
The Impact of Dissolved Organic Matter on Arsenic Mobilization from Goethite in the Presence of Silicic Acid and Phosphate under Reducing Conditions

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Site and Characteristics of the Peat

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C [g kg−1] | Si [mg kg−1] | Fe [mg kg−1] | P [mg kg−1] | As [mg kg−1] |
|---|---|---|---|---|
| 491.0 | 848.0 | 200.0 | 200.0 | 0.53 |
2.2. Synthesis of Goethite
2.3. Incubation Batch Preparation
2.4. Sample Collection
| Treatments 1 | As | PO4−3-P | H4SiO4-Si | No Fe | Medium Fe | High Fe | |
|---|---|---|---|---|---|---|---|
| Fe Conc.–Treatments | (n = 3) | [44 µg L−1] * | [13 mg g−1] ** | [21.7 mg g−1] | [-] | [4.4 mg g−1] | [44.6 mg g−1] |
| nFe-C | ✓ | ✓ | ✓ | ||||
| nFe-PO4 | ✓ | ✓ | ✓ | ✓ | |||
| nFe-H4SiO4 | ✓ | ✓ | ✓ | ✓ | |||
| nFe-H4SiO4+PO4 | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| mFe-C | ✓ | ✓ | ✓ | ||||
| mFe-PO4 | ✓ | ✓ | ✓ | ✓ | |||
| mFe-H4SiO4 | ✓ | ✓ | ✓ | ✓ | |||
| mFe-H4SiO4+PO4 | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| hFe-C | ✓ | ✓ | ✓ | ||||
| hFe-PO4 | ✓ | ✓ | ✓ | ✓ | |||
| hFe-H4SiO4 | ✓ | ✓ | ✓ | ✓ | |||
| hFe-H4SiO4+PO4 | ✓ | ✓ | ✓ | ✓ | ✓ |
2.5. Analytical Methods
Aqueous Samples
2.6. Statistical Analysis
3. Results
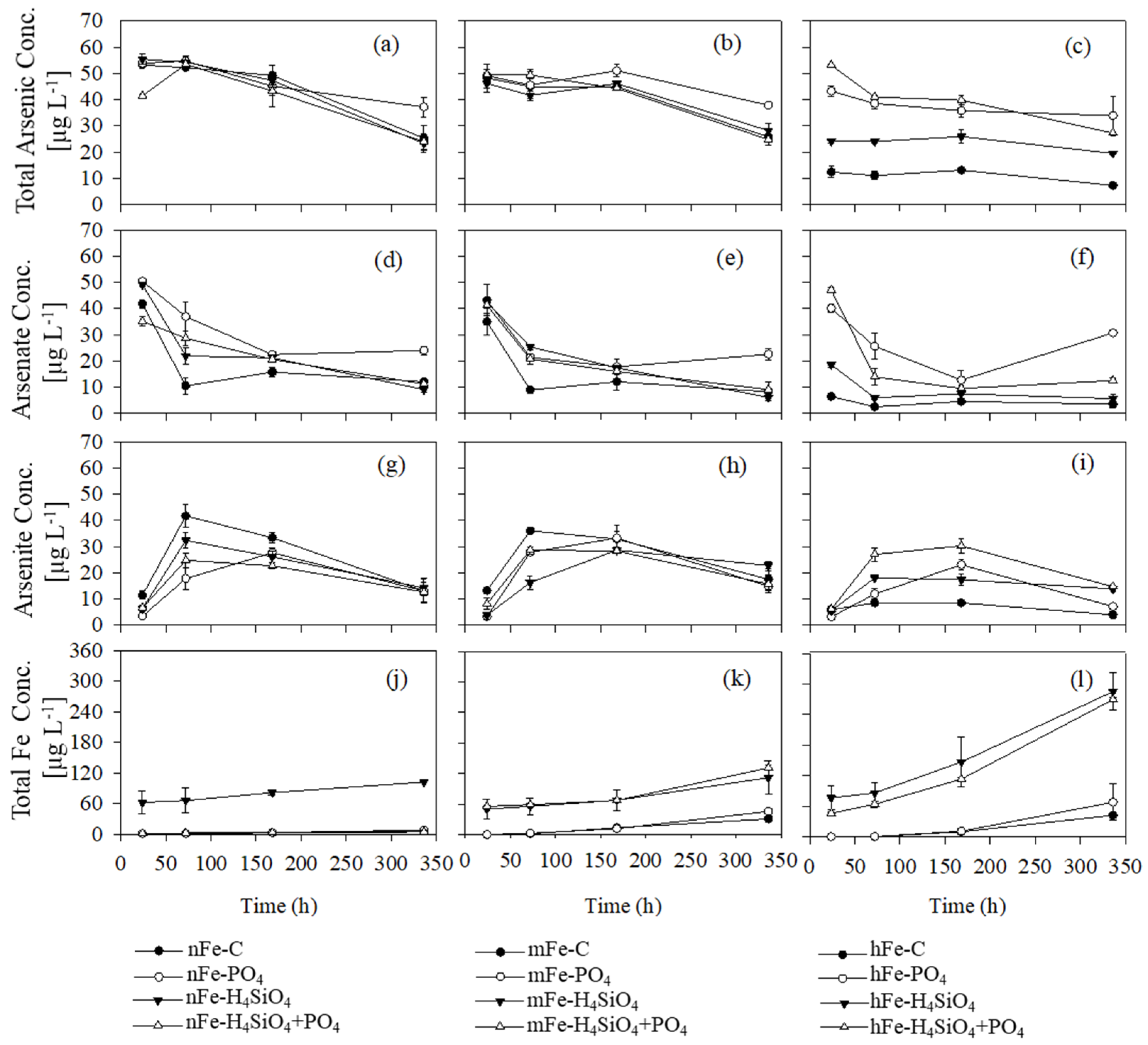
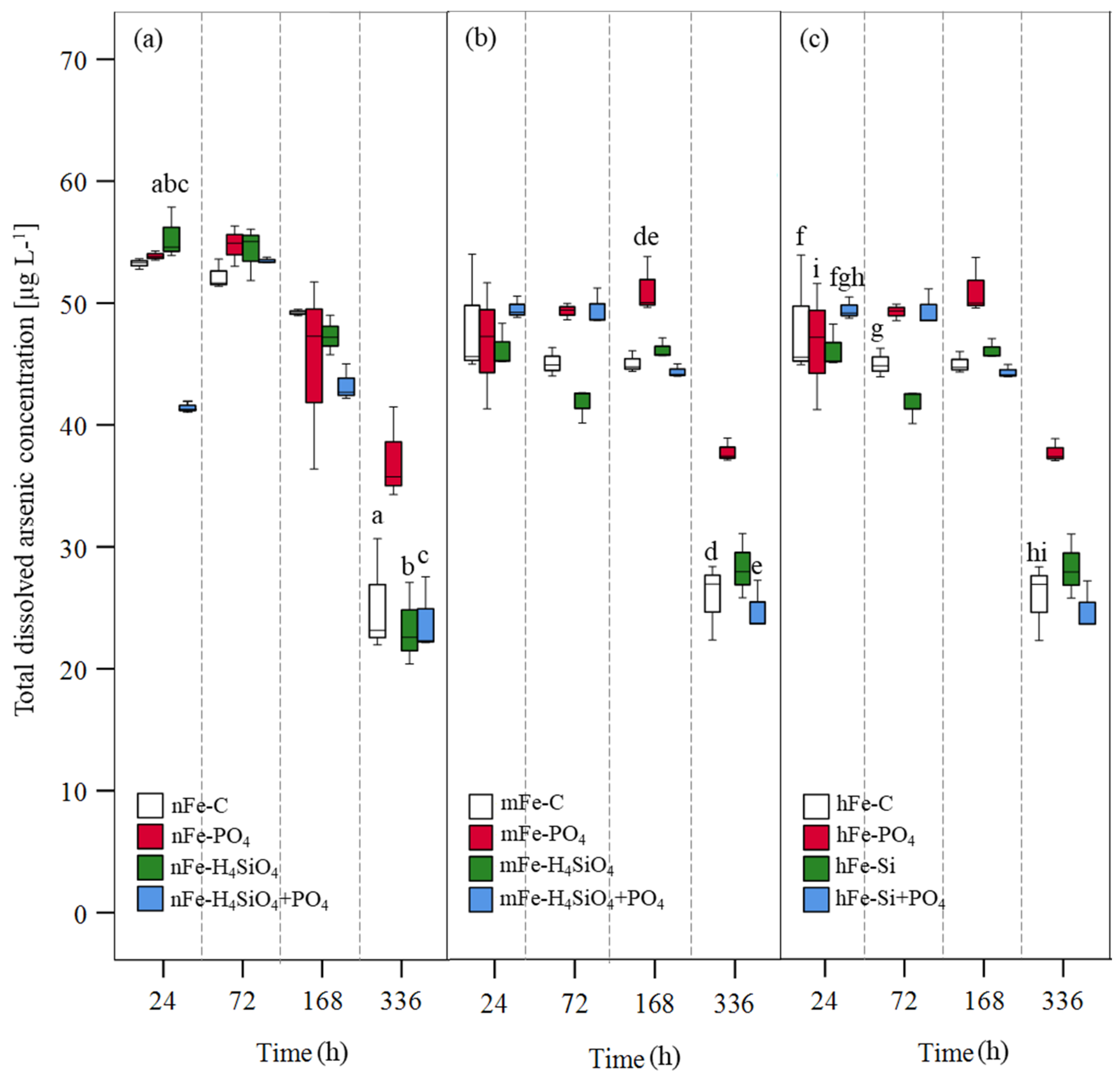
3.1. Solute Total Arsenic Turnover in Presence of Varying Amounts of Goethite
3.2. Arsenate Concentration and Arsenite Formation


3.3. Dissolved Iron
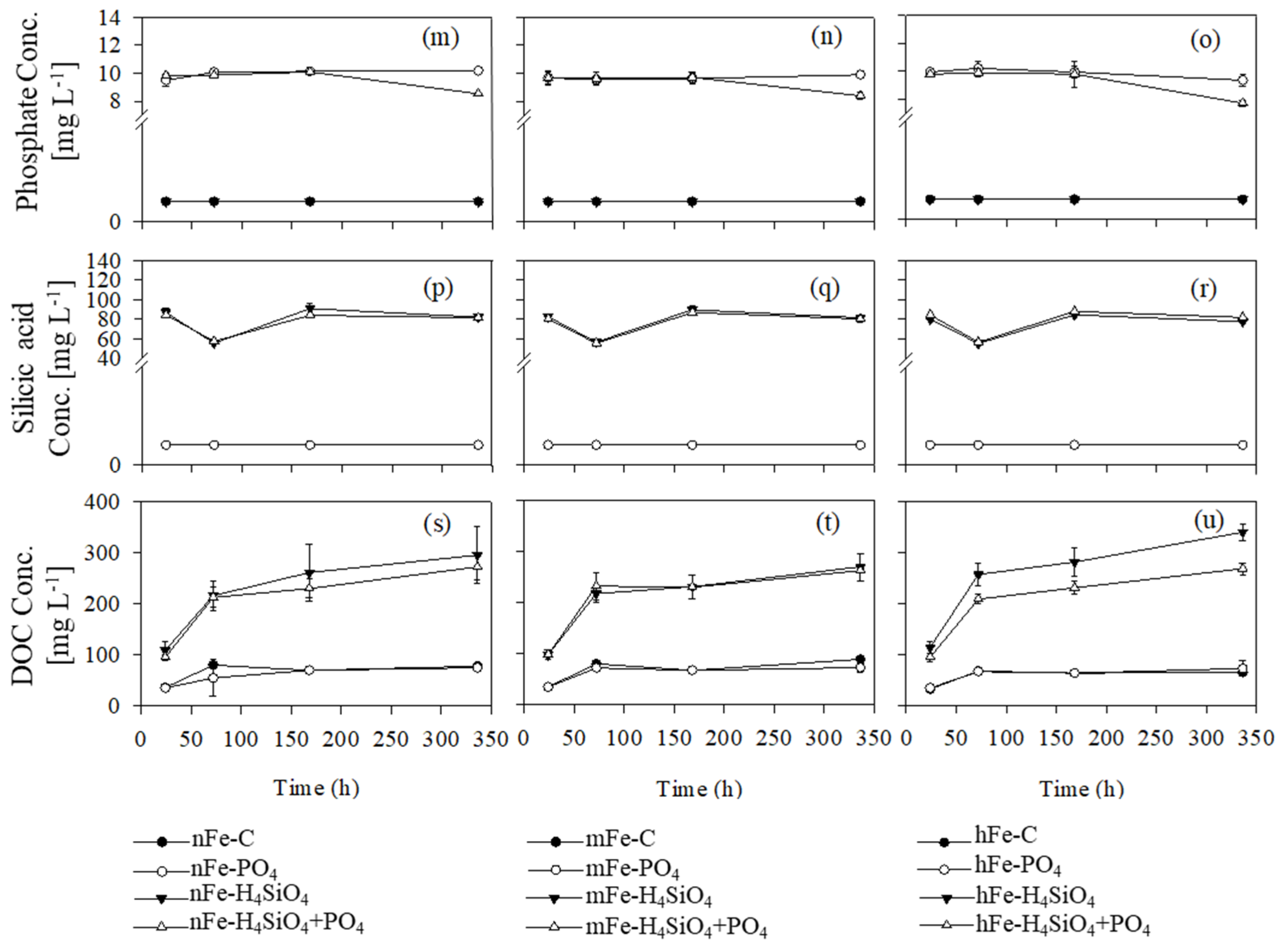
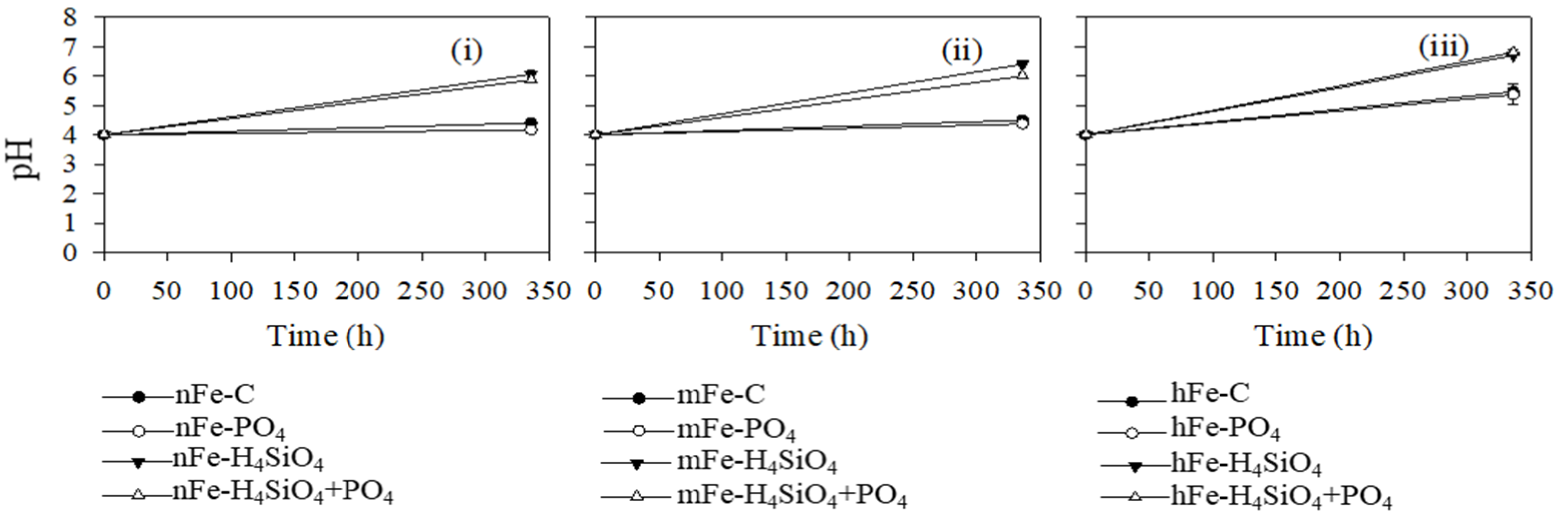
3.4. Phosphate, Silicic Acid, and Dissolved Organic Matter

4. Discussion
4.1. The Effects of Phosphate, Silicic Acid, and DOM on Arsenic Mobilization
4.2. Silicic Acid and Phosphate Interactions with Fe

4.3. Silicic Acid Effects on DOM Concentration
4.4. The Role of DOM on Arsenic Mobilization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer (IARC). Some Drinking-Water Disinfectants and Contaminants, Including Arsenic; World Health Organization: Lyon, France, 2004; p. 526. [Google Scholar]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. J. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef]
- Argos, M.; Kalra, T.; Rathouz, P.J.; Chen, Y.; Pierce, B.; Parvez, F.; Islam, T.; Ahmed, A.; Rakibuz-Zaman, M.; Hasan, R.; et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): A prospective cohort study. Lancet 2010, 376, 252–258. [Google Scholar] [CrossRef]
- Kumarathilaka, P.; Seneweera, S.; Meharg, A.; Bundschuh, J. Arsenic speciation dynamics in paddy rice soil-water environment: Sources, physico-chemical, and biological factors—A review. Water Res. 2018, 140, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M.; Bibi, I.; Shahid, M.; Shaheen, S.M.; Shakoor, M.B.; Bashir, S.; Younas, F.; Rinklebe, J.; Niazi, N.K. Biogeochemical cycling, speciation and transformation pathways of arsenic in aquatic environments with the emphasis on algae. Compr. Anal. Chem. 2019, 85, 15–51. [Google Scholar]
- Schwertmann, U.; Cornell, R.M. The Iron Oxides: Structure, Properties, Reactions, Occurrence and Uses; Wiley-VCH: Weinheim, Germany, 1996. [Google Scholar]
- Cornell, R.M. The influence of some divalent cations on the transformation of ferrihydrite to more crystalline products. Clay Miner. 1988, 23, 329–332. [Google Scholar] [CrossRef]
- Lenoble, V.; Bouras, O.; Deluchat, V.; Serpaud, B.; Bollinger, J.-C. Arsenic adsorption onto pillared clays and iron oxides. J. Colloid Interface Sci. 2002, 255, 52–58. [Google Scholar] [CrossRef]
- Aftabtalab, A.; Rinklebe, J.; Shaheen, S.M.; Niazi, N.K.; Moreno-Jiménez, E.; Schaller, J.; Knorr, K.-H. A review on the interactions of arsenic, iron (oxy)(hydr)oxides, and dissolved organic matter in soils, sediments, and groundwater in a ternary system. Chemosphere 2021, 286, 131790. [Google Scholar] [CrossRef]
- Bauer, M.; Blodau, C. Mobilization of arsenic by dissolved organic matter from iron oxides, soils and sediments. Sci. Total Environ. 2006, 354, 179–190. [Google Scholar] [CrossRef]
- Borch, T.; Kretzschmar, R.; Kappler, A.; van Cappellen, P.; Ginder-Vogel, M.; Voegelin, A.; Campbell, K. Biogeochemical Redox Processes and their Impact on Contaminant Dynamics. Environ. Sci. Technol. 2010, 44, 15–23. [Google Scholar] [CrossRef]
- Fendorf, S.; Michael, H.A.; Van Geen, A. Spatial and temporal variations of groundwater arsenic in South and Southeast Asia. Science 2010, 328, 1123–1127. [Google Scholar] [CrossRef]
- Mladenov, N.; Zheng, Y.; Miller, M.P.; Nemergut, D.R.; Legg, T.; Simone, B.; Hageman, C.; Rahman, M.M.; Ahmed, K.M.; McKnight, D.M. Dissolved organic matter sources and consequences for iron and arsenic mobilization in Bangladesh aquifers. Environ. Sci. Technol. 2010, 44, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Rinklebe, J.; Shaheen, S.M.; Yu, K. Release of As, Ba, Cd, Cu, Pb, and Sr under pre-definite redox conditions in different rice paddy soils originating from the U.S.A. and Asia. Geoderma 2016, 270, 21–32. [Google Scholar] [CrossRef]
- Sharma, P.; Ofner, J.; Kappler, A. Formation of binary and ternary colloids and dissolved complexes of organic matter, Fe and As. Environ. Sci. Technol. 2010, 44, 4479–4485. [Google Scholar] [CrossRef]
- Goldberg, S. Competitive adsorption of arsenate and arsenite on oxides and clay minerals Soil. Sci. Soc. Am. J. 2002, 66, 413–421. [Google Scholar] [CrossRef]
- Shaheen, S.M.; Rinklebe, J.; Frohne, T.; White, J.R.; DeLaune, R.D. Redox effects on release kinetics of arsenic, cadmium, cobalt, and vanadium in Wax Lake Deltaic freshwater marsh soils. Chemosphere 2016, 150, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Mejia, J.; He, S.; Yang, Y.; Ginder-Vogel, M.; Roden, E.E. Dual Role of Humic Substances As Electron Donor and Shuttle for Dissimilatory Iron Reduction. Environ. Sci. Technol. 2018, 52, 5691–5699. [Google Scholar] [CrossRef]
- Verbeeck, M.; Thiry, Y.; Smolders, E. Soil organic matter affects arsenic and antimony sorption in anaerobic soils. Environ. Pollut. 2020, 257, 113566. [Google Scholar] [CrossRef]
- Kappler, A.; Benz, M.; Schink, B.; Brune, A. Electron shuttling via humic acids in microbial iron(III) reduction in a freshwater sediment. FEMS Microbiol. Ecol. 2004, 47, 85–92. [Google Scholar] [CrossRef]
- Aeschbacher, M.; Vergari, D.; Schwarzenbach, R.; Sander, M. Electrochemical analysis of proton and electron transfer equilibria of the reducible moieties in humic acids. Environ. Sci. Technol. 2011, 45, 8385–8394. [Google Scholar] [CrossRef]
- Yang, C.; HE, X.-S.; Xi, B.-D.; Huang, C.-H.; Cui, D.-Y.; Gao, R.-T.; Tan, W.-B.; Zhang, H. Characteristic Study of Dissolved Organic Matter for Electron Transfer Capacity during Initial Landfill Stage. Chin. J. Anal. Chem. 2016, 44, 1568–1574. [Google Scholar] [CrossRef]
- Thanabalasingam, P.; Pickering, W.F. Arsenic sorption by humic acids. Environ. Pollut. Ser. B Chem. Phys. 1986, 12, 233–246. [Google Scholar] [CrossRef]
- Xu, H.; Allard, B.; Grimvall, A. Influence of pH and organic substance on the adsorption of As(V) on geologic materials. Water Air Soil Pollut. 1988, 40, 293–305. [Google Scholar] [CrossRef]
- Wu, X.; Bowers, B.; Kim, D.; Lee, B.; Jun, Y.-S. Dissolved Organic Matter Affects Arsenic Mobility and Iron(III) (hydr)oxide Formation: Implications for Managed Aquifer Recharge. Environ. Sci. Technol. 2019, 53, 14357–14367. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Korfiatis, G.P.; Bang, S.; Bang, K.W. Combined effects of anions on arsenic removal by iron hydroxides. Toxicol. Lett. 2002, 133, 103–111. [Google Scholar] [CrossRef]
- Gao, X.; Root, R.A.; Farrell, J.; Ela, W.; Chorover, J. Effect of silicic acid on arsenate and arsenite retention mechanisms on 6-L ferrihydrite: A spectroscopic and batch adsorption approach. Appl. Geochem. 2013, 38, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Youngran, J.; Fan, M.; Van Leeuwen, J.; Belczyk, J.F. Effect of competing solutes on arsenic(V) adsorption using iron and aluminum oxides. J. Environ. Sci. 2007, 19, 910–919. [Google Scholar] [CrossRef]
- Wang, S.; Mulligan, C.N. Effect of natural organic matter on arsenic release from soils and sediments into groundwater. Environ. Geochem. Health 2006, 28, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Rutten, S.; Eikelboom, M.; Waal, L.D.; Bruning, H.; Bhattacharya, P.; Van der Wal, A. Impact of phosphate, silicate and natural organic matter on the size of Fe(III) precipitates and arsenate co-precipitation efficiency in calcium containing water. Sep. Purif. Technol. 2020, 235, 116117. [Google Scholar] [CrossRef]
- Wu, X.; Burnell, S.; Neil, C.W.; Kim, D.; Zhang, L.; Jung, H.; Jun, Y.-S. Effects of Phosphate, Silicate, and Bicarbonate on Arsenopyrite Dissolution and Secondary Mineral Precipitation. ACS Earth Space Chem. 2020, 4, 515–525. [Google Scholar] [CrossRef]
- Agethen, S.; Sander, M.; Waldemer, C.; Knorr, K.-H. Plant rhizosphere oxidation reduces methane production and emission in rewetted peatlands. Soil Biol. Biochem. 2018, 125, 125–135. [Google Scholar] [CrossRef]
- Schwertmann, U.; Cornell, R.M. Iron Oxides in the Laboratory: Preparation and Characterization; Wiley-VCH: Weinheim, Germany, 1991; Volume 11, p. 137S. [Google Scholar]
- Giménez, M.; Martínez, J.; De Pablo, M.; Rovira, L.D. Arsenic sorption onto natural hematite, magnetite, and goethite. J. Hazard. Mater. 2007, 141, 575–580. [Google Scholar] [CrossRef]
- Dixit, S.; Hering, J.G. Comparison of Arsenic(V) and Arsenic(III) Sorption onto Iron Oxide Minerals: Implications for Arsenic Mobility. Environ. Sci. Technol. 2003, 37, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jimenez, E.; Beesley, L.; Lepp, N.W.; Dickinson, N.M.; Hartley, W.; Clemente, R. Field sampling of soil pore water to evaluate trace element mobility and associated environmental risk. Environ. Pollut. 2011, 159, 3078–3085. [Google Scholar] [CrossRef] [PubMed]
- Alava, P.; Tack, F.; Du Laing, G.; Van Wiele, T. HPLC-ICP-MS method development to monitor arsenic speciation changes by human gut microbiota. Biomed. Chromatogr. BMC 2012, 26, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Riley, J.P.A. Modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta 1962, 27, 31–36. [Google Scholar] [CrossRef]
- Tamura, H.; Goto, K.; Yotsuyanagi, T.; Nagayama, M. Spectrophotometric determination of iron(II) with 1,10-phenanthroline in the presence of large amounts of iron(III). Talenta 1974, 21, 314–318. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Guidelines for Drinking Water Quality, 2nd ed.; WHO: Geneva, Switzerland, 1993; Volume 1. [Google Scholar]
- DeVore, C.L.; Rodriguez-Freire, L.; Mehdi-Ali, A.; Ducheneaux, C.; Artyushkova, K.; Zhou, Z.; Latta, D.E.; Lueth, V.W.; Gonzales, M.; Lewis, J.; et al. Effect of bicarbonate and phosphate on arsenic release from mining-impacted sediments in the Cheyenne River watershed, South Dakota, USA. Environ. Sci. Processes Impacts 2019, 21, 456–468. [Google Scholar] [CrossRef]
- Seyfferth, L.A.; Fendorf, S. Silicate mineral impacts on the uptake and storage of arsenic and plant nutrients in rice (Oryza sativa L.). Environ. Sci. Technol. 2012, 46, 13176–13183. [Google Scholar] [CrossRef]
- Icopini, G.A.; Brantley, S.L.; Heaney, P.J. Kinetics of silica oligomerization and nanocolloid formation as a function of pH andionic strength at 25 °C. Geochim. Cosmochim. Acta 2005, 69, 293–303. [Google Scholar] [CrossRef]
- Schaller, J.; Puppe, D.; Kaczorek, D.; Ellerbrock, R.; Sommer, M. Silicon Cycling in Soils Revisited. Plants 2021, 10, 295. [Google Scholar] [CrossRef]
- Schaller, J.; Fauchere, S.; Joss, H.; Obst, M.; Goeckede, M.; Planer-Friedrich, B.; Peiffer, S.; Gilfedder, B.; Elberling, B. Silicon increases the phosphorus availability of Arctic soils. Sci. Rep. 2019, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Swedlund, P.J.; Webster, J.G. Adsorption and polymerisation of silicic acid on ferrihydrite, and its effect on arsenic adsorption. Water Res. 1999, 33, 3413–3422. [Google Scholar] [CrossRef]
- Christl, I.; Brechbühl, Y.; Graf, M.; Kretzschmar, R. Polymerization of silicate on hematite surfaces and its influence on arsenic sorption. Environ. Sci. Technol. 2012, 46, 13235–13243. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jimenez, E.; Esteban, E.; Peñalosa, J.M. The fate of arsenic in soil-plant systems. In Reviews of Environmental Contamination and Toxicology; Springer: New York, NY, USA, 2012; pp. 1–37. [Google Scholar]
- Bowell, R.J. Sorption of arsenic by iron oxides and oxyhydroxides in soils. J. Appl. Geochem. 1994, 9, 279–286. [Google Scholar] [CrossRef]
- Manning, B.A.; Goldberg, S. Modeling Competitive Adsorption of Arsenate with Phosphate and Molybdate on Oxide Minerals. Soil Sci. Soc. Am. J. 1996, 60, 121. [Google Scholar] [CrossRef]
- Hongshao, Z.; Stanforth, R. Competitive Adsorption of Phosphate and Arsenate on Goethite. Environ. Sci. Technol. 2001, 35, 4753–4757. [Google Scholar] [CrossRef] [PubMed]
- Reithmaier, G.-M.S.; Knorr, K.-H.; Arnhold, S.; Planer-Friedrich, B.; Schaller, J. Enhanced silicon availability leads to increased methane production, nutrient and toxicant mobility in peatlands. Sci. Rep. 2017, 7, 8728. [Google Scholar] [CrossRef] [PubMed]
- Mamindy-Pajany, Y.; Hurel, C.; Marmier, N.; Roméo, M. Arsenic adsorption onto hematite and goethite. Comptes Rendus Chim. 2009, 12, 876–881. [Google Scholar] [CrossRef]
- Pierce, M.L.; Moore, C.B. Adsorption of arsenite and arsenate on amorphous iron hydroxide. Water Res. 1982, 16, 1247–1253. [Google Scholar] [CrossRef]
- Singh, P.; Zhang, W.; Muir, D.M.; Robins, R.G. The effect of silicate on the adsorption of arsenate on coprecipitated ferrihydrite. Arsenic Metallurgy. In Proceedings of the Symposium Held During the TMS Annual Meeting, San Francisco, CA, USA, 13–17 February 2005; pp. 129–135. [Google Scholar]
- Klotzbücher, T.; Treptow, C.; Kaiser, K.; Klotzbücher, A.; Mikutta, R. Sorption competition with natural organic matter as mechanism controlling silicon mobility in soil. Sci. Rep. 2020, 10, 11225. [Google Scholar] [CrossRef]
- Chen, X.; Zeng, X.-C.; Wang, J.; Deng, Y.; Ma, T.; Guoji, E.; Mu, Y.; Yang, Y.; Li, H.; Wang, X. Microbial communities involved in arsenic mobilization and release from the deep sediments into groundwater in Jianghan plain, Central China. Sci. Total Environ. 2017, 579, 989–999. [Google Scholar] [CrossRef]
- El-Naggar, A.; Lee, M.-H.; Hur, J.; Lee, Y.H.; Igalavithana, A.D.; Shaheen, S.M.; Ryu, C.; Rinklebe, J.; Tsang, D.C.W.; Ok, Y.S. Biochar-induced metal immobilization and soil biogeochemical process: An integrated mechanistic approach. Sci. Total Environ. 2020, 698, 134112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.P.; DeLaune, R.D.; Patrick, W.H.; Masscheleyn, P.H. Soil Redox and pH Effects on Methane Production in a Flooded Rice Soil. Soil Sci. Soc. Am. J. 1993, 57, 382–385. [Google Scholar] [CrossRef]
- Le Mer, J.; Roger, P. Production, oxidation, emission and consumption of methane by soils: A review. Eur. J. Soil Biol. 2001, 37, 25–50. [Google Scholar] [CrossRef]
- Thomas, M.A. The Association of Arsenic with Redox Conditions, Depth and Ground-Water Age in the Glacial Aquifer System; Science Investigation Report 2007-5036; U.S. Geological Survey: Reston, VA, USA, 2007. [Google Scholar]
- Sorg, T.J.; Chen, A.S.C.; Wang, L. Arsenic species in drinking water wells in the USA with high arsenic concentrations. Water Res. 2014, 48, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Frohne, T.; Rinklebe, J.; Diaz-Bone, R.A.; Du Laing, G. Controlled variation of redox conditions in a floodplain soil: Impact on metal mobilization and biomethylation of arsenic and antimony. Geoderma 2011, 160, 414–424. [Google Scholar] [CrossRef]
- Frohne, T.; Rinklebe, J.; Diaz-Bone, R.A. Contamination of Floodplain Soils along the Wupper River, Germany, with As, Co, Cu, Ni, Sb, and Zn and the Impact of Pre-definite Redox Variations on the Mobility of These Elements. Soil Sediment Contam. Int. J. 2014, 23, 779–799. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Xia, D.; Jiang, X.; Fu, D.; Shen, L.; Wang, H.; Li, Q.B. Enhanced bioreduction of iron and arsenic in sediment by biochar amendment influencing microbial community composition and dissolved organic matter content and composition. J. Hazard. Mater. 2016, 311, 20–29. [Google Scholar] [CrossRef]
- Scott, M.J.; Morgan, J.J. Reactions at Oxide Surfaces. 1. Oxidation of As(III) by Synthetic Birnessite. Environ. Sci. Technol. 1995, 29, 1898–1905. [Google Scholar] [CrossRef]
- Klüpfel, L.; Piepenbrock, A.; Kappler, A.; Sander, M. Humic substances as fully regenerable electron acceptors in recurrently anoxic environments. Nat. Geosci. 2014, 7, 195–200. [Google Scholar] [CrossRef]
- Shaheen, S.M.; El-Naggar, A.; Antoniadis, V.; Moghanm, F.; Zhang, Z.; Tsang, D.; Ok, Y.S.; Rinklebe, J. Release of toxic elements in fishpond sediments under dynamic redox conditions: Assessing the potential environmental risk for a safe management of fisheries systems and degraded waterlogged sediments. J. Environ. Manag. 2020, 255, 109778. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, C.; Wu, J.; Liu, X.; Xu, J. Impact of organic matter addition on pH change of paddy soils. J. Soils Sediments 2012, 13, 12–23. [Google Scholar] [CrossRef]
- McArthur, J.; Ghosal, U.; Sikdar, P.; Ball, J. Arsenic in Groundwater: The Deep Late Pleistocene Aquifers of the Western Bengal Basin. Environ. Sci. Technol. 2016, 50, 3469–3476. [Google Scholar] [CrossRef] [PubMed]
- Gustave, W.; Yuan, Z.-F.; Sekar, R.; Ren, Y.-X.; Liu, J.-Y.; Zhang, J.; Chen, Z. Soil organic matter amount determines the behavior of iron and arsenic in paddy soil with microbial fuel cells. Chemosphere 2019, 237, 124459. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.M.; Nordstrom, D.K. Arsenic speciation and sorption in natural environments. Rev. Mineral. Geochem. 2014, 79, 185–216. [Google Scholar] [CrossRef]
- Keller, N.S.; Stefánsson, A.; Sigfússon, B. Arsenic speciation in natural sulfidic geothermal waters. Geochim. Cosmochim. Acta 2014, 142, 15–26. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aftabtalab, A.; Moreno-Jiménez, E.; Henschel, J.; Nowak, S.; Schaller, J.; Knorr, K.-H. The Impact of Dissolved Organic Matter on Arsenic Mobilization from Goethite in the Presence of Silicic Acid and Phosphate under Reducing Conditions. Water 2022, 14, 2975. https://doi.org/10.3390/w14192975
Aftabtalab A, Moreno-Jiménez E, Henschel J, Nowak S, Schaller J, Knorr K-H. The Impact of Dissolved Organic Matter on Arsenic Mobilization from Goethite in the Presence of Silicic Acid and Phosphate under Reducing Conditions. Water. 2022; 14(19):2975. https://doi.org/10.3390/w14192975
Chicago/Turabian StyleAftabtalab, Adeleh, Eduardo Moreno-Jiménez, Jonas Henschel, Sascha Nowak, Jörg Schaller, and Klaus-Holger Knorr. 2022. "The Impact of Dissolved Organic Matter on Arsenic Mobilization from Goethite in the Presence of Silicic Acid and Phosphate under Reducing Conditions" Water 14, no. 19: 2975. https://doi.org/10.3390/w14192975
APA StyleAftabtalab, A., Moreno-Jiménez, E., Henschel, J., Nowak, S., Schaller, J., & Knorr, K.-H. (2022). The Impact of Dissolved Organic Matter on Arsenic Mobilization from Goethite in the Presence of Silicic Acid and Phosphate under Reducing Conditions. Water, 14(19), 2975. https://doi.org/10.3390/w14192975