
“Sea Anemone”-like CeFe Oxides for High-Efficient Phosphate Removal

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CeFe-Based Adsorbents
2.3. Characterization
2.4. Adsorption Experiments
3. Results and Discussion
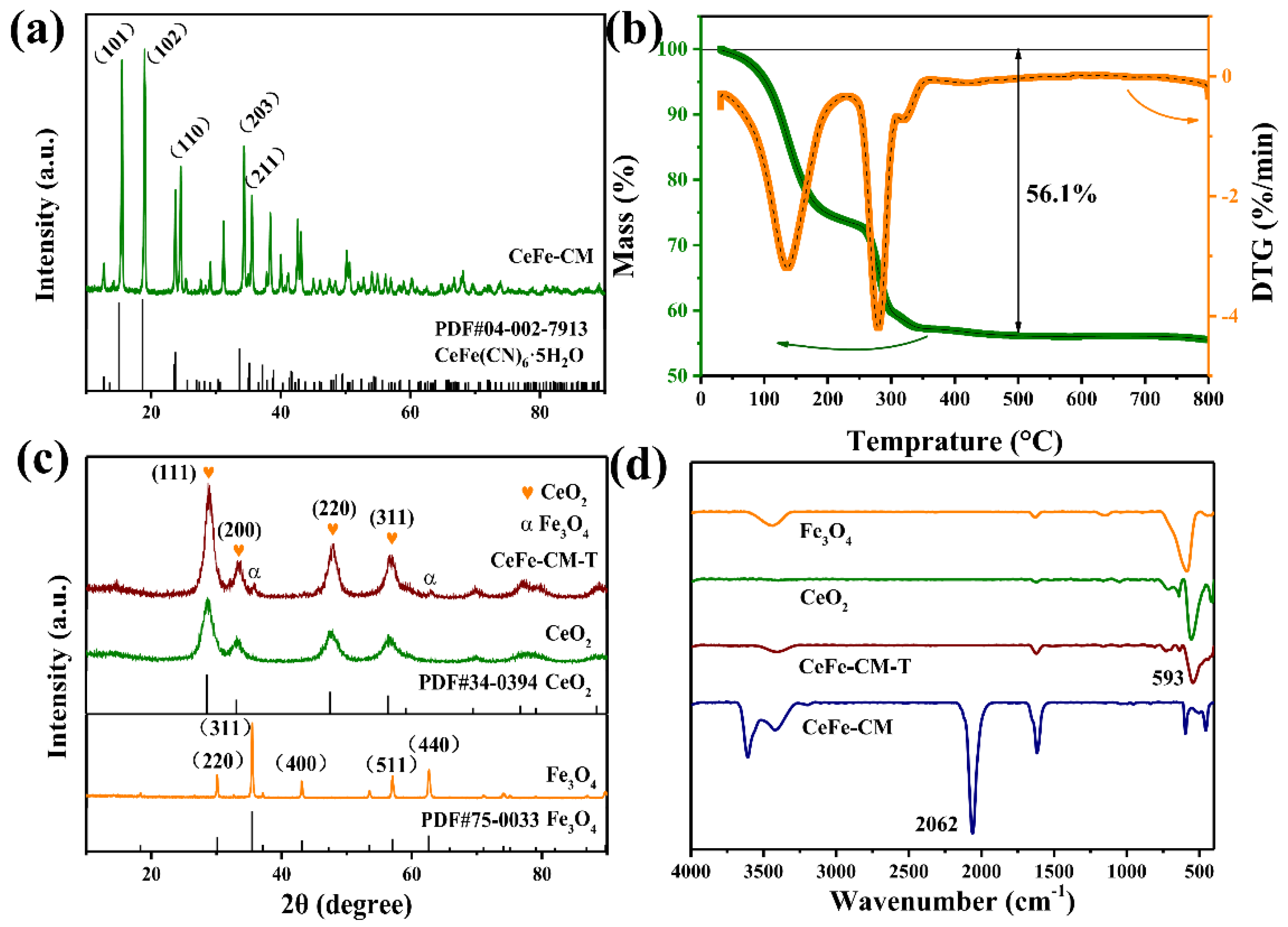
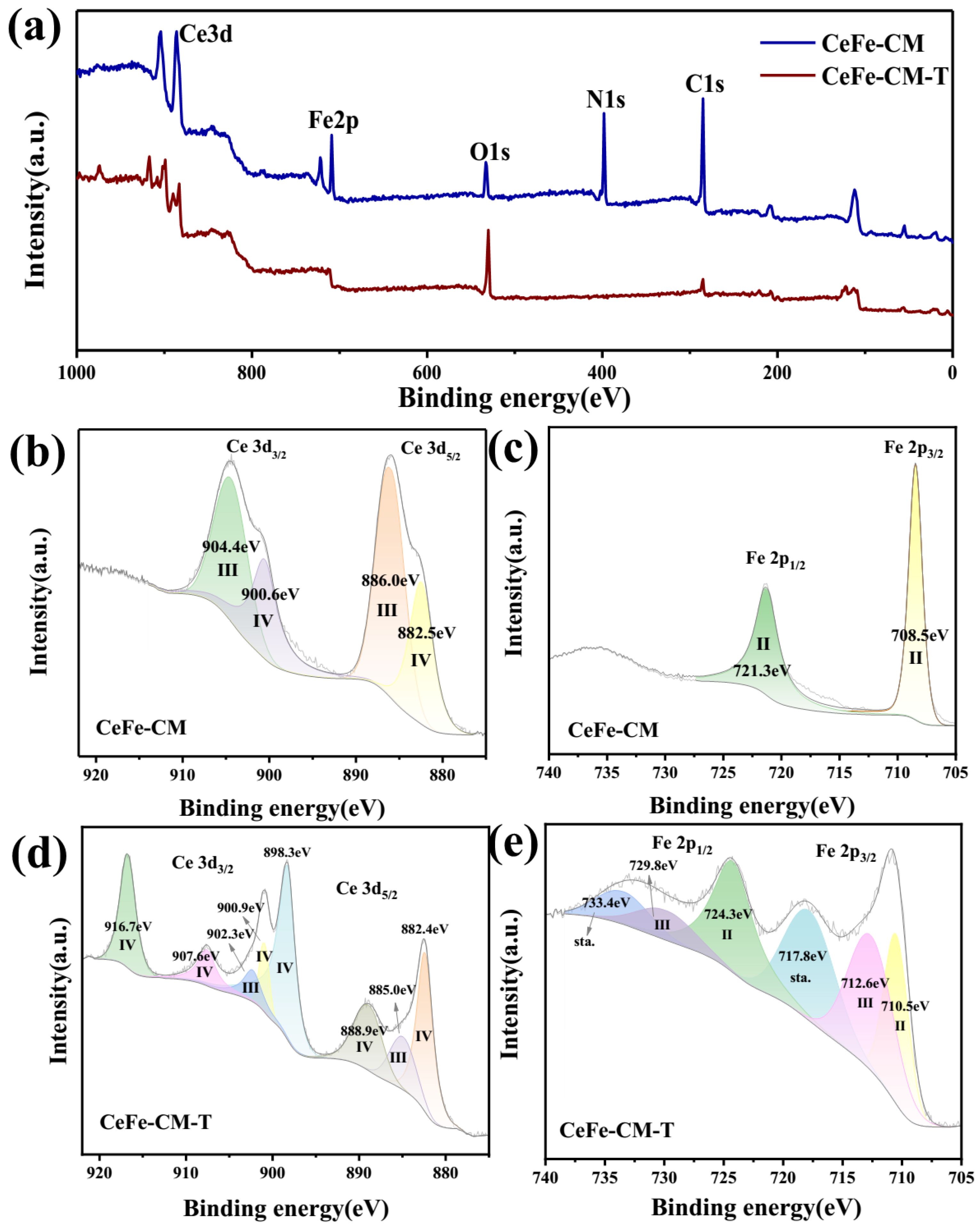
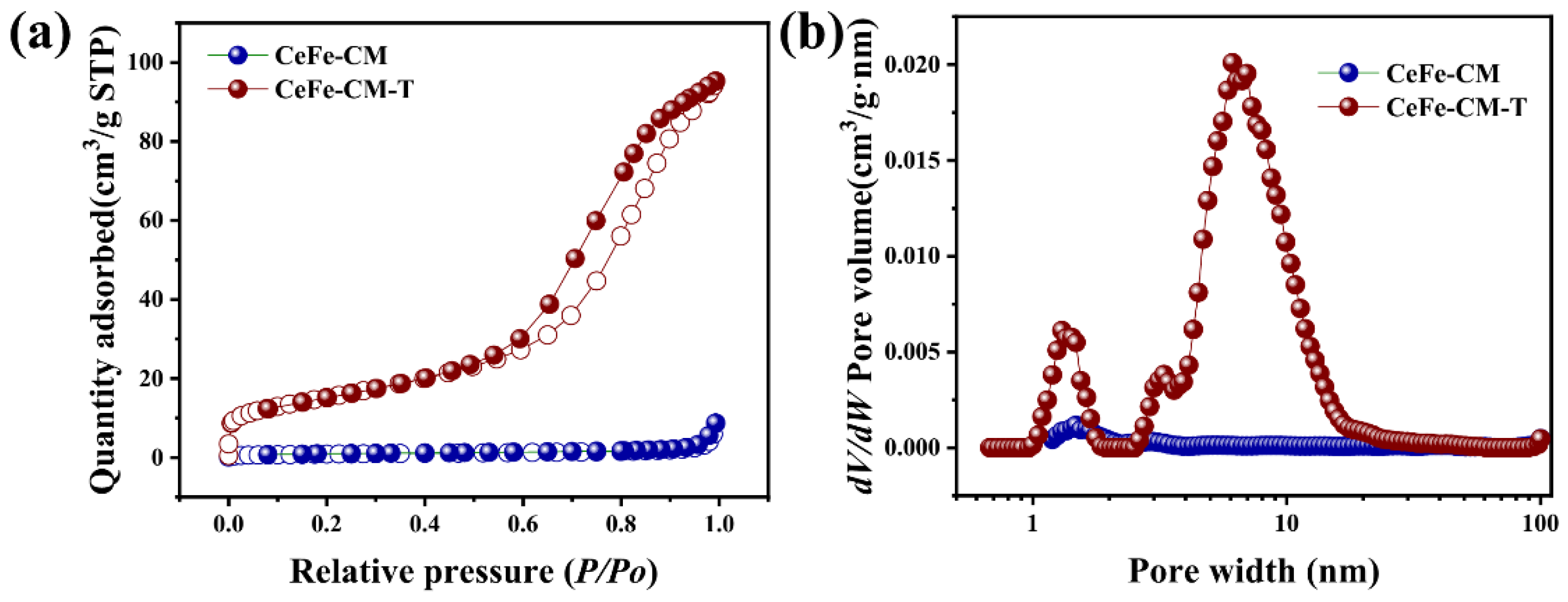
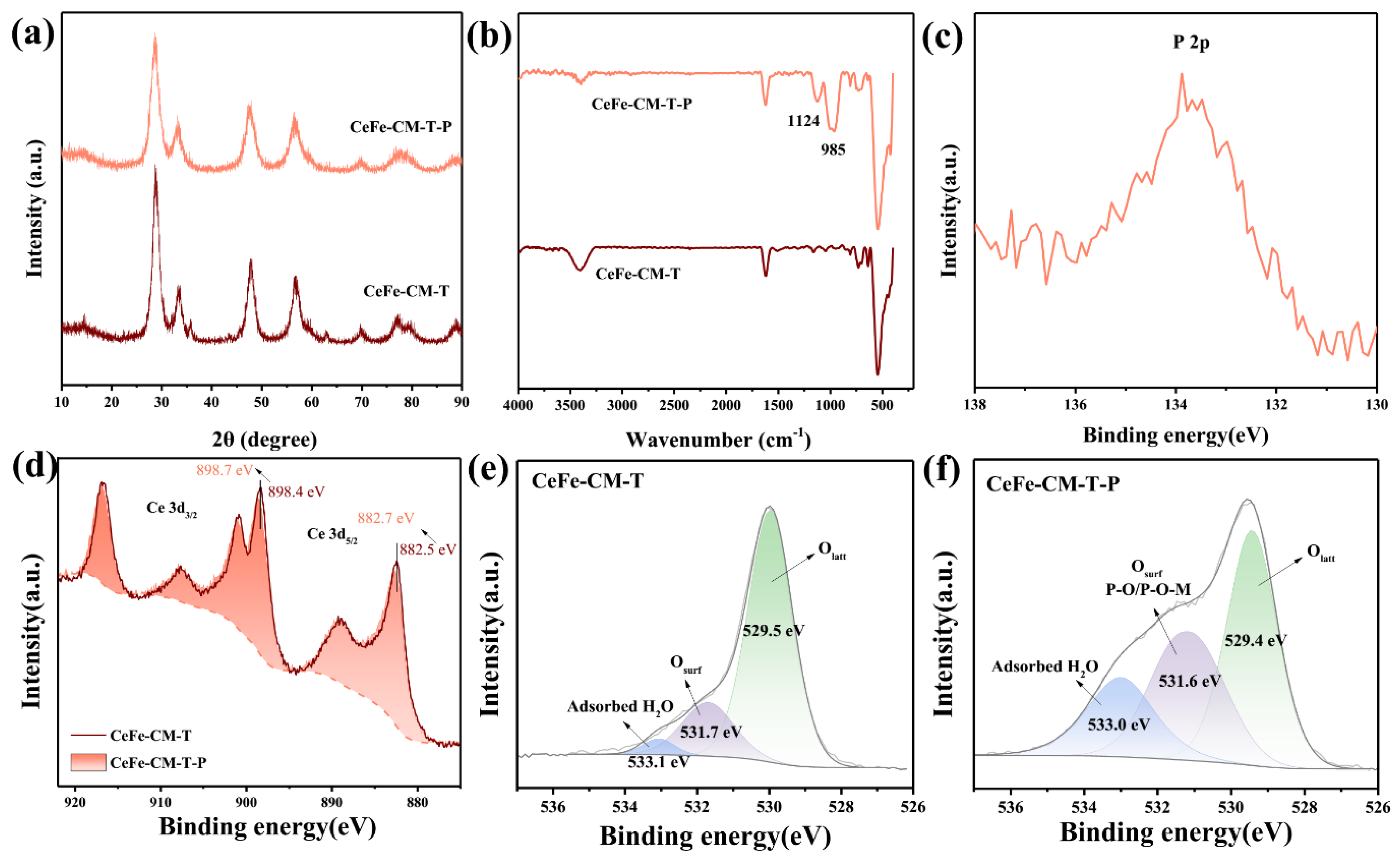
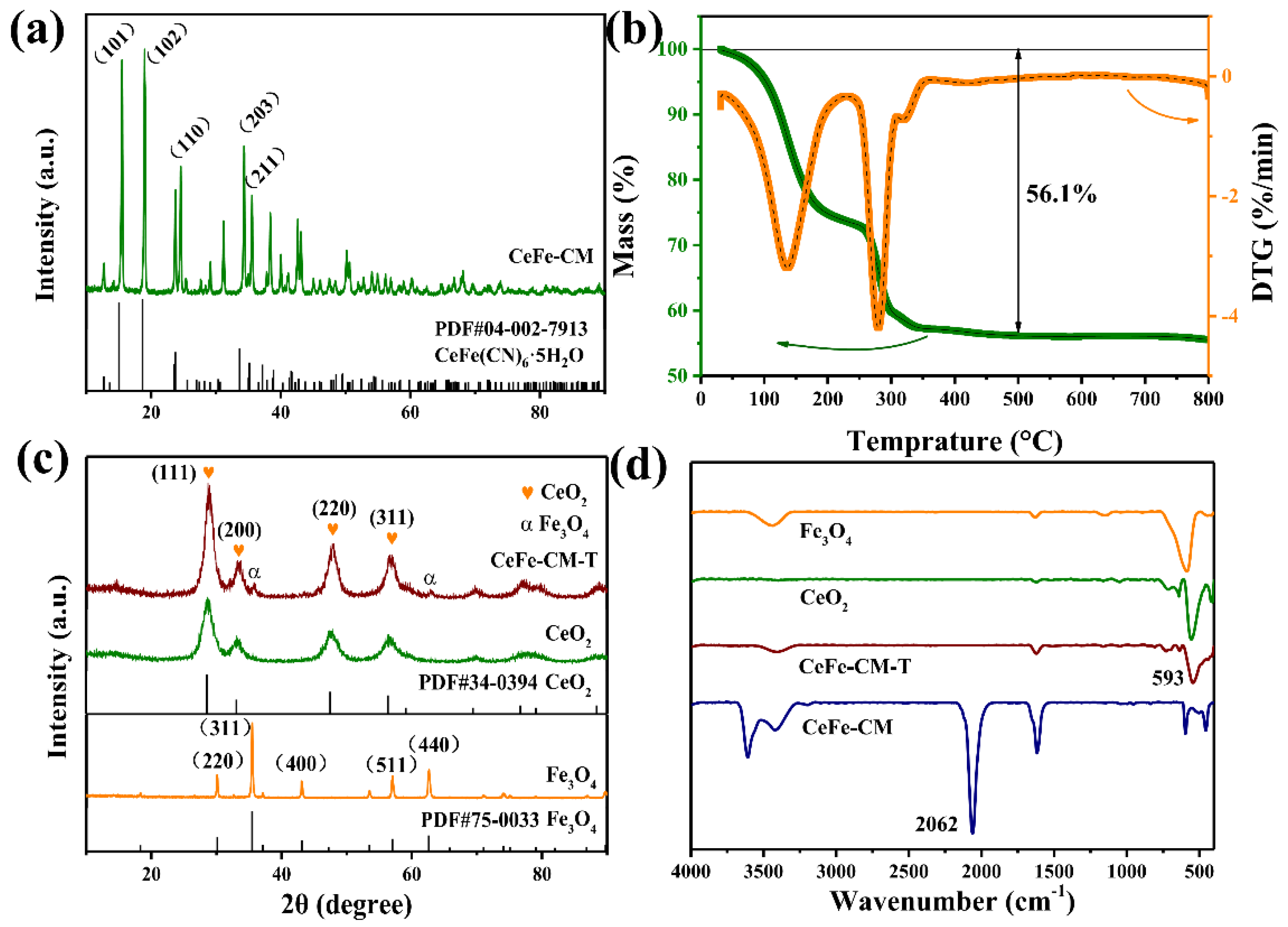
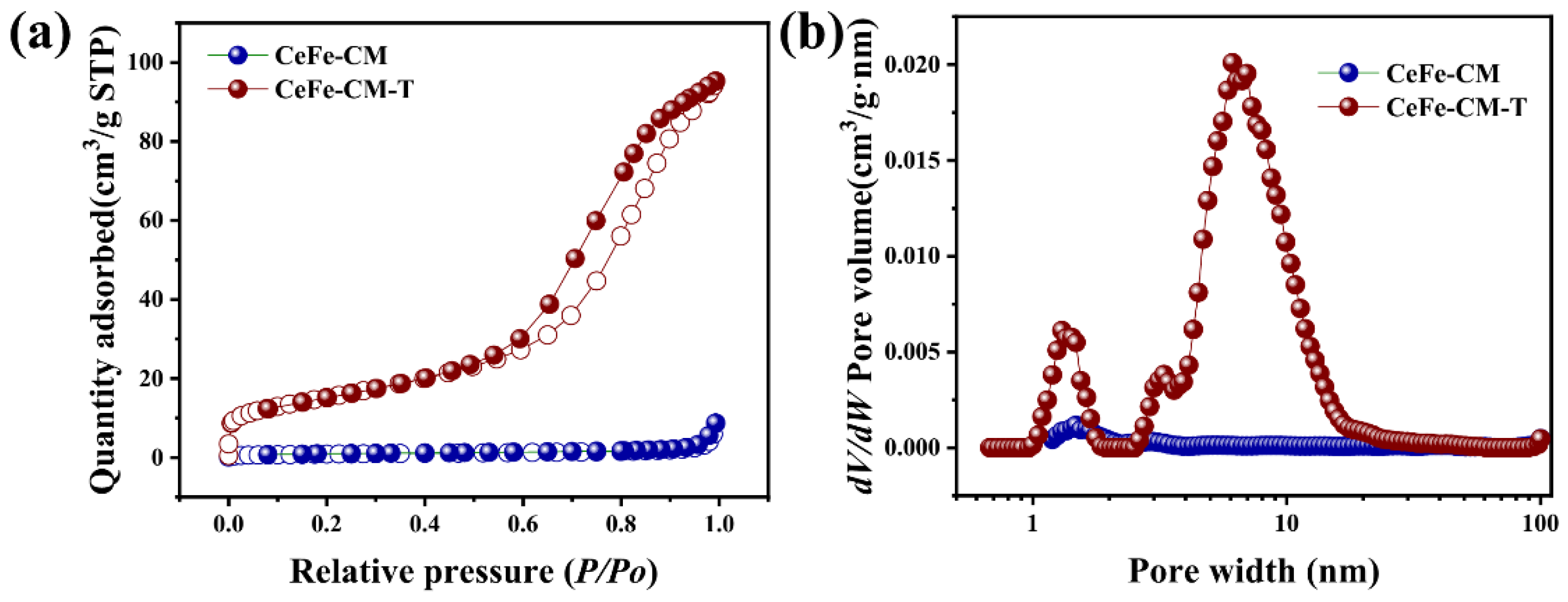
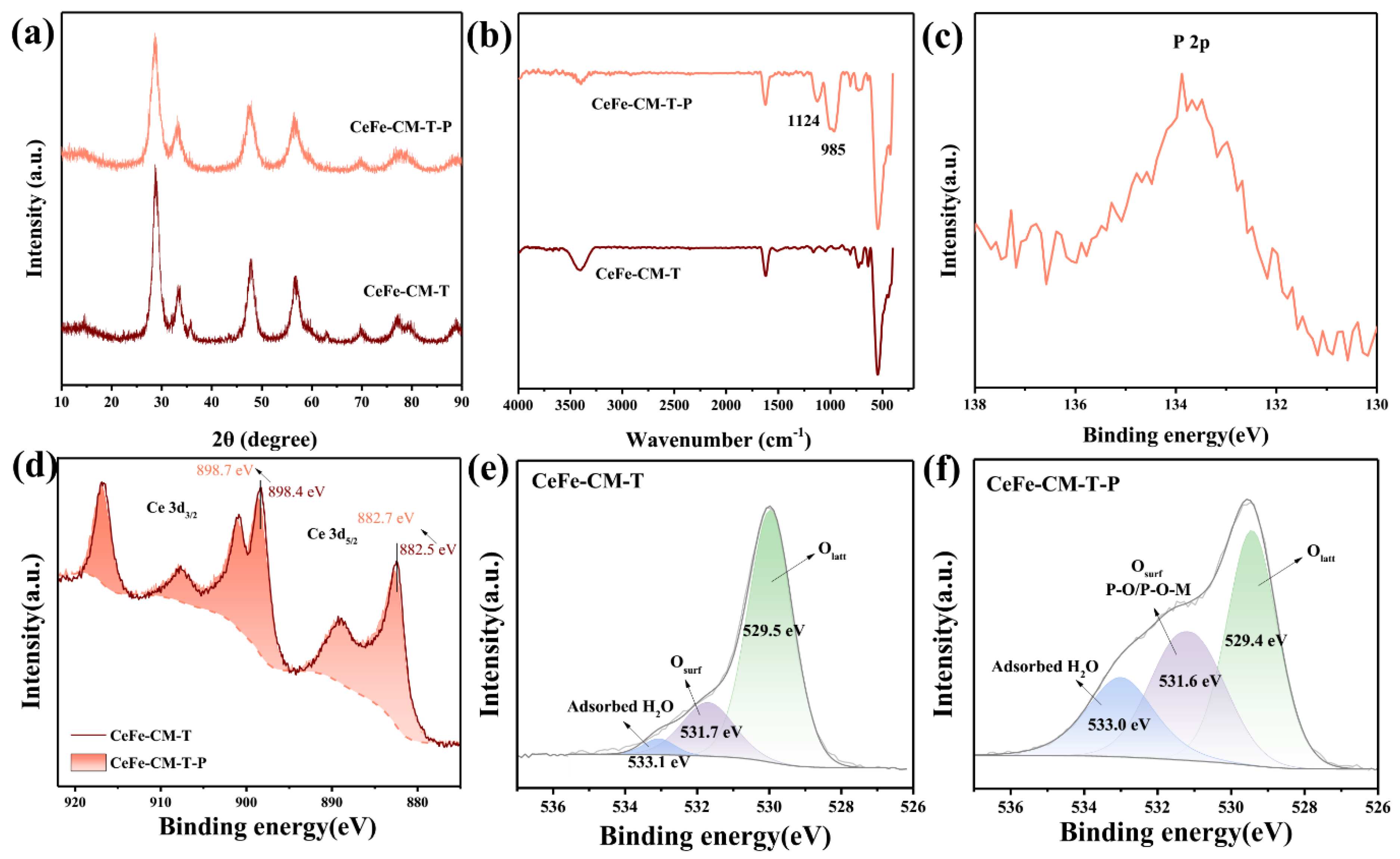
3.1. Morphology and Structure of CeFe-CM and CeFe-CM-T
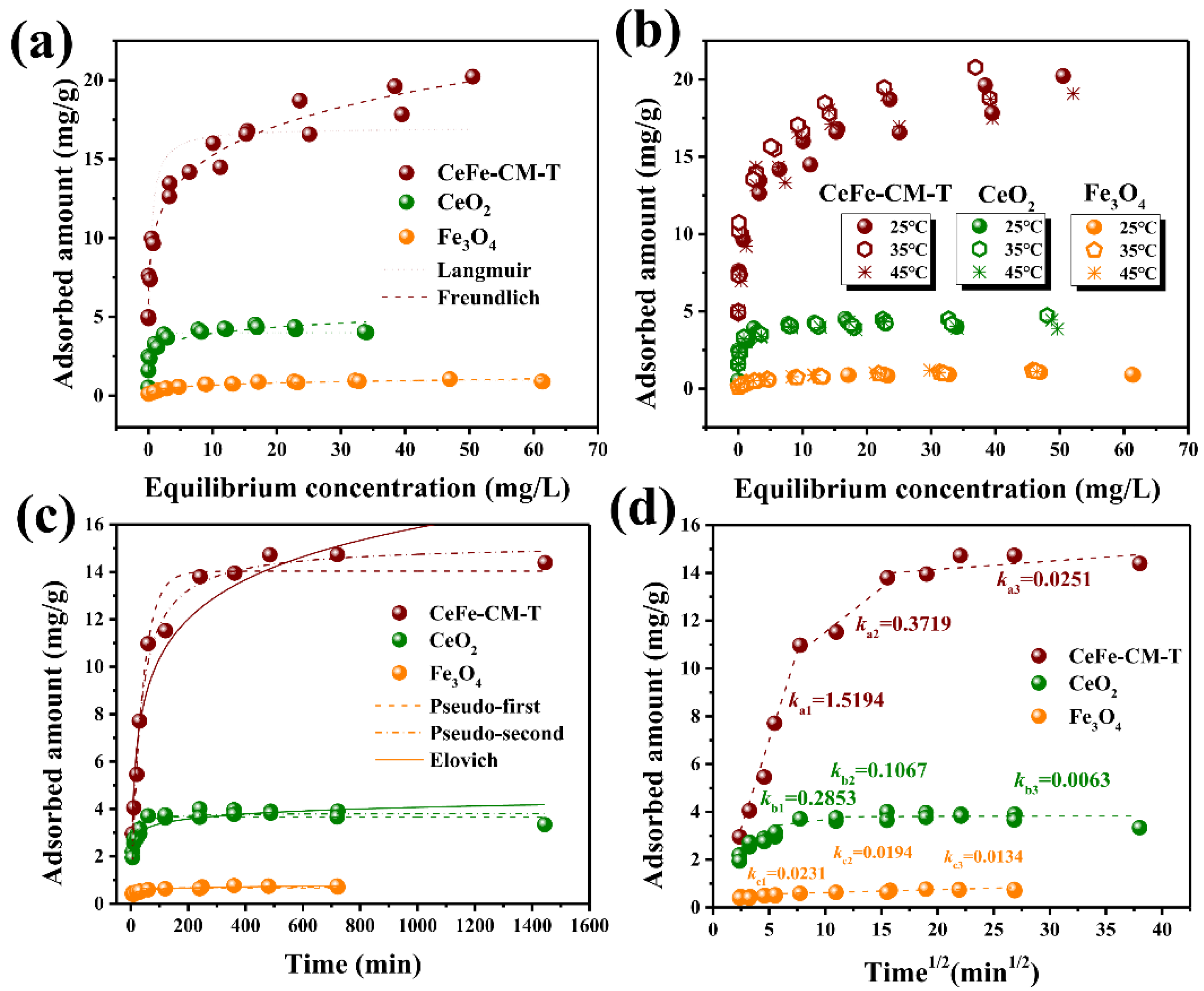
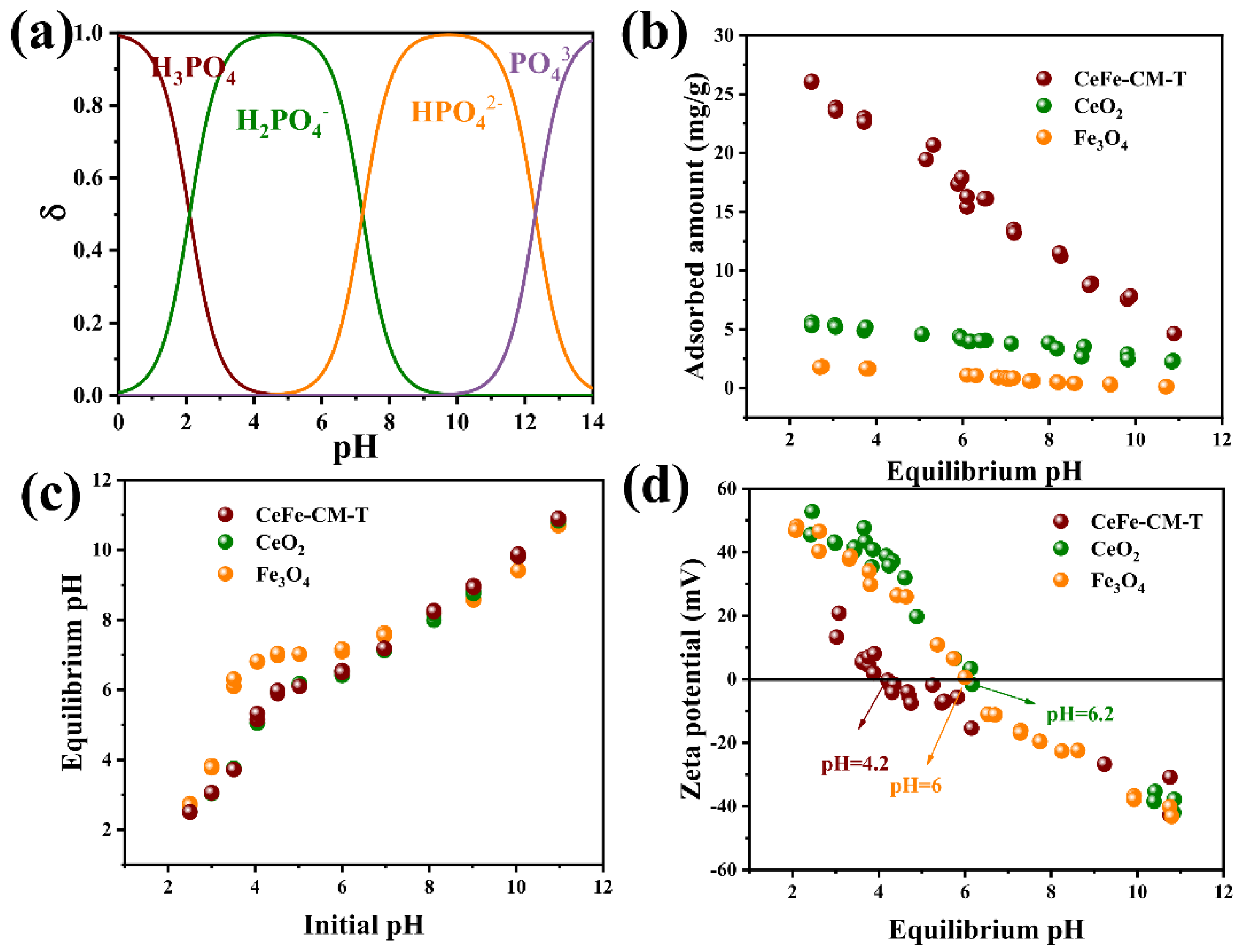
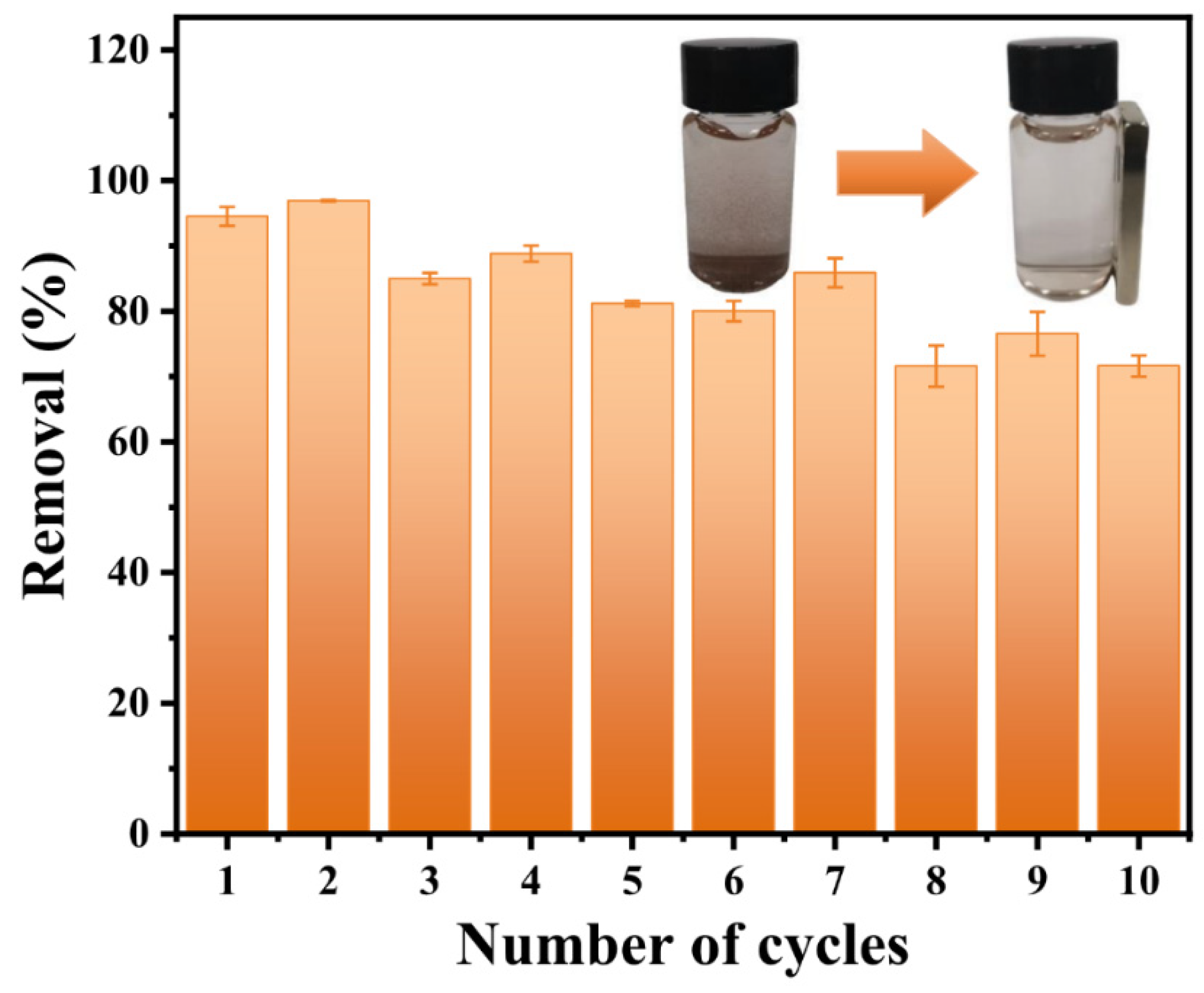
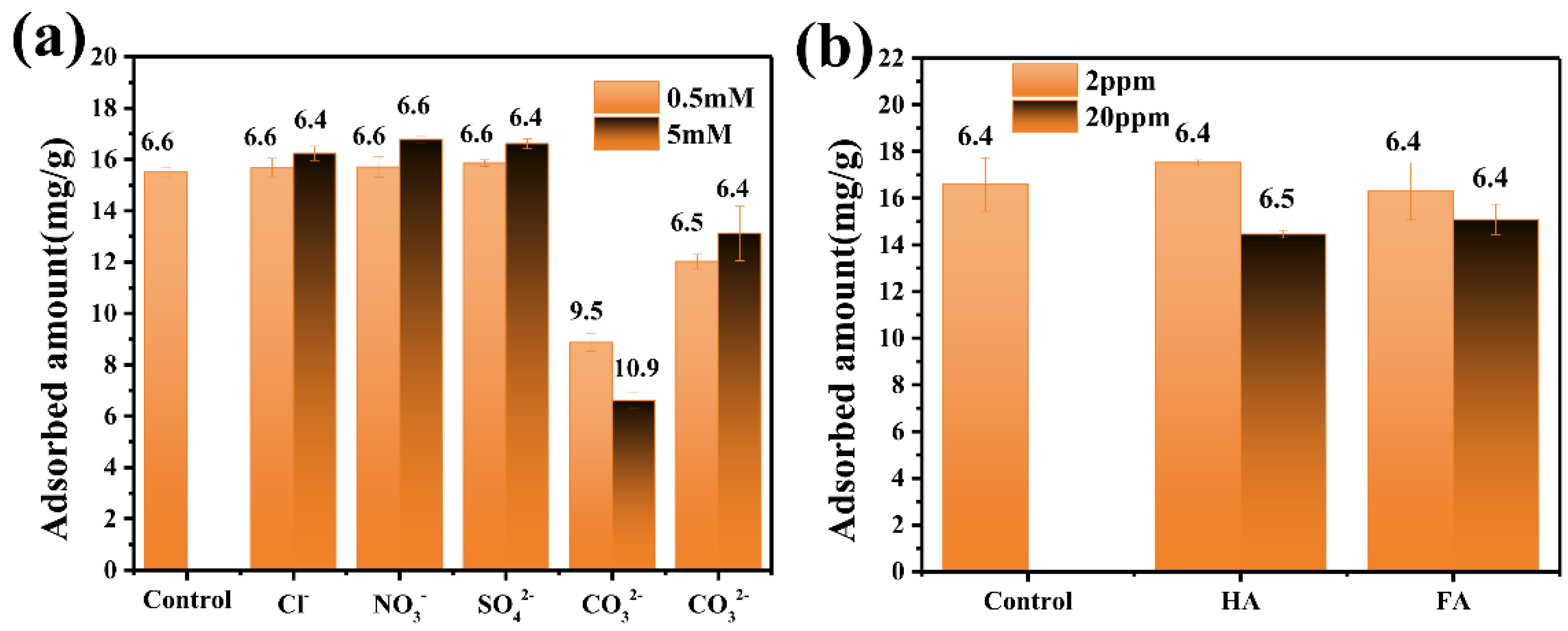
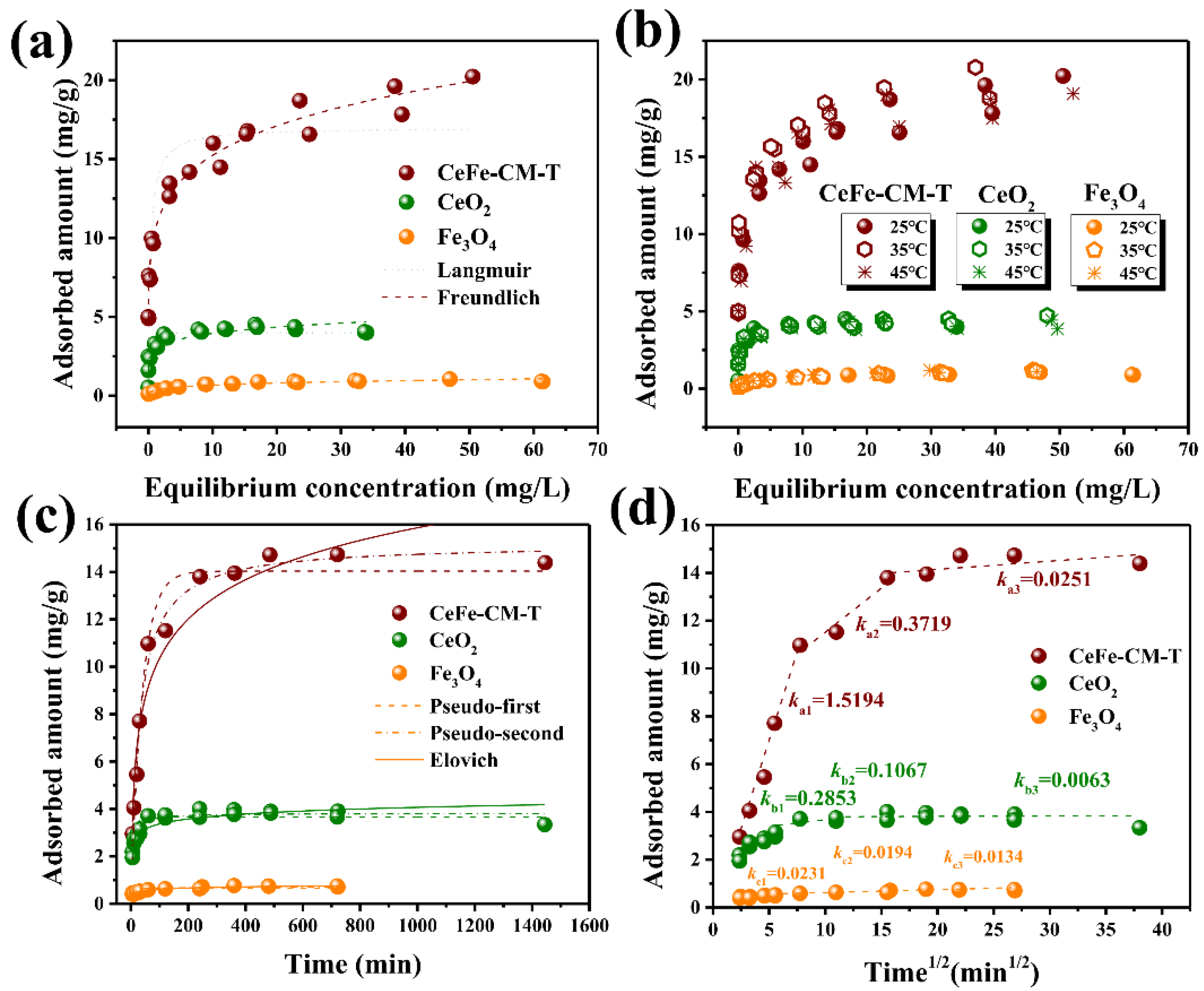
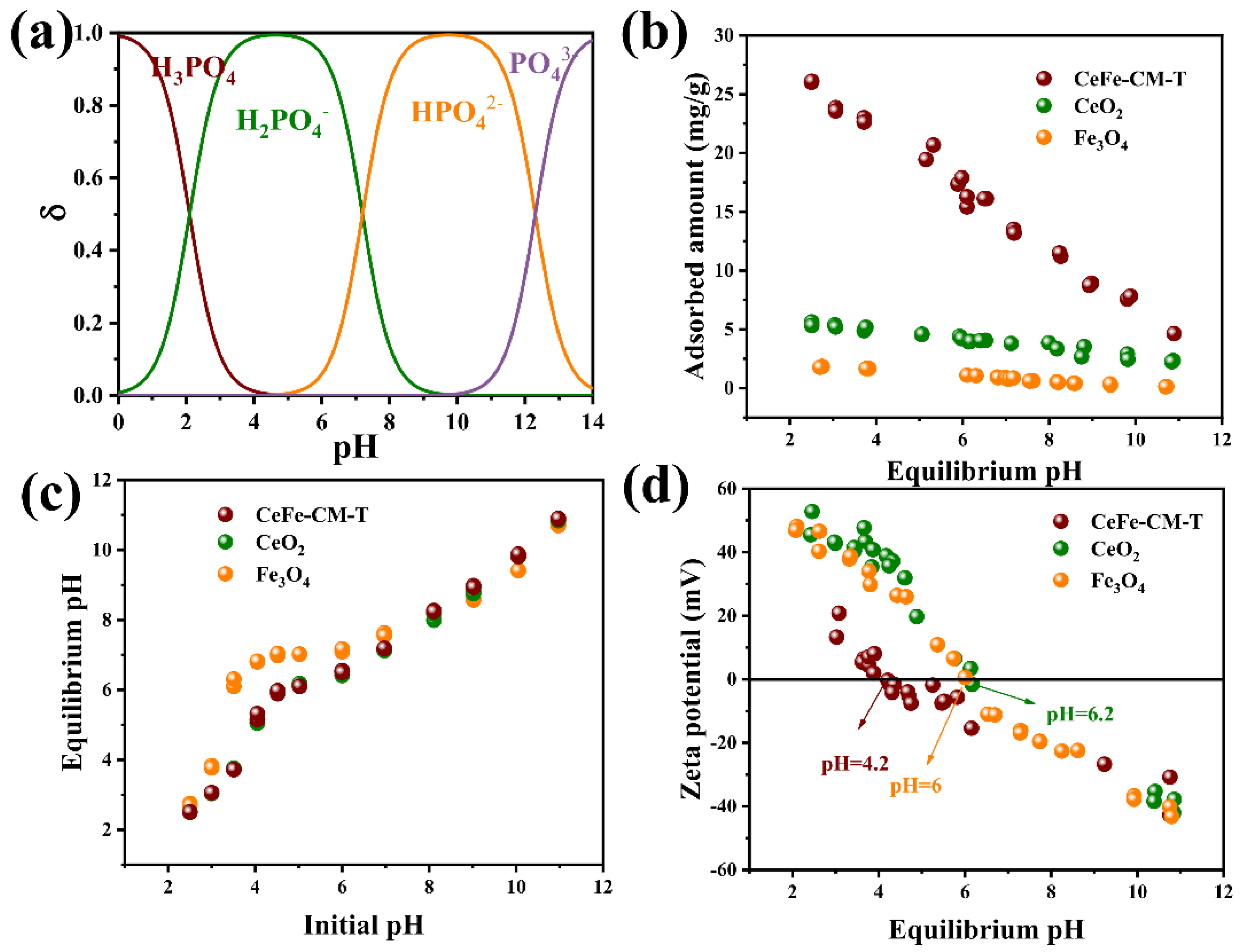
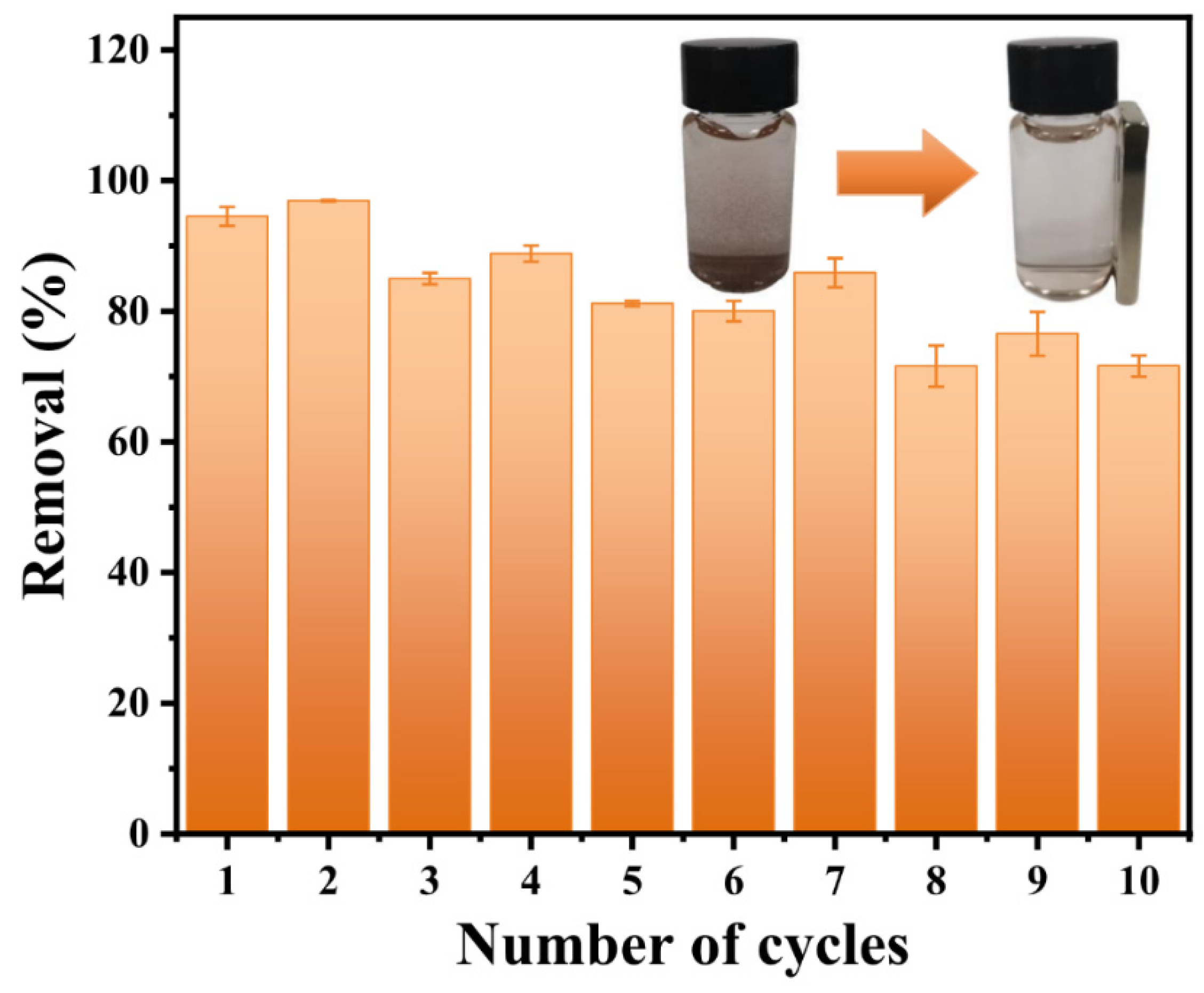
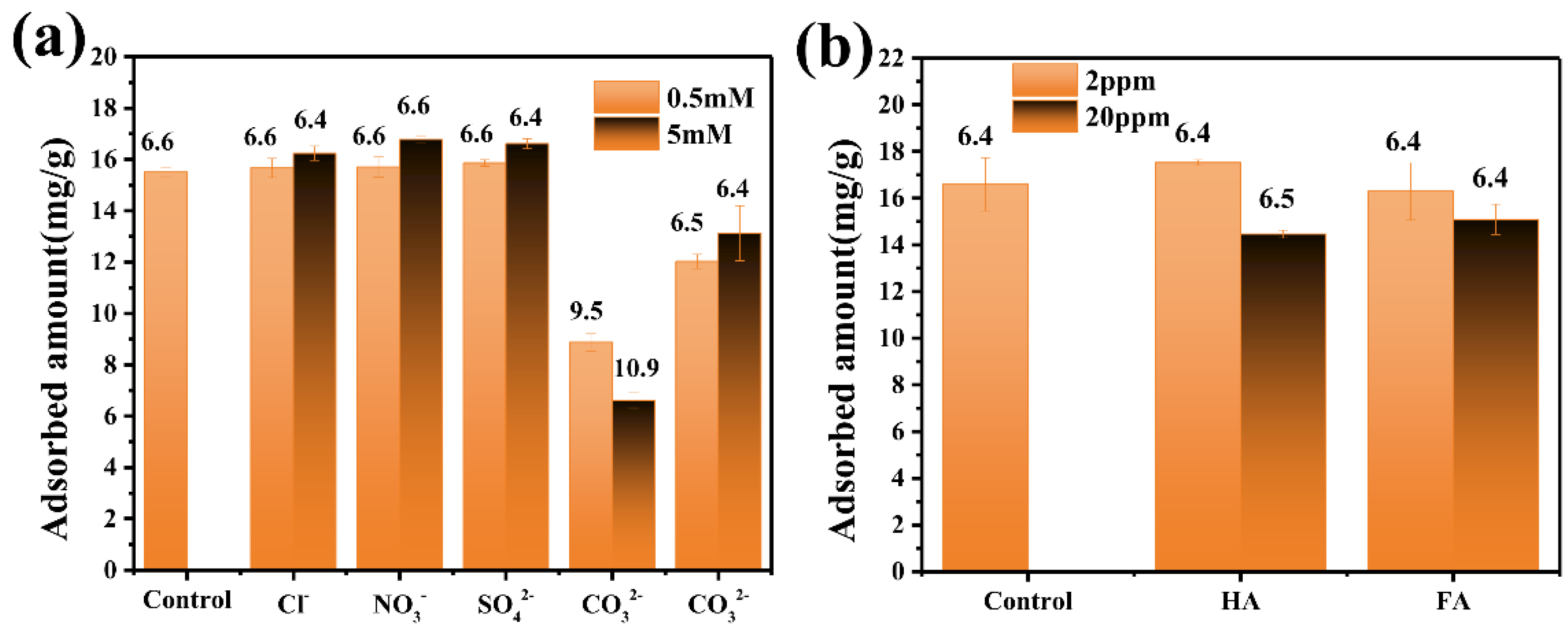
3.2. Phosphate Adsorption Performance of CeFe-Based Adsorbents
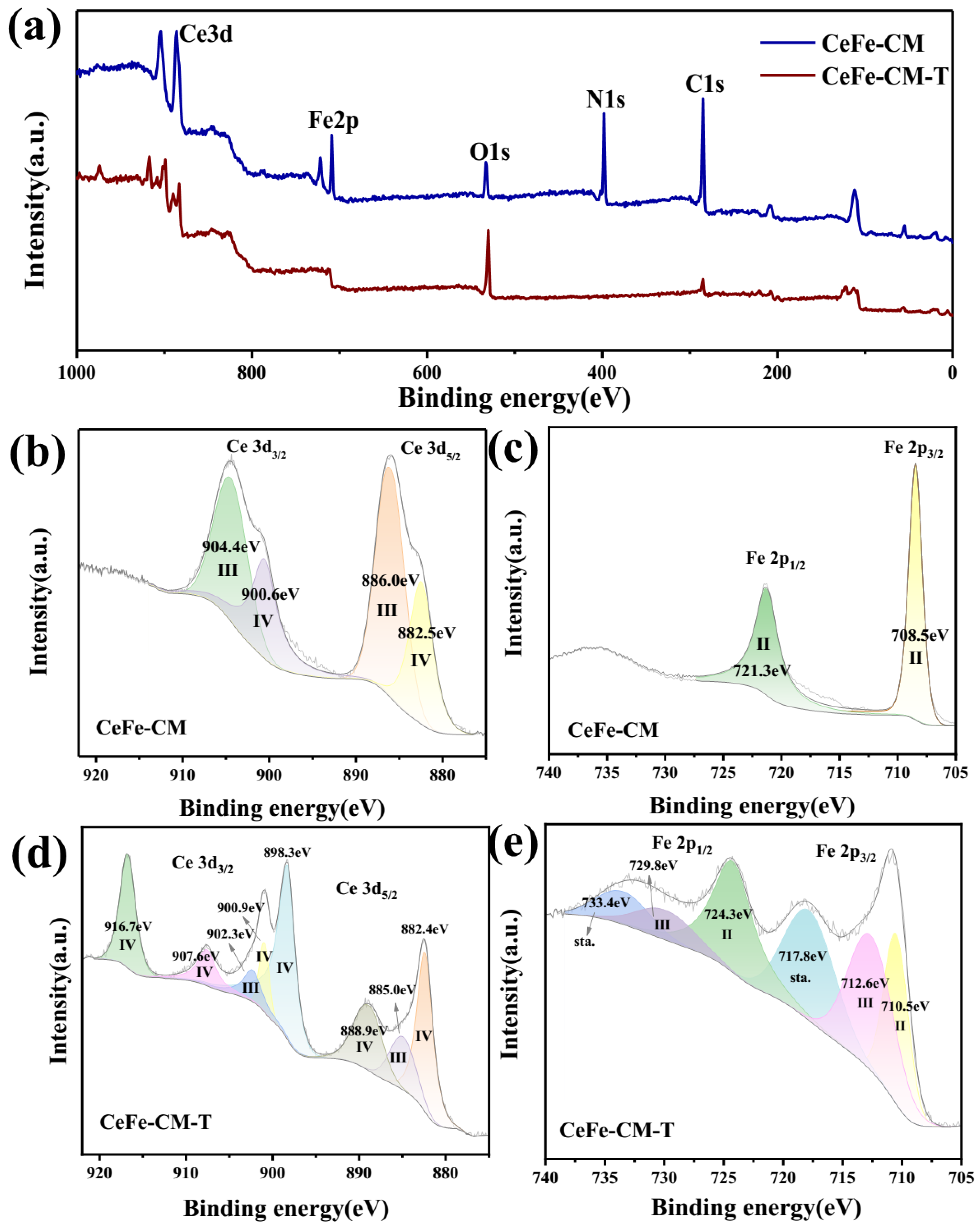
3.3. Phosphate Adsorption Mechanism on CeFe-Based Adsorbent
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, J.; Pei, C.; Yang, Y.; Lai, B.; Sun, Y.; Yang, L. The structural design and valence state control of cerium-based metal-organic frameworks for their highly efficient phosphate removal. J. Clean. Prod. 2021, 321, 128778. [Google Scholar] [CrossRef]
- Downing, J.A.; Polasky, S.; Olmstead, S.M.; Newbold, S.C. Protecting local water quality has global benefits. Nat. Commun. 2021, 12, 2709. [Google Scholar] [CrossRef]
- Wu, B.; Wan, J.; Zhang, Y.; Pan, B.; Lo, I.M.C. Selective phosphate removal from water and wastewater using sorption: Process fundamentals and removal mechanisms. Environ. Sci. Technol. 2020, 54, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, X.; Ballent, W.; Mayer, B.K. Biological phosphorus recovery: Review of current progress and future needs. Water Environ. Res. 2017, 89, 2122–2135. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ngo, H.H.; Guo, W.; Liu, Y.; Li, J.; Liu, Y.; Zhang, X.; Jia, H. Insight into chemical phosphate recovery from municipal wastewater. Sci. Total Environ. 2017, 576, 159–171. [Google Scholar] [CrossRef]
- Zhang, S.; Ding, J.; Tian, D. Incorporation of MIL-101 (Fe or Al) into chitosan hydrogel adsorbent for phosphate removal: Performance and mechanism. J. Solid State Chem. 2022, 306, 122709. [Google Scholar] [CrossRef]
- Jiao, G.-J.; Ma, J.; Li, Y.; Jin, D.; Ali, Z.; Zhou, J.; Sun, R. Recent advances and challenges on removal and recycling of phosphate from wastewater using biomass-derived adsorbents. Chemosphere 2021, 278, 130377. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chi, L.; Wang, X.; Sui, Y.; Wang, Y.; Arandiyan, H. Review of metal (hydr)oxide and other adsorptive materials for phosphate removal from water. J. Environ. Chem. Eng. 2018, 6, 5269–5286. [Google Scholar] [CrossRef]
- Liu, R.; Chi, L.; Wang, X.; Wang, Y.; Sui, Y.; Xie, T.; Arandiyan, H. Effective and selective adsorption of phosphate from aqueous solution via trivalent-metals-based amino-MIL-101 MOFs. Chem. Eng. J. 2019, 357, 159–168. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; He, C.; Lyu, W.; Zhang, W.; Yan, W.; Yang, L. Development of rare earth element doped magnetic biochars with enhanced phosphate adsorption performance. Colloid. Surface. Asp. 2019, 561, 236–243. [Google Scholar] [CrossRef]
- Deng, H.; Yu, X. Adsorption of fluoride, arsenate and phosphate in aqueous solution by cerium impregnated fibrous protein. Chem. Eng. J. 2012, 184, 205–212. [Google Scholar] [CrossRef]
- Bacelo, H.; Pintor, A.M.A.; Santos, S.C.R.; Boaventura, R.A.R.; Botelho, C.M.S. Performance and prospects of different adsorbents for phosphorus uptake and recovery from water. Chem. Eng. J. 2020, 381, 122566. [Google Scholar] [CrossRef]
- Zhang, K.; Hu, L.; Wang, C.; Zhang, K. Middle-low-temperature oxidation and adsorption of arsenic from flue gas by Fe-Ce-based composite catalyst. Chemosphere 2022, 288, 132425. [Google Scholar] [CrossRef] [PubMed]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasireo, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, X.; Chen, X.; Huang, C.; Yang, S. Biochar-loaded Ce3+-enriched ultra-fine ceria nanoparticles for phosphate adsorption. J. Hazard. Mater. 2020, 396, 122626. [Google Scholar] [CrossRef]
- He, J.; Xu, Y.; Wang, W.; Hu, B.; Wang, Z.; Yang, X.; Wang, Y.; Yang, L. Ce(III) nanocomposites by partial thermal decomposition of Ce-MOF for effective phosphate adsorption in a wide pH range. Chem. Eng. J. 2020, 379, 122431. [Google Scholar] [CrossRef]
- Wu, B.; Lo, I.M.C. Surface functional group engineering of CeO2 particles for enhanced phosphate adsorption. Environ. Sci. Technol. 2020, 54, 4601–4608. [Google Scholar] [CrossRef]
- Feng, Y.; Lu, H.; Liu, Y.; Xue, L.; Dionysiou, D.D.; Yang, L.; Xing, B. Nano-cerium oxide functionalized biochar for phosphate retention: Preparation, optimization and rice paddy application. Chemosphere 2017, 185, 816–825. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Wang, X.; Yin, X.; Luo, N.; Khayambashi, A.; Wei, Y. The synthesis and characterization of hydrous cerium oxide nanoparticles loaded on porous silica micro-sphere as novel and efficient adsorbents to remove phosphate radicals from water. Micropor. Mesopor. Mater. 2019, 279, 73–81. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, H.B.; Madhavi, S.; Hng, H.H.; Lou, X.W.D. Formation of Fe2O3 microboxes with hierarchical shell structures from metal-organic frameworks and their lithium storage properties. J. Am. Chem. Soc. 2012, 134, 17388–17391. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Qi, Z.; Zhang, L.; Zhang, Y.; Huang, H.; Peng, Y. Designing ZIF-8/hydroxylated MWCNT nanocomposites for phosphate adsorption from water: Capability and mechanism. Chem. Eng. J. 2020, 394, 124992. [Google Scholar] [CrossRef]
- Li, S.; Lei, T.; Jiang, F.; Liu, M.; Wang, Y.; Wang, S.; Yang, X. Tuning the morphology and adsorption capacity of Al-MIL-101 analogues with Fe3+ for phosphorus removal from water. J. Colloid Interf. Sci. 2020, 560, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Y.; Li, F.; Shen, C.; Kaneti, Y.V.; Yamauchi, Y.; Yuliarto, B.; Chen, B.; Wang, C.-C. Defect-rich hierarchical porous UiO-66 (Zr) for tunable phosphate removal. Environ. Sci. Technol. 2021, 55, 13209–13218. [Google Scholar] [CrossRef]
- Zhang, Y.; Kang, X.; Guo, P.; Tan, H.; Zhang, S.-H. Studies on the removal of phosphate in water through adsorption using a novel Zn-MOF and its derived materials. Arab. J. Chem. 2022, 15, 103955. [Google Scholar] [CrossRef]
- Dong, P.; Jing, X.; Li, Y.; Shen, Y.; Li, Q.; Fang, Q. “Twin lotus flower” adsorbents derived from LaFe cyanometallate for high-performance phosphorus removal. Sep. Purif. Technol. 2022, 291, 120924. [Google Scholar] [CrossRef]
- Alexandrov, E.V.; Virovets, A.V.; Blatov, V.A.; Peresypkina, E. Topological Motifs in Cyanometallates: From Building Units to Three-Periodic Frameworks. Chem. Rev. 2015, 115, 12286–12319. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, J.J.; Liberka, M.; Zychowicz, M.; Chorazy, S. Diverse physical functionalities of rare-earth hexacyanidometallate frameworks and their molecular analogues. Inorg. Chem. Front. 2021, 8, 452–483. [Google Scholar] [CrossRef]
- Nai, J.; Lou, X.W. Hollow structures based on Prussian blue and its analogs for electrochemical energy storage and conversion. Adv. Mater. 2019, 31, 1706825. [Google Scholar] [CrossRef]
- Litvinova, Y.M.; Gayfulin, Y.M.; Leusen, J.; Samsonenko, D.G.; Lazarenko, V.A.; Zubavichus, Y.V.; Kögerler, P.; Mironov, Y.V. Metal-organic frameworks based on polynuclear lanthanide complexes and octahedral rhenium clusters. Inorg. Chem. Front. 2019, 6, 1518–1526. [Google Scholar] [CrossRef]
- Li, Y.; Jing, X.; Li, Q.; Shen, Y.; Fang, Q. Well-defined bimetal oxides derived from Prussian blue analogues with regulable active sites for phosphate removal. J. Colloid Interf. Sci. 2022, 622, 390–401. [Google Scholar] [CrossRef]
- Peng, B.; Cui, J.; Wang, Y.; Liu, J.; Zheng, H.; Jin, L.; Zhang, X.; Zhang, Y.; Wu, Y. CeO2-x/C/rGO nanocomposites derived from Ce-MOF and graphene oxide as robust platform for highly sensitive uric acid detection. Nanoscale 2018, 10, 1939–1945. [Google Scholar] [CrossRef]
- Schelter, E.J. Cerium under the lens. Nat. Chem. 2013, 5, 348. [Google Scholar] [CrossRef]
- Qu, F.; Cao, A.; Yang, Y.; Mahmud, S.; Su, P.; Yang, J.; He, Z.; Lai, Q.; Zhu, L.; Tu, Z.; et al. Hierarchically superhydrophilic poly(vinylidene fluoride) membrane with self-cleaning fabricated by surface mineralization for stable separation of oily wastewater. J. Membr. Sci. 2021, 640, 119864. [Google Scholar] [CrossRef]
- Jiao, G.; Ma, J.; Zhang, Y.; Jin, D.; Li, Y.; Hu, C.; Guo, Y.; Wang, Z.; Zhou, J.; Sun, R. Nitrogen-doped lignin-derived biochar with enriched loading of CeO2 nanoparticles for highly efficient and rapid phosphate capture. Int. J. Biol. Macromol. 2021, 182, 1484–1494. [Google Scholar] [CrossRef]
- Wang, N.; Feng, J.; Chen, J.; Wang, J.; Yan, W. Adsorption mechanism of phosphate by polyaniline/TiO2 composite from wastewater. Chem. Eng. J. 2017, 316, 33–40. [Google Scholar] [CrossRef]
- Huang, Y.; Lee, X.; Grattieri, M.; Yuan, M.; Cai, R.; Macazo, F.C.; Minteer, S.D. Modified biochar for phosphate adsorption in environmentally relevant conditons. Chem. Eng. J. 2020, 380, 122375. [Google Scholar] [CrossRef]
- He, J.; Xu, Y.; Shao, P.; Yang, L.; Sun, Y.; Yang, Y.; Cui, F.; Wang, W. Modulation of coordinative unsaturation degree and valence state for cerium-based adsorbent to boost phosphate adsorption. Chem. Eng. J. 2020, 394, 124912. [Google Scholar] [CrossRef]
- Abdellaoui, Y.; Oualid, H.A.; Hsini, A.; Ibrahimi, B.E.; Laabd, M.; Ouardi, M.E.; Giacoman-Vallejos, G.; Gamero-Melo, P. Synthesis of zirconium-modified merlinoite from fly ash for enhanced removal of phosphate in aqueous medium: Experimental studies supported by Monte Carlo/SA simulaitons. Chem. Eng. J. 2021, 404, 126600. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, B.; Shan, C.; Gao, X. Enhanced phosphate removal by nanosized hydrated La(III) oxide confined in cross-linked polystyrene networks. Environ. Sci. Technol. 2016, 50, 1447–1454. [Google Scholar] [CrossRef]
- Liu, M.; Li, S.; Tang, N.; Wang, Y.; Yang, X.; Wang, S. Highly efficient capture of phosphate from water via cerium-doped metal-organic frameworks. J. Clean. Prod. 2020, 265, 121782. [Google Scholar] [CrossRef]
- Xiang, C.; Ji, Q.; Zhang, G.; Wang, H.; Qu, J. In situ creation of oxygen vacancies in porous bimetallic La/Zr sorbent for aqueous phosphate: Hierarchical pores control mass transport and vacancy sites determine interaction . Environ. Sci. Technol. 2020, 54, 437–445. [Google Scholar] [CrossRef]
- Xiong, W.; Tong, J.; Yang, Z.; Zeng, G.; Zhou, Y.; Wang, D.; Song, P.; Xu, R.; Zhang, C.; Cehng, M. Adsorption of phosphate from aqueous solution using iron-zirconium modified activated carbon nanofiber: Performance and mechanism. J. Colloid. Interf. Sci. 2017, 493, 17–23. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, X.; Dong, P.; Min, H.; Luo, J.; Huang, W.; Wang, X.; Li, Q.; Fang, Q. “Sea Anemone”-like CeFe Oxides for High-Efficient Phosphate Removal. Water 2022, 14, 2445. https://doi.org/10.3390/w14152445
Tan X, Dong P, Min H, Luo J, Huang W, Wang X, Li Q, Fang Q. “Sea Anemone”-like CeFe Oxides for High-Efficient Phosphate Removal. Water. 2022; 14(15):2445. https://doi.org/10.3390/w14152445
Chicago/Turabian StyleTan, Xiaoying, Pingping Dong, Hongping Min, Jinxue Luo, Wenhai Huang, Xiaodong Wang, Qingqing Li, and Qile Fang. 2022. "“Sea Anemone”-like CeFe Oxides for High-Efficient Phosphate Removal" Water 14, no. 15: 2445. https://doi.org/10.3390/w14152445
APA StyleTan, X., Dong, P., Min, H., Luo, J., Huang, W., Wang, X., Li, Q., & Fang, Q. (2022). “Sea Anemone”-like CeFe Oxides for High-Efficient Phosphate Removal. Water, 14(15), 2445. https://doi.org/10.3390/w14152445