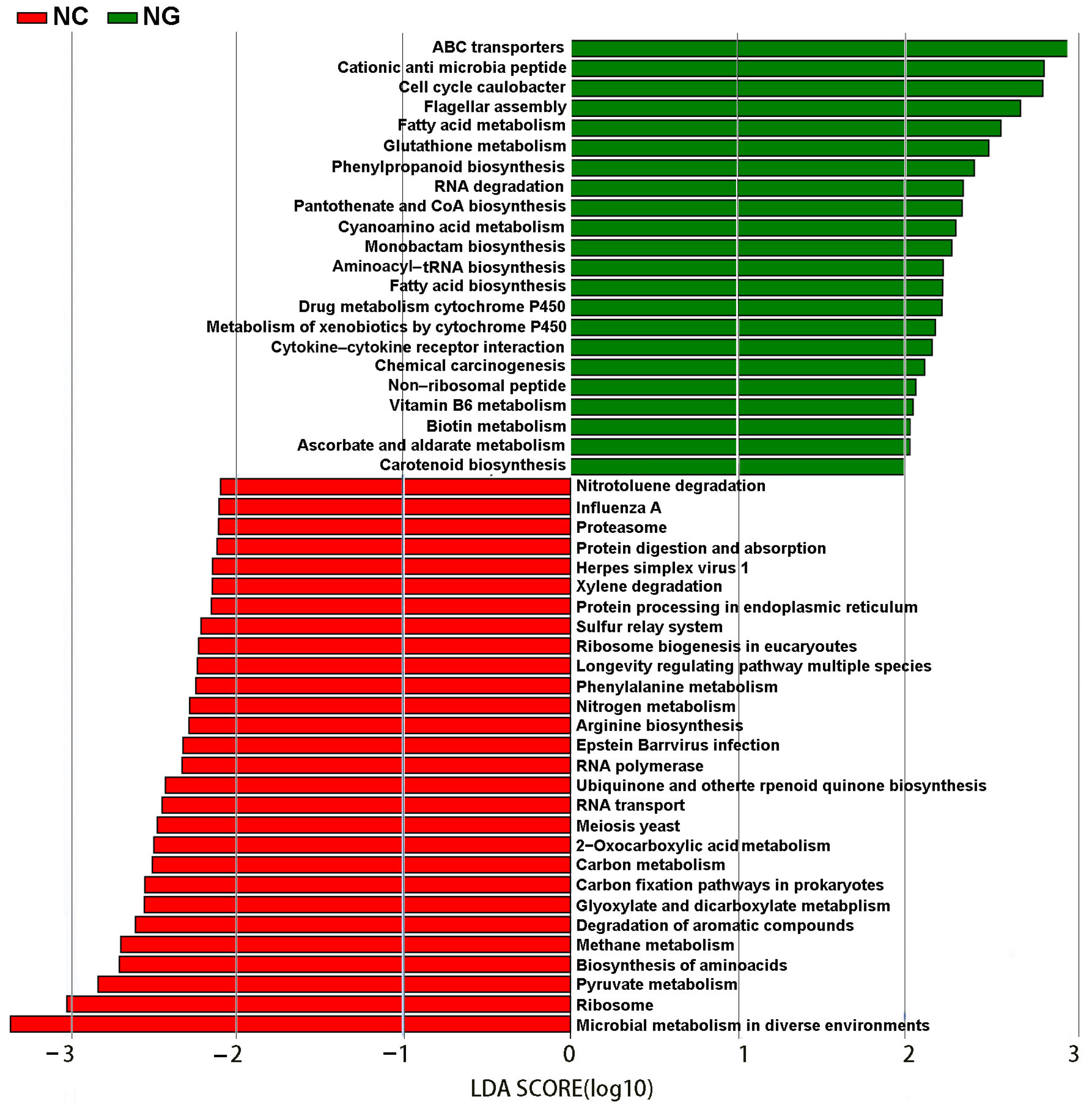
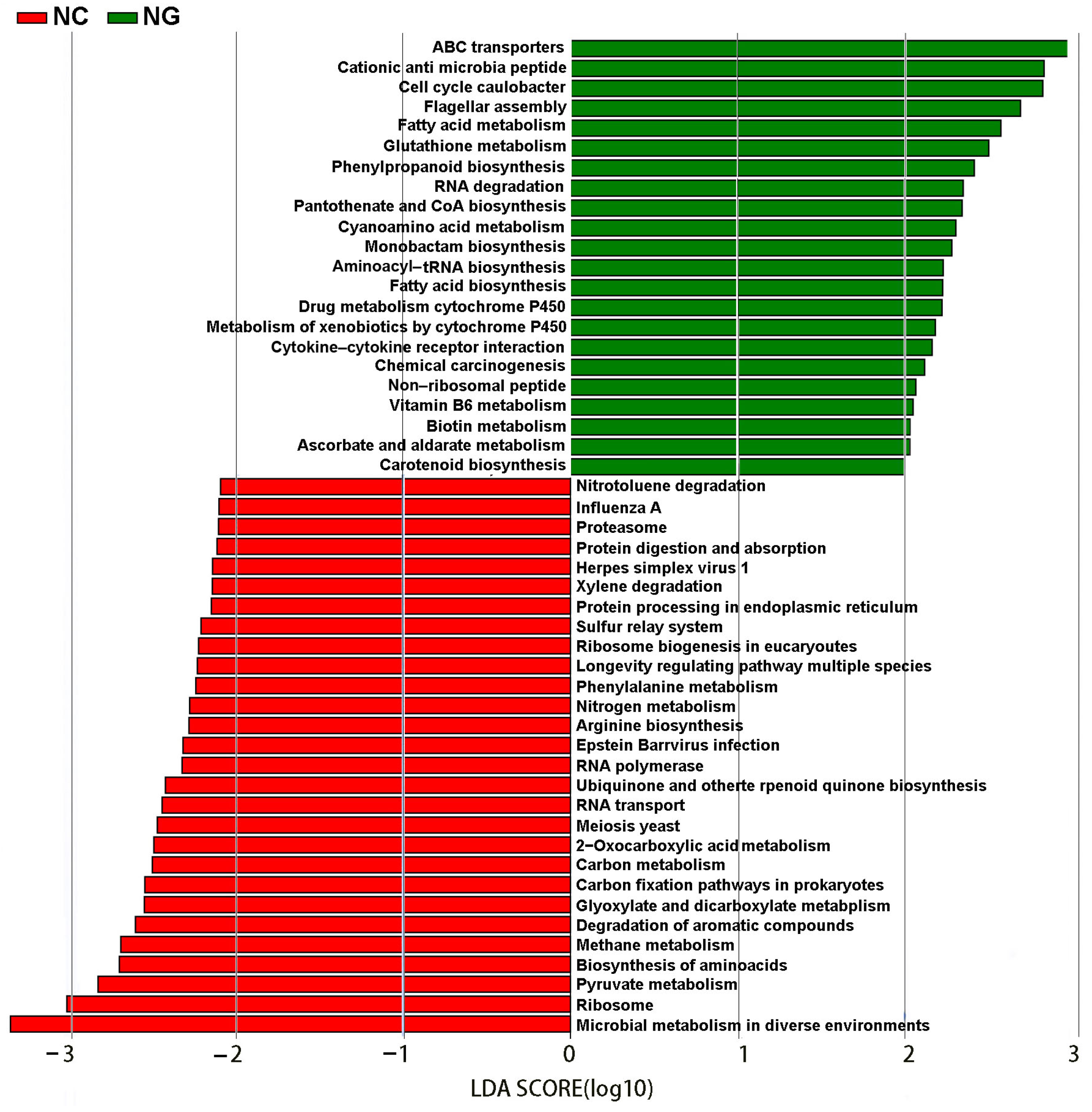
3.4. Differences in Metabolic Pathways between Rhizosphere and Non-Rhizosphere Soil Microorganisms
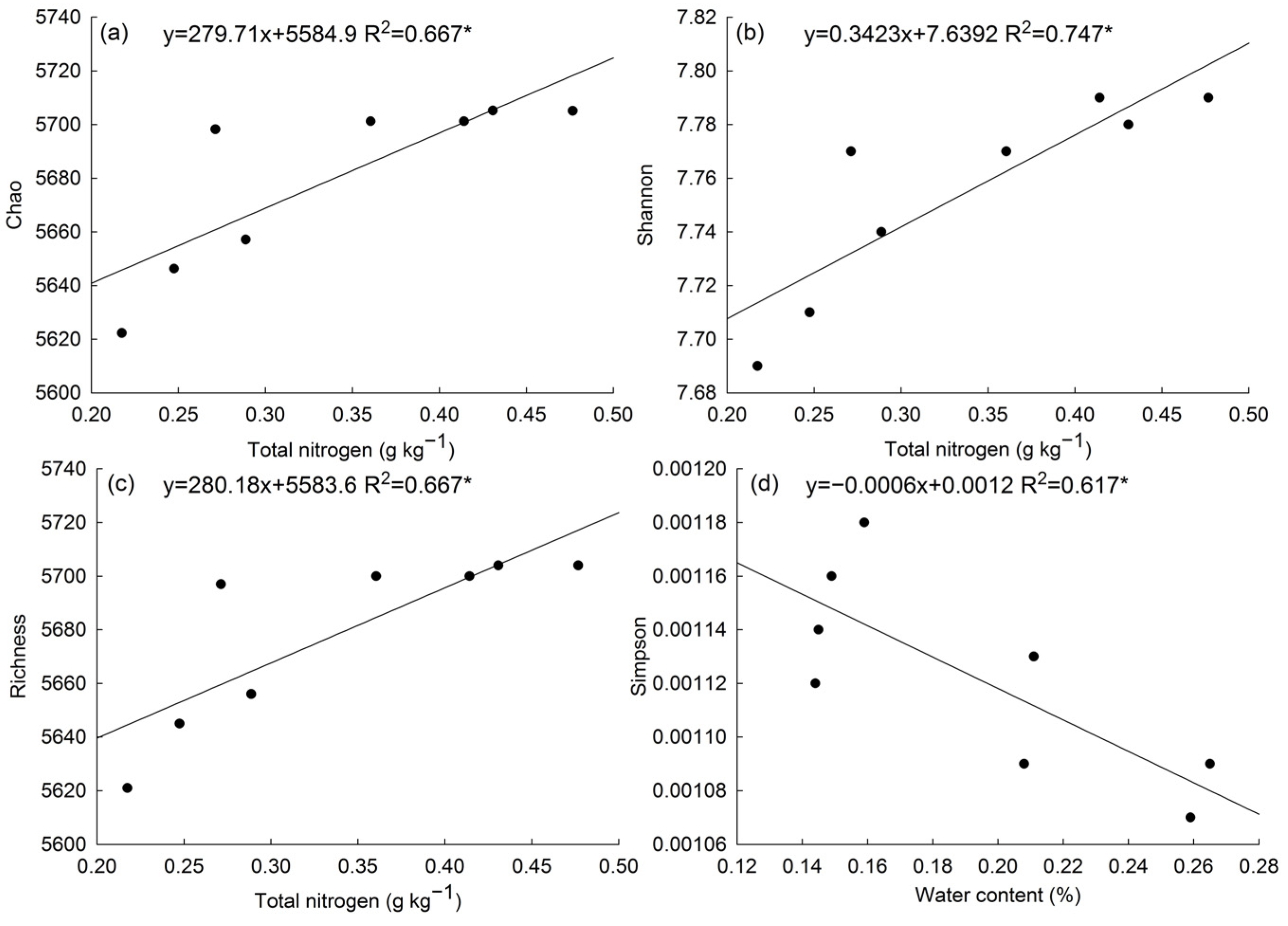
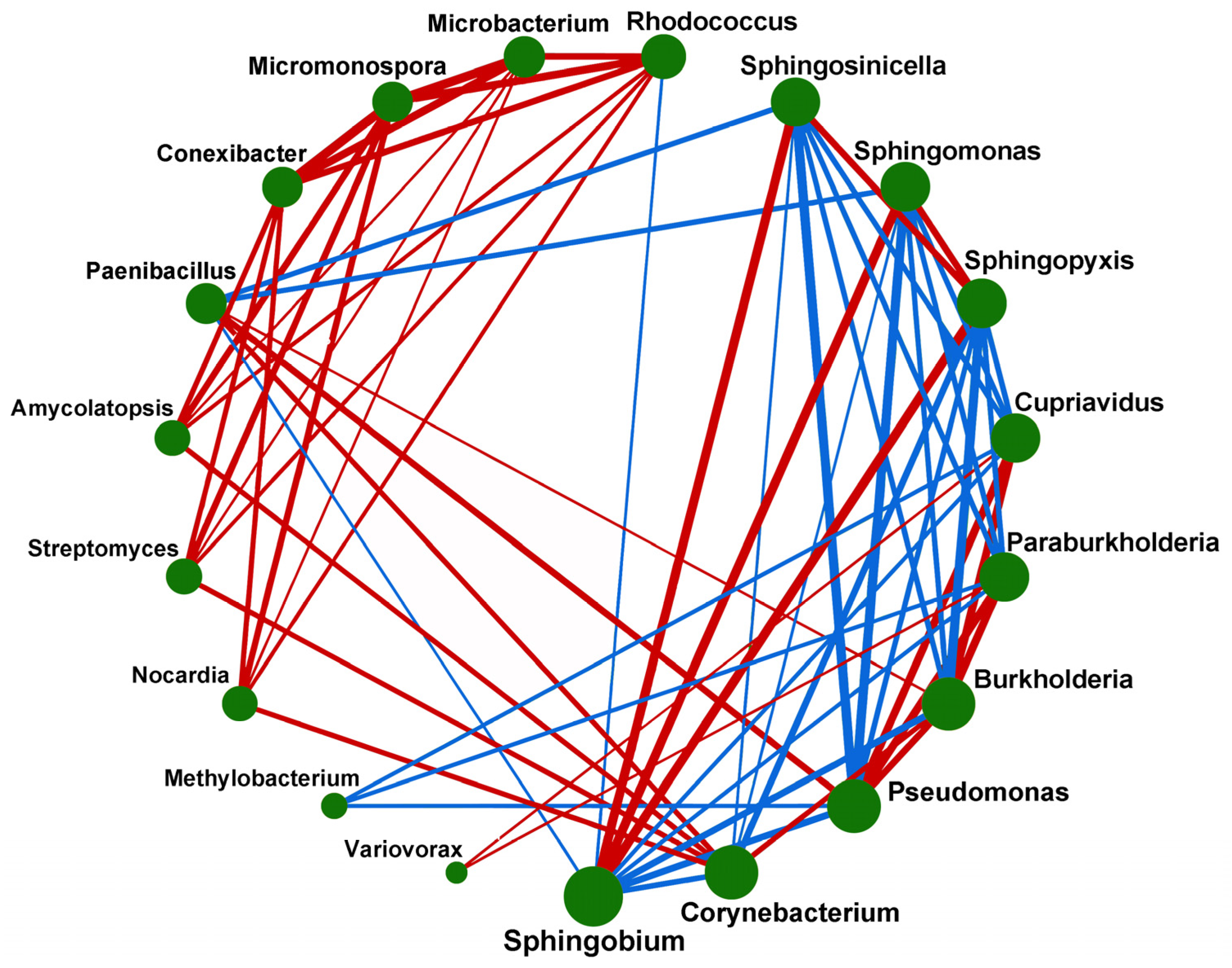
3.5. Correlation of Microorganisms of Poa alpigena L. Soils on Bird Island
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, N.N.; Sun, G.; Liang, J.; Wang, E.T.; Shi, C.G.; He, J.; Hu, X.; Zhao, C.Z.; Wu, N. Response of ammonium oxidizers to the application of nitrogen fertilizer in an alpine meadow on the Qinghai-Tibetan Plateau. Appl. Soil Ecol. 2018, 124, 266–274. [Google Scholar] [CrossRef]
- Zhou, T.Q.; Kong, W.D.; Chen, H. Advances in microbial research on Tibetan grassland soil. Chin. J. Ecol. 2022. Available online: https://kns.cnki.net/kcms/detail/21.1148.Q.20220520.1948.043.html (accessed on 15 November 2022). (In Chinese).
- Liu, J.B.; Kong, W.D.; Zhang, G.S.; Khan, A.; Guo, G.X.; Zhu, C.M.; Wei, X.J.; Kang, S.C.; Morgan-Kiss, R.M. Diversity and succession of autotrophic microbial community in high elevation soils along deglaciation chronosequence. FEMS Microbiol. Ecol. 2016, 92, fiw160. [Google Scholar] [CrossRef] [Green Version]
- Marilley, L.; Vogt, G.; Blanc, M.; Aragno, M. Bacterial diversity in the bulk soil and rhizosphere fractions of Lolium perenne and Trifolium repens as revealed by PCR restriction analysis of 16S rDNA. Plant Soil 1998, 198, 219–224. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-Growth-Promoting Rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Grayston, S.J.; Wang, S.Q.; Campbell, C.D.; Anthony, C.E. Selective influence of plant species on microbial diversity in the rhizosphere. Soil Biol. Biochem. 1998, 30, 369–378. [Google Scholar] [CrossRef]
- Jia, J.J.; Wang, Y.F.; Lu, Y.; Sun, K.; Lyu, S.D.; Gao, Y. Driving mechanisms of gross primary productivity geographical patterns for Qinghai-Tibet Plateau lake systems. Sci. Total Environ. 2021, 791, 148286–148299. [Google Scholar] [CrossRef]
- Zhai, W.T.; Chen, D.D.; Li, Q.; Zhao, L.; Liu, Z.; Xu, S.X.; Dong, Q.M.; Zhao, X.Q. Effect of grazing intensity on carbon metabolic characteristics of soil microbial communities in an alpine steppe in the regions around Qinghai Lake. Chin. J. Appl. Environ. Biol. 2017, 23, 685–692. (In Chinese) [Google Scholar]
- Wang, X.Y.; Li, Y.Q.; Gong, X.W.; Niu, Y.Y.; Chen, Y.P.; Shi, X.P.; Li, W.; Liu, J. Changes of soil organic carbon stocks from the 1980s to 2018 in northern China’s agro-pastoral ecotone. Catena 2020, 194, 104722. [Google Scholar] [CrossRef]
- Shi, H.X.; Hou, X.Y.; Shi, S.L.; Wu, X.H.; Yang, T.T.; Li, P. Poa alpigena response traits affected by grazing and enclosuresin an alpine meadow on the Qinghai-Tibet Plateau. Acta Ecol. Sin. 2016, 36, 3601–3608. (In Chinese) [Google Scholar]
- Wei, L.N.; Zhang, C.P.; Dong, Q.M.; Yu, Y.; Yang, X.X. Characterization of the complete chloroplast genome of Poa pratensis L. cv. Qinghai (Gramineae). Mitochondrial DNA Part B Resour. 2020, 5, 532–533. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.J.; Ni, Y.; Liao, L.X.; Xiao, Y.; Guo, Y.J. Poa pratensis ECERIFERUM1 (PpCER1) is involved in wax alkane biosynthesis and plant drought tolerance. Plant Physiol. Biochem. 2021, 159, 312–321. [Google Scholar] [CrossRef]
- Dong, W.K.; Ma, X.; Jiang, H.Y.; Zhao, C.X.; Ma, H.L. Physiological and transcriptome analysis of Poa pratensis var. anceps cv. Qinghai in response to cold stress. BMC Plant Biol. 2020, 20, 362. [Google Scholar] [CrossRef]
- Riley, D.; Barber, S.A. Effect of ammonium and nitrate fertilization on phosphorus uptake as related to root-Induced pH changes at the root-soil interface. Soil Sci. Soc. Amer. Proc. 1971, 35, 301–306. [Google Scholar] [CrossRef]
- Liu, F.D.; Mo, X.; Kong, W.J.; Song, Y. Soil bacterial diversity, structure, and function of Suaeda salsa in rhizosphere and non-rhizosphere soils in various habitats in the Yellow River Delta, China. Sci. Total Environ. 2020, 740, 140144. [Google Scholar] [CrossRef]
- Han, G.M.; Song, F.Q.; Zhang, Z.J.; Ni, W.; He, S.E.; Tian, X.J. An economic and efficient method for further purification of crude DNA extracted from forest soils. J. Forestry Res. 2010, 21, 246–250. [Google Scholar] [CrossRef]
- Hollister, E.B.; Engledow, A.S.; Hammett, A.M.; Provin, T.L.; Wilkinson, H.H.; Gentry, T.J. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 2010, 4, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.S.; Sheng, H.Y.; Luo, S.S.; Hu, Y.M.; Yu, L.L. Characteristics of prokaryotic microbial community structure and molecular ecological network in four habitat soils around Lake Qinghai. Ecol. Environ. Sci. 2021, 30, 1393–1403. (In Chinese) [Google Scholar]
- Yang, H.J.; Wang, Q.; Wan, Z.X.; Zhang, Z.Y.; Chen, D.D.; Tan, J. Structure and diversity of microbial communities in the rhizosphere and non-rhizosphere soil in areas with invasive Solidago canadensis L. J. Biosaf. 2021, 30, 235–243. (In Chinese) [Google Scholar]
- Zhang, P.; Cui, Z.Y.; Guo, M.Q.; Xi, R.C. Characteristics of the soil microbial community in the forestland of Camellia oleifera. PeerJ 2020, 8, 9117. [Google Scholar] [CrossRef]
- Frey, S.D.; Knorr, M.; Parrent, J.L.; Simpson, R.T. Chronic nitrogen enrichment affects the structure and function of the soil microbial community in temperate hardwood and pine forests. For. Ecol. Manag. 2004, 196, 159–171. [Google Scholar] [CrossRef]
- Beimforde, C.; Feldberg, K.; Nylinder, S.; Rikkinen, J.; Tuovila, H.; Dörfelt, H.; Gube, M.; Jackson, D.J.; Reitner, J.; Seyfullah, L.J.; et al. Estimating the Phanerozoic history of the Ascomycota lineages: Combining fossil and molecular data. Mol. Phylogenet Evol. 2014, 78, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cao, Y.R.; Wiese, J.; Tang, S.K.; Xu, L.H.; Imhoff, J.F.; Jiang, C.L. Streptomyces sparsus sp. nov., isolated from a saline and alkaline soil. Int. J. Syst. Evol Microbiol. 2011, 61, 1601–1605. [Google Scholar] [CrossRef] [Green Version]
- Harbison, A.B.; Garson, M.A.; Lamit, L.J.; Basiliko, N.; Bräuer, S.L. A novel isolate and widespread abundance of the candidate alphaproteobacterial order (Ellin 329), in southern Appalachian peatlands. FEMS Microbiol. Lett. 2016, 363, fnw151. [Google Scholar] [CrossRef] [Green Version]
- Urbanová, M.; Šnajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Marilley, L.; Aragno, M. Phylogenetic diversity of bacterial communities differing in degree of proximity of Loliumperenne and Trifolium repens roots. Appl. Soil Ecol. 1999, 13, 127–136. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, B.W.; Sun, R.D.; Zhang, Y.Y.; Song, J.Y.; Zhang, G.J.; Xu, L.; Mu, R.L. Characteristics of culturable microbial communities in rhizosphere /nonrhizosphere soil of Ligularia virgaurea in alpine meadow elevation gradient. Acta Ecol. Sin. 2021, 41, 4853–4863. (In Chinese) [Google Scholar]
- Zhang, K.; Bao, W.K.; Yang, B.; Hu, B. The effects of understory vegetation on soil microbial community composition and structure. Chin. J. Appl. Environ. Biol. 2017, 23, 1178–1184. (In Chinese) [Google Scholar]
- Yang, Y.; Liu, B.R. Distribution of soil nutrient and microbial biomass in rhizosphere versus non-rhizosphere area of different plant species in desertified steppe. Acta Ecol. Sin. 2015, 35, 7562–7570. [Google Scholar]
- Wang, Z.H.; Jiang, X.J. Contrasting responses of the microbial community structure and functional traits to soil pH in Purple Soils. Environ. Sci. 2022, 43, 3876–3883. [Google Scholar]
- Palit, K.; Rath, S.; Chatterjee, S.; Das, S. Microbial diversity and ecological interactions of microorganisms in the mangrove ecosystem: Threats, vulnerability, and adaptations. Environ. Sci. Pollut. Res. 2022, 29, 32467–32512. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Naylor, D.; Dong, Z.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 4284–4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenstein, K.; Dawson, R.J.P.; Locher, K.P. Struct and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T. Structural dynamics of ABC transporters: Molecular simulation studies. Biochem. Soc. Trans. 2021, 49, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, X.; Wang, H.; Liu, T.; Cheng, J.; Jiang, H. Discovery and modification of cytochrome P450 for plant natural products biosynthesis. Synth. Syst. Biotechnol. 2020, 5, 187–199. [Google Scholar] [CrossRef]
- Waring, R.H. Waring Cytochrome P450: Genotype to phenotype. Xenobiotica 2020, 50, 9–18. [Google Scholar] [CrossRef]
- Murayama, N.; Yamazaki, H. Metabolic activation and deactivation of dietary-derived coumarin mediated by cytochrome P450 enzymes in rat and human liver preparations. J. Toxicol. Sci. 2021, 46, 371–378. [Google Scholar] [CrossRef]
- Nie, J.T.; Wang, H.S.; Zhang, W.L.; Teng, X.; Yu, C.; Cai, R.; Wu, G. Characterization of lncRNAs and mRNAs involved in powdery mildew resistance in Cucumber. Phytopathology 2021, 111, 1613–1624. [Google Scholar] [CrossRef]
- Ettwig, K.F.; Butler, M.K.; Le, P.D.; Pelletier, E.; Mangenot, S.; Kuypers, M.M.; Schreiber, F.; Dutilh, B.E.; Zedelius, J.; de Beer, D. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 2010, 464, 543–548. [Google Scholar] [CrossRef]
- Phale, P.S.; Basu, A.; Majhi, P.D.; Deveryshetty, J.; Vamsee-Krishna, C.; Shrivastava, R. Metabolic diversity in bacterial degradation of aromatic compounds. OMICS J. Integr. Biol. 2007, 11, 252–279. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbrück, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Knight, R.; Joint, I.; et al. Defining seasonal marine microbial community dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.R.; Huang, Y.C.; Yang, X.R.; Xue, W.J.; Zhang, X.; Zhang, Y.H.; Pang, J.; Liu, Y.M.; Liu, Z.Q. Burkholderia sp. Y4 inhibits cadmium accumulation in rice by increasing essential nutrient uptake and preferentially absorbing cadmium. Chemosphere 2020, 252, 126603. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.Y.; Lv, Y.K.; Liu, Y.X.; Ren, R.P. Removal of nitrogen by heterotrophic nitrification-aerobic denitrification of a novel metal resistant bacterium Cupriavidus sp. S1. Bioresour. Technol. 2016, 220, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Finkel, O.M.; Salas-González, I.; Castrillo, G.; Conway, J.M.; Law, T.F.; Teixeira, P.J.P.L.; Wilson, E.D.; Fitzpatrick, C.R.; Jones, C.D.; Dangl, J.L. A single bacterial genus maintains root growth in a complex microbiome. Nature 2020, 587, 103–133. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Properties | NC | NG |
|---|---|---|
| Water content (%) | 0.24 ± 0.02 a | 0.15 ± 0.01 b |
| pH | 8.91 ± 0.17 a | 8.59 ± 0.30 a |
| Electronic conductivity (ms cm−1) | 0.05 ± 0.01 a | 0.06 ± 0.02 a |
| Total carbon (g kg−1) | 5.12 ± 0.40 a | 5.14 ± 0.45 a |
| Total nitrogen (g kg−1) | 0.42 ± 0.05 a | 0.26 ± 0.03 b |
| Project Name | NC | NG |
|---|---|---|
| Number of clean reads (couple) | 36,702,067.5 ± 1074957.9 a | 36,367,431.5 ± 2157852.8 a |
| Unclassified reads (%) | 57.13 ± 0.72 a | 54.72 ± 1.55 b |
| Classified reads (%) | 42.88 ± 0.72 b | 45.28 ± 1.55 a |
| Chordate reads (%) | 6.23 ± 0.06 a | 6.20 ± 0.27 a |
| Microbial reads (%) | 36.50 ± 1.00 b | 38.94 ± 2.00 a |
| Bacterial reads (%) | 28.80 ± 0.54 a | 31.22 ± 1.91 a |
| Fungal reads (%) | 1.42 ± 0.04 a | 1.42 ± 0.06 a |
| Viral reads (%) | 0.16 ± 0.00 a | 0.16 ± 0.00 a |
| Protozoan reads (%) | 0.18 ± 0.00 a | 0.18 ± 0.01 a |
| Alpha Diversity Indexes | NC | NG |
|---|---|---|
| Richness index | 5702.00 ± 2.31 a | 5654.75 ± 31.73 b |
| Chao 1 index | 5703.18 ± 2.28 a | 5655.98 ± 31.69 b |
| Shannon index | 7.78 ± 0.01 a | 7.73 ± 0.01 b |
| Simpson index | 1.10 × 10−3 ± 2.52 × 10−5 b | 1.15 × 10−3 ± 2.58 × 10−5 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Che, Z.; Cao, Y.; Qi, L.; Chen, K.; Wang, H. Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis. Water 2023, 15, 239. https://doi.org/10.3390/w15020239
Li L, Che Z, Cao Y, Qi L, Chen K, Wang H. Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis. Water. 2023; 15(2):239. https://doi.org/10.3390/w15020239
Chicago/Turabian StyleLi, Lingling, Zihan Che, Yanhong Cao, Lulu Qi, Kelong Chen, and Hengsheng Wang. 2023. "Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis" Water 15, no. 2: 239. https://doi.org/10.3390/w15020239