
MinION Nanopore Sequencing Accelerates Progress towards Ubiquitous Genetics in Water Research

 , , , , ,
, , , , ,
Abstract
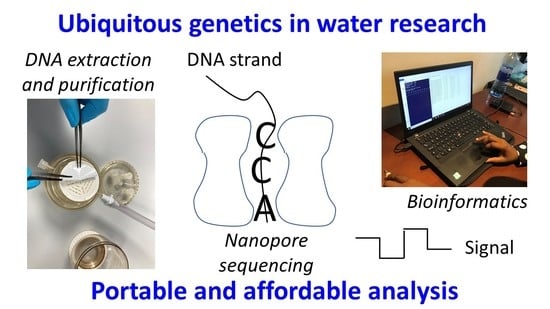
:
1. Introduction

2. Methodology
3. Literature Review
3.1. Trends in MinION Applications in Water Research
3.2. Sample Preparation
3.2.1. Biomass Collection and Concentration
3.2.2. Extraction and Quantification of Genetic Material
3.2.3. Sequencing Library Preparation
Shotgun versus Amplicon Sequencing
End Repair and Ligation of Sequencing Adaptors
Amplification of Genes of Interest
Strategies for Obtaining Long-Read Fragments of Genetic Material
Reverse Transcription of RNA into cDNA
Ligation of Hairpin Adaptors
Multiplexing of Samples
3.2.4. Portability of Sample Preparation Methods
3.3. Sequencing
3.4. Bioinformatics
3.4.1. Basecalling
3.4.2. Demultiplexing and Adaptor Trimming
3.4.3. Sequencing Data Visualization and Quality Control
3.4.4. Biological Interpretation
EPI2ME
Taxonomic Classification
Functional Analysis
Consensus Sequences and Genome Assembly
3.5. Data Visualization and Statistical Analysis
3.6. Data Management
3.7. Quality Control
3.7.1. Blank Samples
3.7.2. Known Samples
3.7.3. Comparison with Other Methods
Other Sequencing Technologies
Complementary Biology Methods
3.7.4. Ethical Considerations
4. Conclusions
- Currently, the MinION is the only low-cost and miniaturized sequencer meeting these two basic requirements for “ubiquitous genetics”.
- Our review supports the utility of the MinION for water research, as evidenced by the diversity of samples analyzed, the variety of research remits, and use for research in countries that lack universal access to safe water and sanitation.
- Despite its fabled portability, most studies used the MinION in a conventional laboratory setting. Nonetheless, a few studies demonstrated fully portable workflows by using the MinION onboard a diving vessel, an ocean-going research ship, and at sewage treatment works.
- Lower nanopore sequencing read accuracy as compared to other platforms still hinders MinION applications beyond research, but such limitations may be overcome with the latest updates to the MinION flow cells and sequencing chemistry. Regardless, the inclusion of positive and negative controls should become standard practice in MinION applications.
- ONT’s EPI2ME platform is a major step towards user-friendly bioinformatics, but a lack of consensus regarding the most appropriate bioinformatic pipeline for water research currently hinders the “democratization of sequencing” and intercomparison of study results.
- A lack of regulatory standards based on the analysis of genetic material in water samples is a “ubiquitous genetics” challenge that the MinION shares with other molecular microbiology methods.
- If next-generation sequencing is to be practiced more widely, the bioinformatic processing and storage of such huge data sets will create enormous IT resource demands with economic and environmental implications.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, S.; Yan, Z.; Hänfling, B.; Zheng, X.; Wang, P.; Fan, J.; Li, J. Methodology of fish eDNA and its applications in ecology and environment. Sci. Total Environ. 2021, 755, 142622. [Google Scholar] [CrossRef] [PubMed]
- Douterelo, I.; Boxall, J.B.; Deines, P.; Sekar, R.; Fish, K.E.; Biggs, C.A. Methodological approaches for studying the microbial ecology of drinking water distribution systems. Water Res. 2014, 65, 134–156. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Davis, B.C.; Milligan, E.; Blair, M.F.; Keenum, I.; Maile-Moskowitz, A.; Pan, J.; Gnegy, M.; Liguori, K.; Gupta, S.; et al. Next generation sequencing approaches to evaluate water and wastewater quality. Water Res. 2021, 194, 116907. [Google Scholar] [CrossRef]
- Mthethwa, N.; Amoah, I.; Reddy, P.; Bux, F.; Kumari, S. A review on application of next-generation sequencing methods for profiling of protozoan parasites in water: Current methodologies, challenges, and perspectives. J. Microbiol. Methods 2021, 187, 106269. [Google Scholar] [CrossRef]
- Wong, K.; Fong, T.-T.; Bibby, K.; Molina, M. Application of enteric viruses for fecal pollution source tracking in environmental waters. Environ. Int. 2012, 45, 151–164. [Google Scholar] [CrossRef]
- Bouhajja, E.; Agathos, S.; George, I.F. Metagenomics: Probing pollutant fate in natural and engineered ecosystems. Biotechnol. Adv. 2016, 34, 1413–1426. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Advent of a new sequencing era: Long-read and on-site sequencing. J. Hum. Genet. 2020, 65, 1. [Google Scholar] [CrossRef]
- Erlich, Y. A vision for ubiquitous sequencing. Genome Res. 2015, 25, 1411–1416. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Mikheyev, A.; Tin, M.M.Y. A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Calus, S.T.; Ijaz, U.Z.; Pinto, A.J. NanoAmpli-Seq: A workflow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform. GigaScience 2018, 7, giy140. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Tyson, J.R.; Loose, M.; Ip, C.L.; Eccles, D.A.; O’Grady, J.; Malla, S.; Leggett, R.M.; Wallerman, O.; Jansen, H.J.; et al. MinION Analysis and Reference Consortium: Phase 2 data release and analysis of R9.0 chemistry. F1000Research 2017, 6, 760. [Google Scholar] [CrossRef]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 long-read sequencing enables near-perfect bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. bioRxiv 2021. [Google Scholar]
- de Lannoy, C.; de Ridder, D.; Risse, J. A sequencer coming of age: De novo genome assembly using MinION reads. F1000Research 2017, 6, 1083. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef]
- Plesivkova, D.; Richards, R.; Harbison, S. A review of the potential of the MinION™ single-molecule sequencing system for forensic applications. WIREs Forensic Sci. 2019, 1, e1323. [Google Scholar] [CrossRef]
- Preul, M.C.; Patel, A.; Belykh, E.; Miller, E.J.; George, L.L.; Martirosyan, N.L.; Byvaltsev, V.A. MinION rapid sequencing: Review of potential applications in neurosurgery. Surg. Neurol. Int. 2018, 9, 157. [Google Scholar] [CrossRef]
- Pavlovic, J.; Cavalieri, D.; Mastromei, G.; Pangallo, D.; Perito, B.; Marvasi, M. MinION technology for microbiome sequencing applications for the conservation of cultural heritage. Microbiol. Res. 2021, 247, 126727. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.C.; Penrice-Randal, R.; Alruwaili, M.; Dong, X.; Pullan, S.T.; Carter, D.P.; Bewley, K.; Zhao, Q.; Sun, Y.; Hartley, C.; et al. Amplicon based MinION sequencing of SARS-CoV-2 and metagenomic characterisation of nasopharyngeal swabs from patients with COVID-19. medRxiv 2020. [Google Scholar]
- Pater, A.A.; Bosmeny, M.S.; White, A.A.; Sylvain, R.J.; Eddington, S.B.; Parasrampuria, M.; Ovington, K.N.; Metz, P.E.; Yinusa, A.O.; Barkau, C.L.; et al. High throughput nanopore sequencing of SARS-CoV-2 viral genomes from patient samples. J. Biol. Methods 2021, 8, e155. [Google Scholar] [PubMed]
- Sample, I. Handheld DNA reader revolutionary and democratising, say scientists. In The Guardian; Guardian News & Media: London, UK, 2015. [Google Scholar]
- CDC. Global Water, Sanitation, & Hygiene (WASH). 2021. Available online: https://www.cdc.gov/healthywater/global/wash_statistics.html#:~:text=An%20estimated%202.2%20billion%20people,access%20to%20basic%20handwashing%20facilities (accessed on 15 June 2022).
- UN. Goal 6: Ensure Access to Water and Sanitation for All. 2020. Available online: https://www.un.org/sustainabledevelopment/water-and-sanitation/ (accessed on 30 March 2020).
- Acharya, K.; Blackburn, A.; Mohammed, J.; Haile, A.T.; Hiruy, A.M.; Werner, D. Metagenomic water quality monitoring with a portable laboratory. Water Res. 2020, 184, 116112. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.; Matejusova, I.; Nguyen, L.; Ruane, N.; Falk, K.; MacQueen, D.J. Nanopore sequencing for rapid diagnostics of salmonid RNA viruses. Sci. Rep. 2018, 8, 16307. [Google Scholar] [CrossRef] [PubMed]
- Morales-Rivera, M.F.; Valenzuela-Miranda, D.; Valenzuela-Muñoz, V.; Nuñez-Acuña, G.; Avendaño-Herrera, R.; Gallardo-Escárate, C. Nanopore sequencing evidenced the presence of fish bacterial pathogens in the sea louse (Caligus rogercresseyi) microbiota collected from distant salmon farms in Chile. Aquaculture 2022, 552, 738026. [Google Scholar] [CrossRef]
- Sauvage, T.; Schmidt, W.E.; Yoon, H.S.; Paul, V.J.; Fredericq, S. Promising prospects of nanopore sequencing for algal hologenomics and structural variation discovery. BMC Genom. 2019, 20, 850. [Google Scholar] [CrossRef]
- Chang, J.J.M.; Ip, Y.C.A.; Bauman, A.G.; Huang, D. MinION-in-ARMS: Nanopore Sequencing to Expedite Barcoding of Specimen-Rich Macrofaunal Samples From Autonomous Reef Monitoring Structures. Front. Mar. Sci. 2020, 7, 448. [Google Scholar] [CrossRef]
- Chang, J.J.M.; Ip, Y.C.A.; Ng, C.S.L.; Huang, D. Takeaways from Mobile DNA Barcoding with BentoLab and MinION. Genes 2020, 11, 1121. [Google Scholar] [CrossRef]
- Semmouri, I.; De Schamphelaere, K.A.; Mees, J.; Janssen, C.R.; Asselman, J. Evaluating the potential of direct RNA nanopore sequencing: Metatranscriptomics highlights possible seasonal differences in a marine pelagic crustacean zooplankton community. Mar. Environ. Res. 2019, 153, 104836. [Google Scholar] [CrossRef]
- Knot, I.E.; Zouganelis, G.D.; Weedall, G.D.; Wich, S.A.; Rae, R. DNA Barcoding of Nematodes Using the MinION. Front. Ecol. Evol. 2020, 8, 100. [Google Scholar] [CrossRef]
- Freitag, H.; de Vries, R.; Paterno, M.; Maestri, S.; Delledonne, M.; Thompson, C.; Lamed, H.; Lambert, R.; Fox, M.; Gonzalez, M.; et al. Hydraena (s.str.) dinarica, new species (Coleoptera: Hydraenidae) along with further records of Hydraena spp. from Durmitor National Park, Montenegro and comments on the DNA barcoding problem with the genus. Biodivers. Data J. 2021, 9, e59892. [Google Scholar] [CrossRef] [PubMed]
- Semmouri, I.; Schamphelaere, K.A.C.D.; Willemse, S.; Vandegehuchte, M.B.; Janssen, C.R.; Asselman, J. Metabarcoding reveals hidden species and improves identification of marine zooplankton communities in the North Sea. ICES J. Mar. Sci. 2021, 78, 3411–3427. [Google Scholar] [CrossRef]
- Reddington, K.; Eccles, D.; O’Grady, J.; Drown, D.M.; Hansen, L.H.; Nielsen, T.K.; Ducluzeau, A.; Leggett, R.M.; Heavens, D.; Peel, N.; et al. Metagenomic analysis of planktonic riverine microbial consortia using nanopore sequencing reveals insight into river microbe taxonomy and function. Gigascience 2020, 9, giaa053. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Escárate, C.; Valenzuela-Muñoz, V.; Núñez-Acuña, G.; Valenzuela-Miranda, D.; Benaventel, B.P.; Sáez-Vera, C.; Urrutia, H.; Novoa, B.; Figueras, A.; Roberts, S.; et al. The wastewater microbiome: A novel insight for COVID-19 surveillance. Sci. Total Environ. 2020, 764, 142867. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Solonenko, N.; Moore, K.; Chittick, L.; Gregory, A.C.; Allen, M.J.; Sullivan, M.B.; Temperton, B. Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 2019, 7, e6800. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Aw, T.G.; Van Bonn, W.; Rose, J.B. Evaluation of a portable nanopore-based sequencer for detection of viruses in water. J. Virol. Methods 2019, 278, 113805. [Google Scholar] [CrossRef]
- Swift, C.L.; Isanovic, M.; Velez KE, C.; Norman, R.S. Community-level SARS-CoV-2 sequence diversity revealed by wastewater sampling. Sci. Total Environ. 2021, 801, 149691. [Google Scholar] [CrossRef]
- Dharmadhikari, T.; Rajput, V.; Yadav, R.; Boargaonkar, R.; Patil, D.; Kale, S.; Kamble, S.P.; Dastager, S.G.; Dharne, M.S. High throughput sequencing based direct detection of SARS-CoV-2 fragments in wastewater of Pune, West India. Sci. Total Environ. 2022, 807, 151038. [Google Scholar] [CrossRef]
- Hamner, S.; Brown, B.L.; Hasan, N.A.; Franklin, M.J.; Doyle, J.; Eggers, M.J.; Colwell, R.R.; Ford, T.E. Metagenomic Profiling of Microbial Pathogens in the Little Bighorn River, Montana. Int. J. Environ. Res. Public Health 2019, 16, 1097. [Google Scholar] [CrossRef]
- Davidov, K.; Iankelevich-Kounio, E.; Yakovenko, I.; Koucherov, Y.; Rubin-Blum, M.; Oren, M. Identification of plastic-associated species in the Mediterranean Sea using DNA metabarcoding with Nanopore MinION. Sci. Rep. 2020, 10, 17533. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, M.; Yadav, R.; Rajput, V.; Dharne, M.S.; Rastogi, G. Metagenomic analysis reveals genetic insights on biogeochemical cycling, xenobiotic degradation, and stress resistance in mudflat microbiome. J. Environ. Manag. 2021, 292, 112738. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, S.; Singh, D. Metagenomic insights into Himalayan glacial and kettle lake sediments revealed microbial community structure, function, and stress adaptation strategies. Extremophiles 2021, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.O.O.; Ndegwa, N.; Alneberg, J.; Johansson, S.; Logue, J.B.; Huss, M.; Käller, M.; Lundeberg, J.; Fagerberg, J.; Andersson, A.F. Stationary and portable sequencing-based approaches for tracing wastewater contamination in urban stormwater systems. Sci. Rep. 2018, 8, 11907. [Google Scholar] [CrossRef]
- Curren, E.; Yoshida, T.; Kuwahara, V.S.; Leong, S.C.Y. Rapid profiling of tropical marine cyanobacterial communities. Reg. Stud. Mar. Sci. 2018, 25, 100485. [Google Scholar] [CrossRef]
- Samson, R.; Shah, M.; Yadav, R.; Sarode, P.; Rajput, V.; Dastager, S.G.; Dharne, M.S.; Khairnar, K. Metagenomic insights to understand transient influence of Yamuna River on taxonomic and functional aspects of bacterial and archaeal communities of River Ganges. Sci. Total Environ. 2019, 674, 288–299. [Google Scholar] [CrossRef]
- Acharya, K.; Khanal, S.; Pantha, K.; Amatya, N.; Davenport, R.J.; Werner, D. A comparative assessment of conventional and molecular methods, including MinION nanopore sequencing, for surveying water quality. Sci. Rep. 2019, 9, 15726. [Google Scholar] [CrossRef]
- Andersen, M.H.; McIlroy, S.J.; Nierychlo, M.; Nielsen, P.H.; Albertsen, M. Genomic insights into Candidatus Amarolinea aalborgensis gen. nov., sp. nov., associated with settleability problems in wastewater treatment plants. Syst. Appl. Microbiol. 2018, 42, 77–84. [Google Scholar] [CrossRef]
- Che, Y.; Xia, Y.; Liu, L.; Li, A.-D.; Yang, Y.; Zhang, T. Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome 2019, 7, 44. [Google Scholar] [CrossRef]
- Chakraborty, J.; Sapkale, V.; Rajput, V.; Shah, M.; Kamble, S.; Dharne, M. Shotgun metagenome guided exploration of anthropogenically driven resistomic hotspots within Lonar soda lake of India. Ecotoxicol. Environ. Saf. 2020, 194, 110443. [Google Scholar] [CrossRef]
- Acharya, K.; Halla, F.F.; Massawa, S.M.; Mgana, S.M.; Komar, T.; Davenport, R.J.; Werner, D. Chlorination effects on DNA based characterization of water microbiomes and implications for the interpretation of data from disinfected systems. J. Environ. Manag. 2020, 276, 111319. [Google Scholar] [CrossRef]
- Poghosyan, L.; Koch, H.; Frank, J.; van Kessel, M.A.; Cremers, G.; van Alen, T.; Jetten, M.S.; Camp, H.J.O.D.; Lücker, S. Metagenomic profiling of ammonia- and methane-oxidizing microorganisms in two sequential rapid sand filters. Water Res. 2020, 185, 116288. [Google Scholar] [CrossRef] [PubMed]
- Ress, J.; Monrrabal, G.; Díaz, A.; Pérez-Pérez, J.; Bastidas, J. Microbiologically influenced corrosion of welded AISI 304 stainless steel pipe in well water. Eng. Fail. Anal. 2020, 116, 104734. [Google Scholar] [CrossRef]
- Chakraborty, J.; Rajput, V.; Sapkale, V.; Kamble, S.; Dharne, M. Spatio-temporal resolution of taxonomic and functional microbiome of Lonar soda lake of India reveals metabolic potential for bioremediation. Chemosphere 2020, 264, 128574. [Google Scholar] [CrossRef] [PubMed]
- Urban, L.; Holzer, A.; Baronas, J.J.; Hall, M.B.; Braeuninger-Weimer, P.; Scherm, M.J.; Kunz, D.J.; Perera, S.N.; Martin-Herranz, D.E.; Tipper, E.T.; et al. Freshwater monitoring by nanopore sequencing. eLife 2021, 10, e61504. [Google Scholar] [CrossRef] [PubMed]
- van der Loos, L.M.; D’hondt, S.; Willems, A.; De Clerck, O. Characterizing algal microbiomes using long-read nanopore sequencing. Algal Res. 2021, 59, 102456. [Google Scholar]
- Yadav, R.; Rajput, V.; Dharne, M. Functional metagenomic landscape of polluted river reveals potential genes involved in degradation of xenobiotic pollutants. Environ. Res. 2020, 192, 110332. [Google Scholar] [CrossRef]
- Yadav, R.; Rajput, V.; Dharne, M. Metagenomic analysis of a mega-city river network reveals microbial compositional heterogeneity among urban and peri-urban river stretch. Sci. Total Environ. 2021, 783, 146960. [Google Scholar] [CrossRef]
- Arumugam, K.; Bessarab, I.; Haryono, M.A.S.; Liu, X.; Zuniga–Montanez, R.E.; Roy, S.; Qiu, G.; Drautz–Moses, D.I.; Law, Y.Y.; Wuertz, S.; et al. Recovery of complete genomes and non-chromosomal replicons from activated sludge enrichment microbial communities with long read metagenome sequencing. NPJ Biofilms Microbiomes 2021, 7, 23. [Google Scholar] [CrossRef]
- Ho, J.Y.; Jong, M.-C.; Acharya, K.; Liew, S.S.X.; Smith, D.R.; Noor, Z.Z.; Goodson, M.L.; Werner, D.; Graham, D.W.; Eswaran, J. Multidrug-resistant bacteria and microbial communities in a river estuary with fragmented suburban waste management. J. Hazard. Mater. 2020, 405, 124687. [Google Scholar] [CrossRef]
- Martin, C.; Stebbins, B.; Ajmani, A.; Comendul, A.; Hamner, S.; Hasan, N.A.; Colwell, R.; Ford, T. Nanopore-based metagenomics analysis reveals prevalence of mobile antibiotic and heavy metal resistome in wastewater. Ecotoxicology 2021, 30, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Mirasbekov, Y.; Abdimanova, A.; Sarkytbayev, K.; Samarkhanov, K.; Abilkas, A.; Potashnikova, D.; Arbuz, G.; Issayev, Z.; Vorobjev, I.A.; Malashenkov, D.V.; et al. Combining Imaging Flow Cytometry and Molecular Biological Methods to Reveal Presence of Potentially Toxic Algae at the Ural River in Kazakhstan. Front. Mar. Sci. 2021, 8, 680482. [Google Scholar] [CrossRef]
- Pantha, K.; Acharya, K.; Mohapatra, S.; Khanal, S.; Amatya, N.; Ospina-Betancourth, C.; Butte, G.; Shrestha, S.D.; Rajbhandari, P.; Werner, D. Faecal pollution source tracking in the holy Bagmati River by portable 16S rRNA gene sequencing. NPJ Clean Water 2021, 4, 12. [Google Scholar] [CrossRef]
- Rajput, V.; Yadav, R.; Dharne, M.S. Metagenomic exploration reveals a differential patterning of antibiotic resistance genes in urban and peri-urban stretches of a riverine system. Environ. Sci. Pollut. Res. 2021, 28, 66477–66484. [Google Scholar] [CrossRef] [PubMed]
- Thongsamer, T.; Neamchan, R.; Blackburn, A.; Acharya, K.; Sutheeworapong, S.; Tirachulee, B.; Pattanachan, P.; Vinitnantharat, S.; Zhou, X.-Y.; Su, J.-Q.; et al. Environmental antimicrobial resistance is associated with faecal pollution in Central Thailand’s coastal aquaculture region. J. Hazard. Mater. 2021, 416, 125718. [Google Scholar] [CrossRef]
- An, X.-L.; Abass, O.K.; Zhao, C.-X.; Xu, M.-R.; Pan, T.; Pu, Q.; Liao, H.; Li, H.; Zhu, Y.-G.; Su, J.-Q. Nanopore sequencing analysis of integron gene cassettes in sewages and soils. Sci. Total Environ. 2022, 817, 152766. [Google Scholar] [CrossRef]
- Dai, D.; Brown, C.; Bürgmann, H.; Larsson, D.G.J.; Nambi, I.; Zhang, T.; Flach, C.-F.; Pruden, A.; Vikesland, P.J. Long-read metagenomic sequencing reveals shifts in associations of antibiotic resistance genes with mobile genetic elements from sewage to activated sludge. Microbiome 2022, 10, 20. [Google Scholar] [CrossRef]
- Hiruy, A.M.; Mohammed, J.; Haileselassie, M.M.; Acharya, K.; Butte, G.; Haile, A.T.; Walsh, C.; Werner, D. Spatiotemporal variation in urban wastewater pollution impacts on river microbiomes and associated hazards in the Akaki catchment, Addis Ababa, Ethiopia. Sci. Total Environ. 2022, 826, 153912. [Google Scholar] [CrossRef]
- Kang, M.; Yang, J.; Kim, S.; Park, J.; Kim, M.; Park, W. Occurrence of antibiotic resistance genes and multidrug-resistant bacteria during wastewater treatment processes. Sci. Total Environ. 2022, 811, 152331. [Google Scholar] [CrossRef]
- Truelove, N.K.; Andruszkiewicz, E.A.; Block, B.A. A rapid environmental DNA method for detecting white sharks in the open ocean. Methods Ecol. Evol. 2019, 10, 1128–1135. [Google Scholar] [CrossRef]
- Hatfield, R.G.; Batista, F.M.; Bean, T.P.; Fonseca, V.G.; Santos, A.; Turner, A.D.; Lewis, A.; Dean, K.J.; Martinez-Urtaza, J. The Application of Nanopore Sequencing Technology to the Study of Dinoflagellates: A Proof of Concept Study for Rapid Sequence-Based Discrimination of Potentially Harmful Algae. Front. Microbiol. 2020, 11, 844. [Google Scholar] [CrossRef] [PubMed]
- Samson, R.; Rajput, V.; Shah, M.; Yadav, R.; Sarode, P.; Dastager, S.G.; Dharne, M.S.; Khairnar, K. Deciphering taxonomic and functional diversity of fungi as potential bioindicators within confluence stretch of Ganges and Yamuna Rivers, impacted by anthropogenic activities. Chemosphere 2020, 252, 126507. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Rajput, V.; Gohil, K.; Khairnar, K.; Dharne, M. Comprehensive metagenomic insights into a unique mass gathering and bathing event reveals transient influence on a riverine ecosystem. Ecotoxicol. Environ. Saf. 2020, 202, 110938. [Google Scholar] [CrossRef] [PubMed]
- ONT. DNA and RNA Sequencing Kits. 2022. Available online: https://nanoporetech.com/products/kits#tabs-0=rna (accessed on 21 May 2022).
- Masago, K.; Fujita, S.; Oya, Y.; Takahashi, Y.; Matsushita, H.; Sasaki, E.; Kuroda, H. Comparison between Fluorimetry (Qubit) and Spectrophotometry (NanoDrop) in the Quantification of DNA and RNA Extracted from Frozen and FFPE Tissues from Lung Cancer Patients: A Real-World Use of Genomic Tests. Medicina 2021, 57, 1375. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Tarumoto, N.; Runtuwene, L.R.; Sakai, J.; Hayashida, K.; Eshita, Y.; Maeda, R.; Tuda, J.; Ohno, H.; Murakami, T.; et al. An innovative diagnostic technology for the codon mutation C580Y in kelch13 of Plasmodium falciparum with MinION nanopore sequencer. Malar. J. 2018, 17, 217. [Google Scholar] [CrossRef]
- Soroka, M.; Wasowicz, B.; Rymaszewska, A. Loop-Mediated Isothermal Amplification (LAMP): The Better Sibling of PCR? Cells 2021, 10, 1931. [Google Scholar] [CrossRef]
- Stahl-Rommel, S.; Jain, M.; Nguyen, H.N.; Arnold, R.R.; Aunon-Chancellor, S.M.; Sharp, G.M.; Castro, C.L.; John, K.K.; Juul, S.; Turner, D.J.; et al. Real-Time Culture-Independent Microbial Profiling Onboard the International Space Station Using Nanopore Sequencing. Genes 2021, 12, 106. [Google Scholar] [CrossRef]
- ONT. Flow Cells. 2022. Available online: https://store.nanoporetech.com/uk/flow-cells.html (accessed on 22 July 2022).
- ONT. Qcat. 2020. Available online: https://github.com/nanoporetech/qcat (accessed on 22 July 2022).
- GitHub. Porechop. 2018. Available online: https://github.com/rrwick/Porechop (accessed on 5 August 2022).
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- ONT. Nanopore Sequencing Data Analysis. 2022. Available online: https://nanoporetech.com/nanopore-sequencing-data-analysis (accessed on 31 May 2022).
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- NCBI. Basic Local Alignment Search Tool. 2022. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 31 May 2022).
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33 (Suppl. S1), D501–D504. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; Souza, M.D.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Minot, S.S.; Krumm, N.; Greenfield, N.B. One Codex: A Sensitive and Accurate Data Platform for Genomic Microbial Identification. bioRxiv 2015, 027607. [Google Scholar]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Loman, N.; Rowe, W.; Rambaut, A. ARTIC Network Bioinformatic Pipeline. 2020. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 4 June 2022).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Srivathsan, A.; Hartop, E.; Puniamoorthy, J.; Lee, W.T.; Kutty, S.N.; Kurina, O.; Meier, R. Rapid, large-scale species discovery in hyperdiverse taxa using 1D MinION sequencing. BMC Biol. 2019, 17, 96. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Maestri, S.; Cosentino, E.; Paterno, M.; Freitag, H.; Garces, J.M.; Marcolungo, L.; Alfano, M.; Njunjić, I.; Schilthuizen, M.; Slik, F.; et al. A Rapid and Accurate MinION-Based Workflow for Tracking Species Biodiversity in the Field. Genes 2019, 10, 468. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.A.; Dai, D.; Pruden, A.; Vikesland, P.; Heath, L.S.; Zhang, L. NanoARG: A web service for detecting and contextualizing antimicrobial resistance genes from nanopore-derived metagenomes. Microbiome 2019, 7, 88. [Google Scholar] [CrossRef]
- CosmosID. Bioinformatics Services—Functional Metagenomics and Metatranscriptomics. 2022. Available online: https://www.cosmosid.com/functional-metagenomics/ (accessed on 31 May 2022).
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.H.S.; Balachandran, K.R.S.; Rangamaran, V.R.; Gopal, D. RemeDB: Tool for Rapid Prediction of Enzymes Involved in Bioremediation from High-Throughput Metagenome Data Sets. J. Comput. Biol. 2020, 27, 1020–1029. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33 (Suppl. S1), D325–D328. [Google Scholar] [CrossRef]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C.; et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 2021, 6, 3–6. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar]
- Bertrand, D.; Shaw, J.; Kalathiyappan, M.; Ng, A.H.Q.; Kumar, M.S.; Li, C.; Dvornicic, M.; Soldo, J.P.; Koh, J.Y.; Tong, C.; et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 2019, 37, 937–944. [Google Scholar] [CrossRef]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2015, 32, 1009–1015. [Google Scholar] [CrossRef]
- RCoreTeam. R: A Language and Environment for Statistical Computing. 2022. Available online: https://www.R-project.org/ (accessed on 4 June 2022).
- RStudioTeam. RStudio: Integrated Development for R. 2022. Available online: http://www.rstudio.com/ (accessed on 4 June 2022).
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Arndt, D.; Xia, J.; Liu, Y.; Zhou, Y.; Guo, A.C.; Cruz, J.A.; Sinelnikov, I.; Budwill, K.; Nesbø, C.L.; Wishart, D.S. METAGENassist: A comprehensive web server for comparative metagenomics. Nucleic Acids Res. 2012, 40, W88–W95. [Google Scholar] [CrossRef]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.T.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef]
- Paliy, O.; Shankar, V. Application of multivariate statistical techniques in microbial ecology. Mol. Ecol. 2017, 25, 1032–1057. [Google Scholar] [CrossRef]
- Uprety, S.; Dangol, B.; Nakarmi, P.; Dhakal, I.; Sherchan, S.P.; Shisler, J.L.; Jutla, A.; Amarasiri, M.; Sano, D.; Nguyen, T.H. Assessment of microbial risks by characterization of Escherichia coli presence to analyze the public health risks from poor water quality in Nepal. Int. J. Hyg. Environ. Health 2020, 226, 113484. [Google Scholar] [CrossRef]
- Zan, R.; Acharya, K.; Blackburn, A.; Kilsby, C.G.; Werner, D. A Mobile Laboratory Enables Fecal Pollution Source Tracking in Catchments Using Onsite qPCR Assays. Water 2022, 14, 1224. [Google Scholar] [CrossRef]
- Surkova, E.; Nikolayevskyy, V.; Drobniewski, F. False-positive COVID-19 results: Hidden problems and costs. Lancet Respir. Med. 2020, 8, 1167–1168. [Google Scholar] [CrossRef]
- WHO. Guidelines for Drinking-Water Quality: Fourth Edition Incorporating the First and Second Addenda; World Health Organization: Geneva, Switzerland, 2022; p. 583. [Google Scholar]
- Tiwari, A.; Oliver, D.; Bivins, A.; Sherchan, S.; Pitkänen, T. Bathing Water Quality Monitoring Practices in Europe and the United States. Int. J. Environ. Res. Public Health 2021, 18, 5513. [Google Scholar] [CrossRef] [PubMed]
- WHO. Quantitative Microbial Risk Assessment: Application for Water Safety Management; World Health Organization: Geneva, Switzerland, 2016; p. 186. [Google Scholar]
- Lucivero, F. Big Data, Big Waste? A Reflection on the Environmental Sustainability of Big Data Initiatives. Sci. Eng. Ethics 2020, 26, 1009–1030. [Google Scholar] [CrossRef] [PubMed]




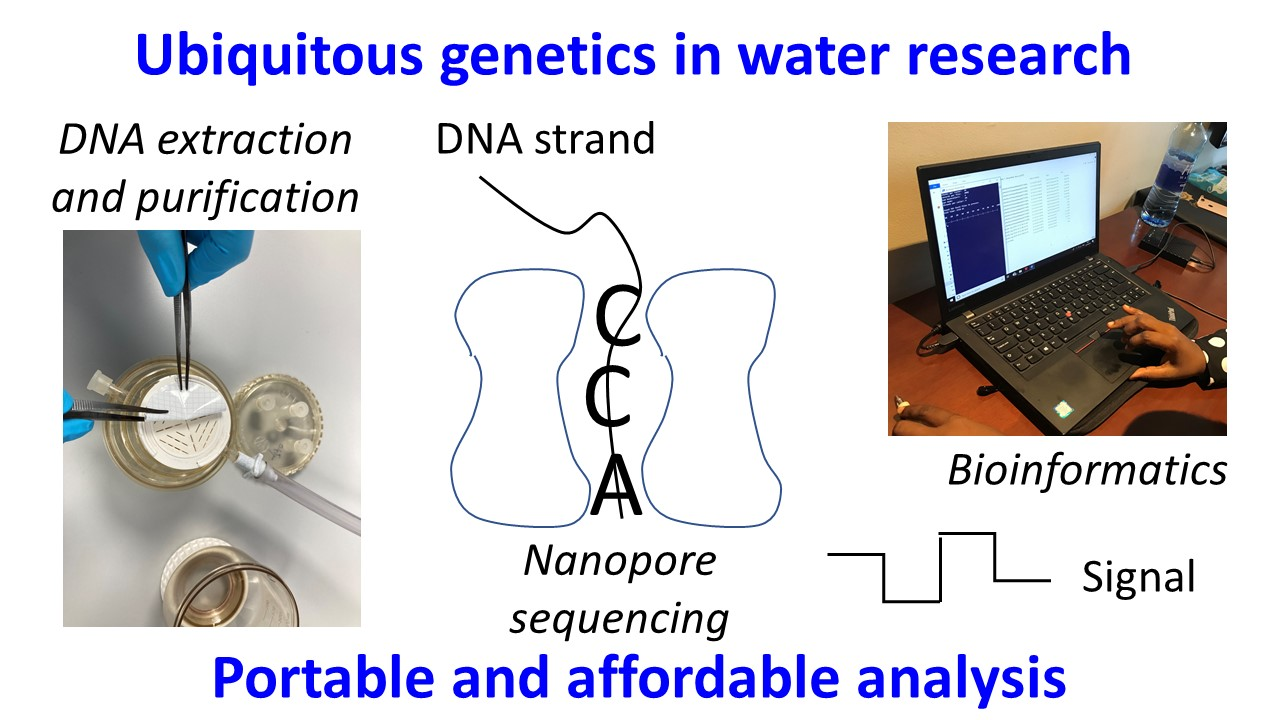{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promises | Challenges |
|---|---|
|
|
| Purpose | Equipment | Weight kg | Dimensions (W × L × H in cm) | Capacity (Samples per Run) | Year 2022 Costs £ incl. VAT |
|---|---|---|---|---|---|
| Centrifugation (C) | Mikro 200 R (Hinderson Biomedical, London, UK) | 28.0 | 28.1 × 55.3 × 26.0 | 30 | 6610 |
| Centrifugation (P) | mySPIN 6 (Life Technologies Ltd, Pailsey, UK) | 0.9 | 10.4 × 12.8 × 15.3 | 6 | 462 |
| Bead milling (C) | FastPrep-24™ 5G ribolyser (MP Biomedicals, Eschwege, Germany) | 23.6 | 47.2 × 38.5 × 49 | 24 | 7344 |
| Bead milling (P) | SuperFastprep-2 (Fisher Scientific, Loughborough, UK) | 1.0 | 33.0 × 8.1 × 11.7 | 2 | 2934 |
| Thermocycling (C) | PCRmax Alpha Cycler 1 Thermal Cycler (Cole-Parmer, Staffordshire, UK) | 15.4 | 43 × 26 × 20 | 96 | 4020 |
| Thermocycling (P) | Mini-PCR mini16 (MiniPCR Bio, Cambridge, MA, USA) | 0.5 | 5.1 × 12.7 × 10.2 | 16 | 792 |
| Gel electrophoresis (C) | Bio-Rad Wide Mini-Sub Cell GT (Bio-rad Laboratories, Watford, UK) | 2.1 | 17.8 × 25.5 × 6.8 (buffer tank) 21 × 24.5 × 6.5 (power supply) | 60 | 1087 |
| Gel electrophoresis (P) | blueGel (MiniPCR Bio, Cambridge, MA, USA) | 0.4 | 23.0 × 10.0 × 7.0 | 9 | 299 |
| Centrifugation, thermocycling, gel electrophoresis (P) | Bento Lab Pro (Bento Bioworks Ltd., London, UK) | 3.5 | 33.0 × 21.4 × 8.1 | 6 (centrifuge) | 1919 |
| Sequencing (C) | MiSeq (Illumina Cambridge Ltd., Cambridge, UK) | 93.6 | 68.6 × 56.5 × 52.3 | 96 | 113,400 |
| Sequencing (C) | Sequel II (Pacific Biosciences, Menlo Park, CA, USA) | 362.0 | 92.7 × 86.4 × 167.6 | 192 | 435,000 |
| Sequencing (P) | MinION (Oxford Nanopore Technologies, Oxford, UK) | 0.1 | 10.5 × 2.3 × 3.3 | 12–96 (with barcodes) | 960 |
| Software | Features |
|---|---|
| MinKNOW | ONT’s software for controlling the MinION. Carries out the data acquisition, starts and stops or fine controls runs, and reports on the status of pores. Includes an option for real-time basecalling. |
| Guppy | ONT’s basecalling software to translate the electronic raw signal output of the MinION into a succession of bases defining the nucleic acid sequence. Includes post-processing features, such as barcoding/demultiplexing, adapter trimming, and alignment. |
| EPI2ME | ONT’s cloud-based platform for onward analysis of nanopore sequences. Includes WIMP for species identification from shotgun sequencing data, 16S taxonomic classification for bacteria, ARMA for identifying genes responsible for antimicrobial resistance (AMR), and a FASTQ custom alignment workflow for matching reads to uploaded references. Requires no command line experience. |
| NanoPack [85] | A set of tools for visualization and processing of nanopore sequencing data. Includes NanoStat to summarize information on read quantity and quality, NanoPlot to produce related figures, NanoComp to compare experiments, and NanoFilt for read filtering and trimming. |
| BLAST [89] | Basic Local Alignment Search Tool. A suite of tools from the National Centre for Biotechnology Information (NCBI) to compare nucleotide or protein sequences to sequence databases. |
| MG-RAST [93] | Metagenomic Rapid Annotations using Subsystems Technology. Pipeline for phylogenetic and functional assignments of metagenomes. Compares sequences to databases. |
| Canu [86] | Software for assembling nanopore sequences. Includes tools to improve the read accuracy, remove dubious regions, order reads into overlapping segments, and generate consensus sequences. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Werner, D.; Acharya, K.; Blackburn, A.; Zan, R.; Plaimart, J.; Allen, B.; Mgana, S.M.; Sabai, S.M.; Halla, F.F.; Massawa, S.M.; et al. MinION Nanopore Sequencing Accelerates Progress towards Ubiquitous Genetics in Water Research. Water 2022, 14, 2491. https://doi.org/10.3390/w14162491
Werner D, Acharya K, Blackburn A, Zan R, Plaimart J, Allen B, Mgana SM, Sabai SM, Halla FF, Massawa SM, et al. MinION Nanopore Sequencing Accelerates Progress towards Ubiquitous Genetics in Water Research. Water. 2022; 14(16):2491. https://doi.org/10.3390/w14162491
Chicago/Turabian StyleWerner, David, Kishor Acharya, Adrian Blackburn, Rixia Zan, Jidapa Plaimart, Ben Allen, Shaaban Mrisho Mgana, Shadrack Mwita Sabai, Franella Francos Halla, Said Maneno Massawa, and et al. 2022. "MinION Nanopore Sequencing Accelerates Progress towards Ubiquitous Genetics in Water Research" Water 14, no. 16: 2491. https://doi.org/10.3390/w14162491