Genome-Resolved Metagenomics and Antibiotic Resistance Genes Analysis in Reclaimed Water Distribution Systems


Abstract
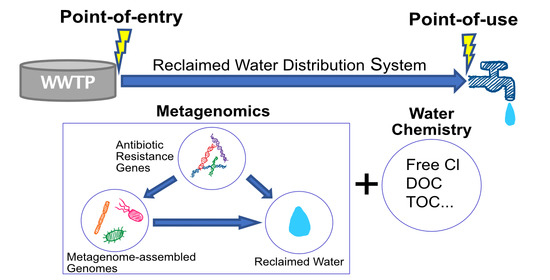
:
1. Introduction
2. Materials and Methods
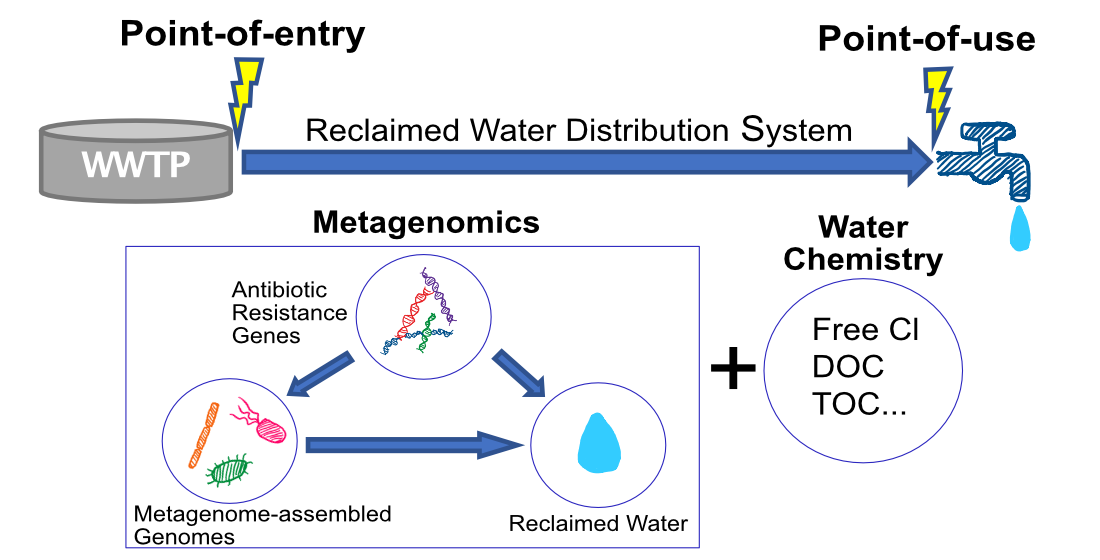
2.1. Datasets Collection and Description
2.2. Identification of Bacterial Community Compositions and Metagenome Assembled Genomes (MAG)
2.3. Relative Abundance and Indices of Replication of Metagenome Assembled Genomes
2.4. Antibiotic Resistance Gene Detection for MAGs and Read Contigs
2.5. Horizontal Gene Transfer Detection for ARGs from MAGs
2.6. Statistical Analyses for Water Chemistry Data
3. Results and Discussion
3.1. Water Quality across Different Utility
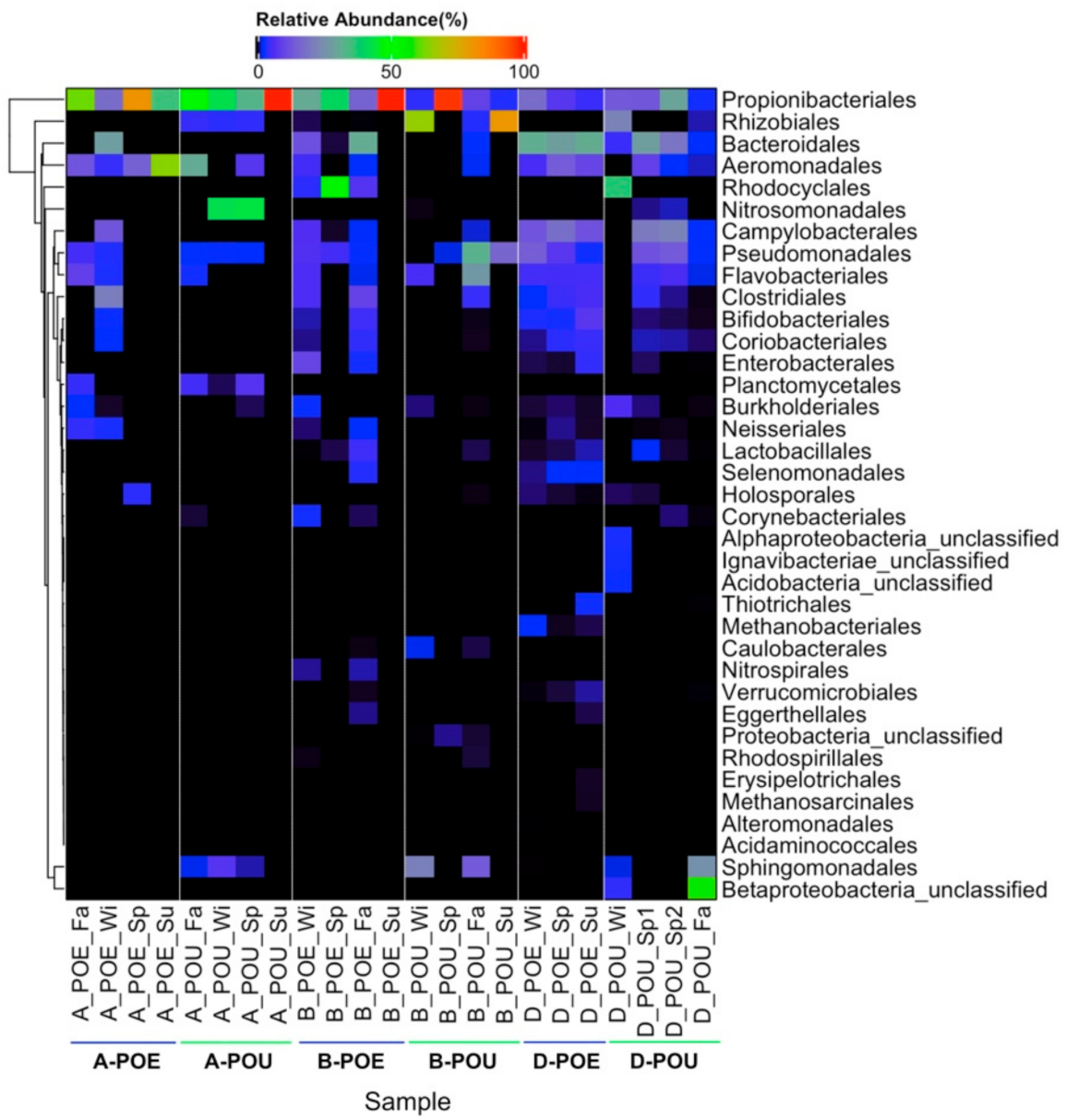
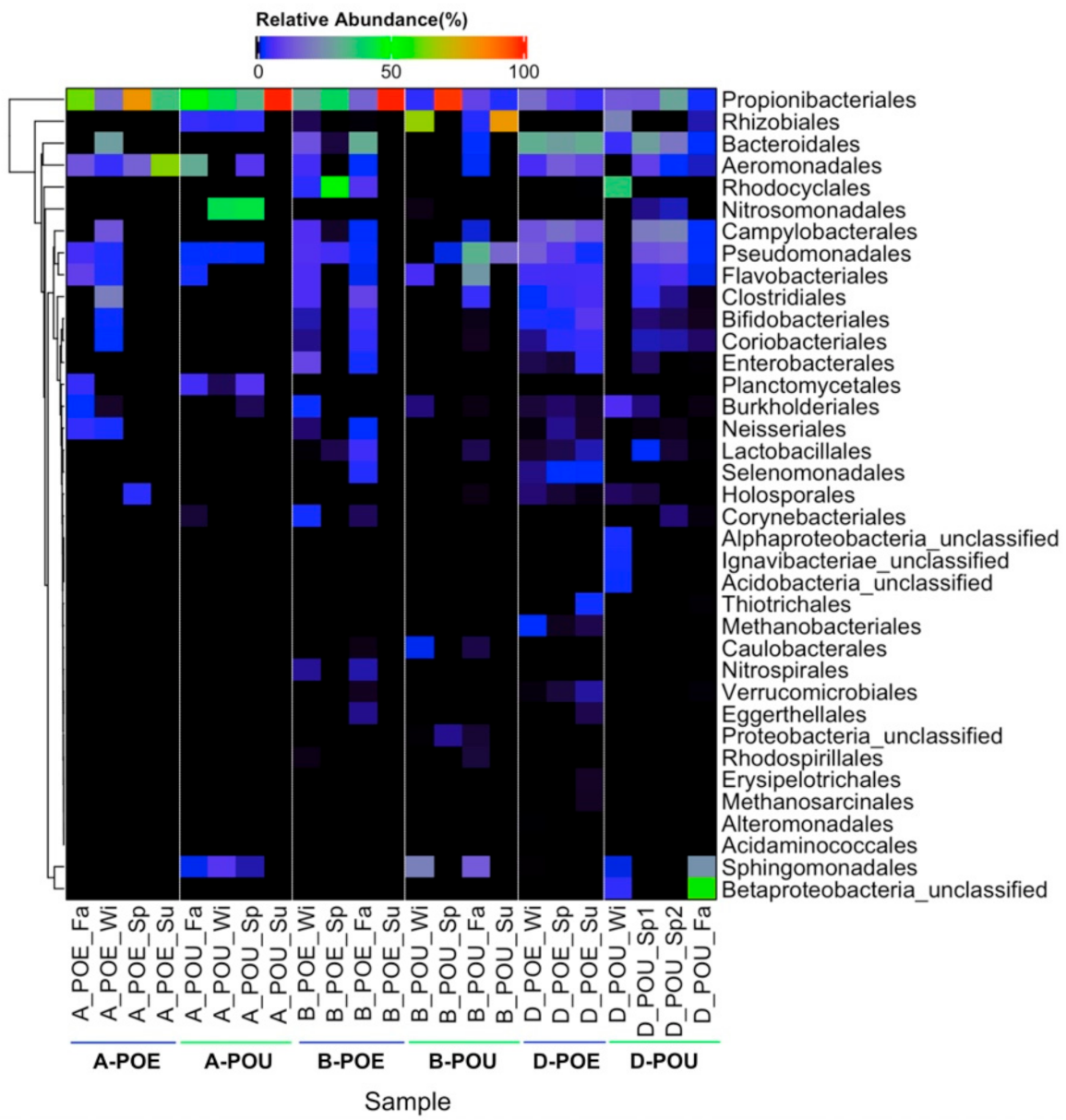
3.2. Bacterial Composition across Different Samples and Sites
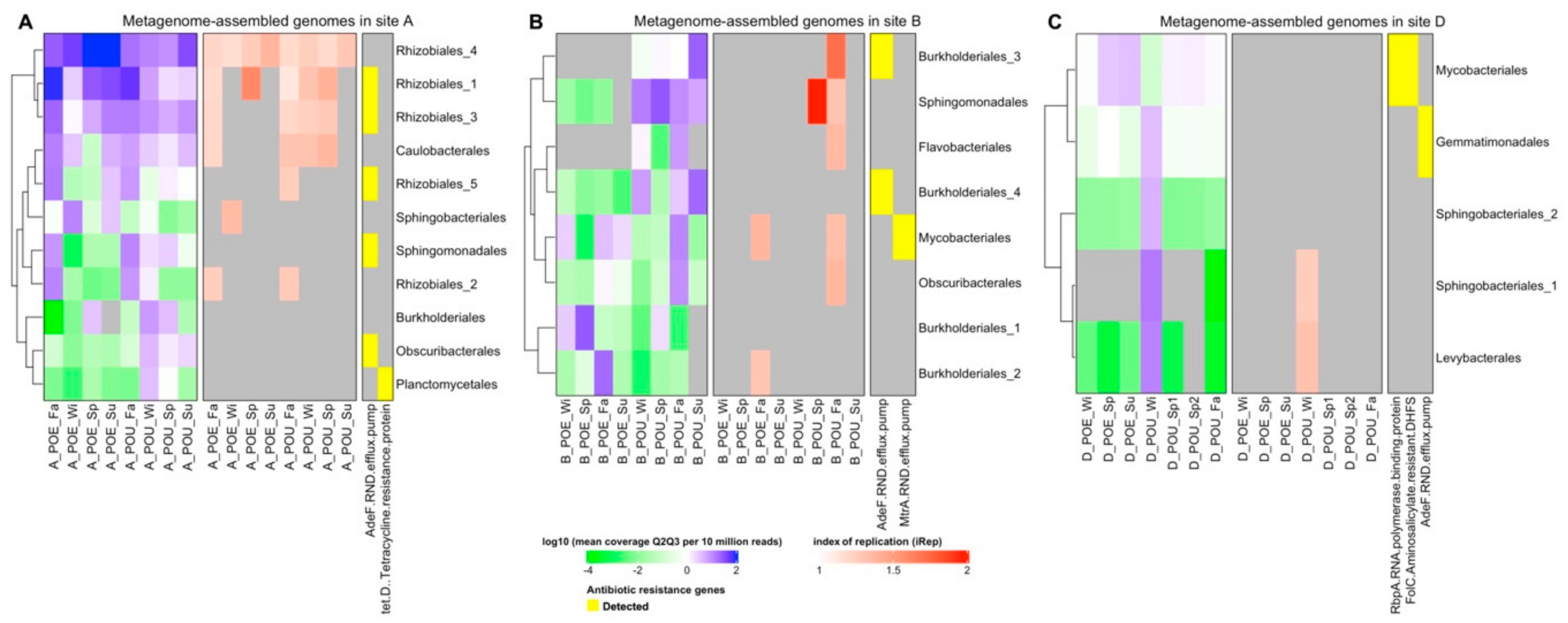
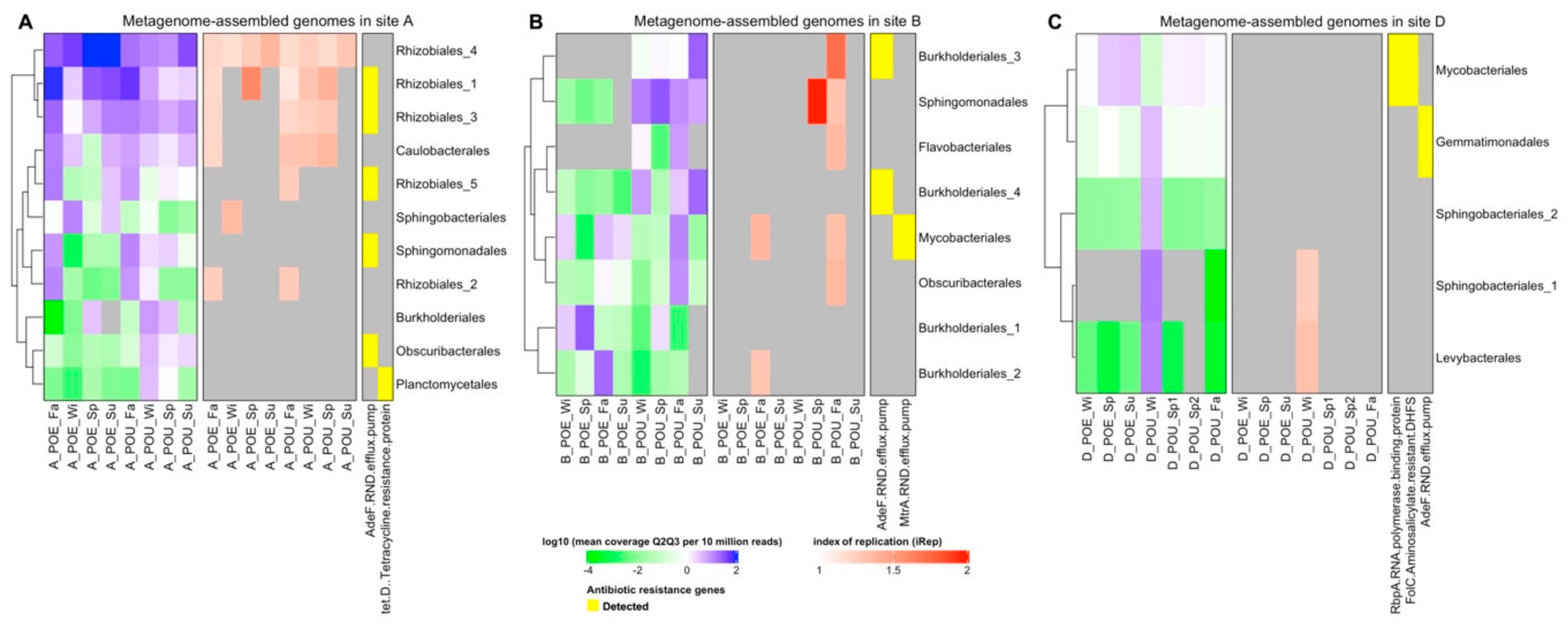
3.3. Metagenome Assembled Genomes (MAG) across Different Samples
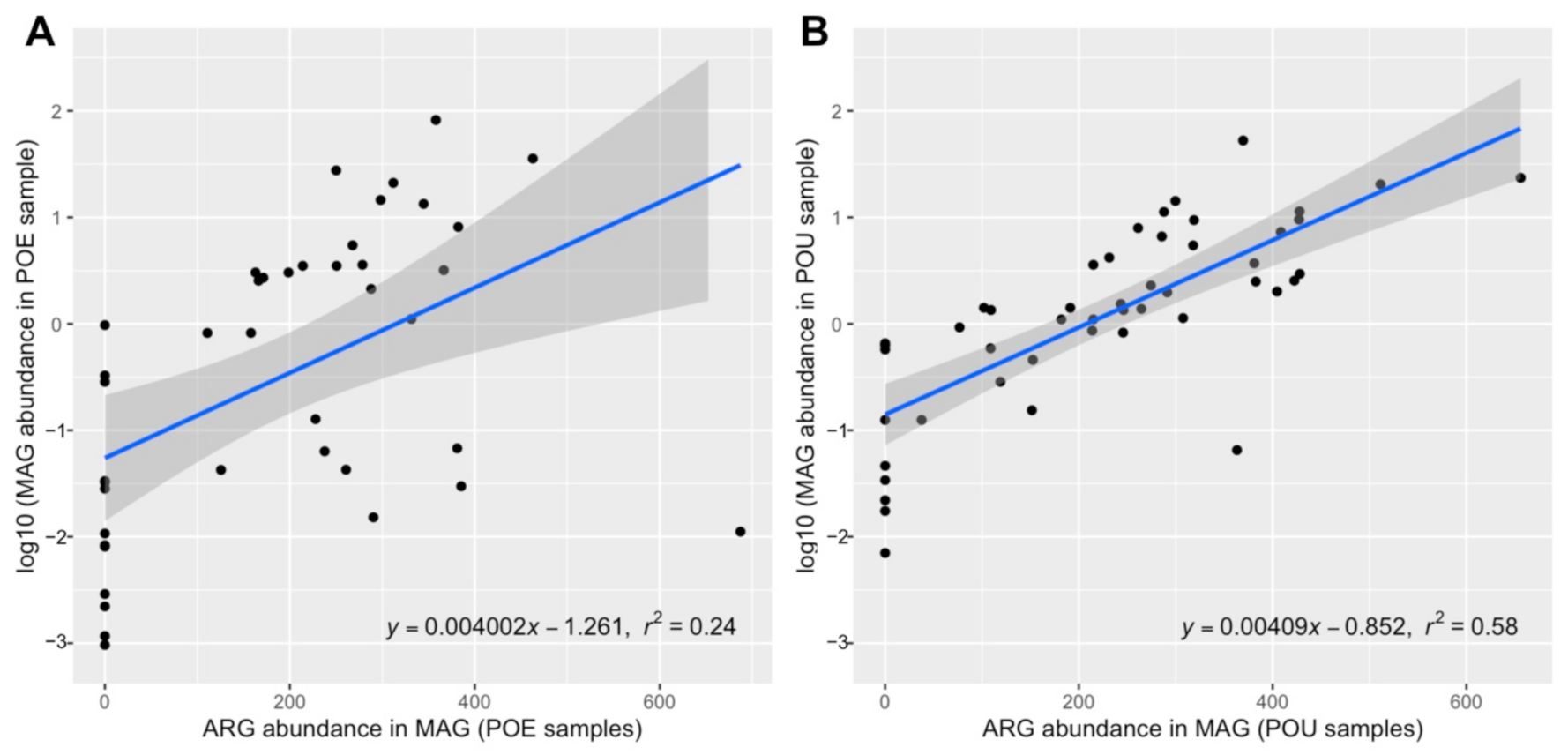
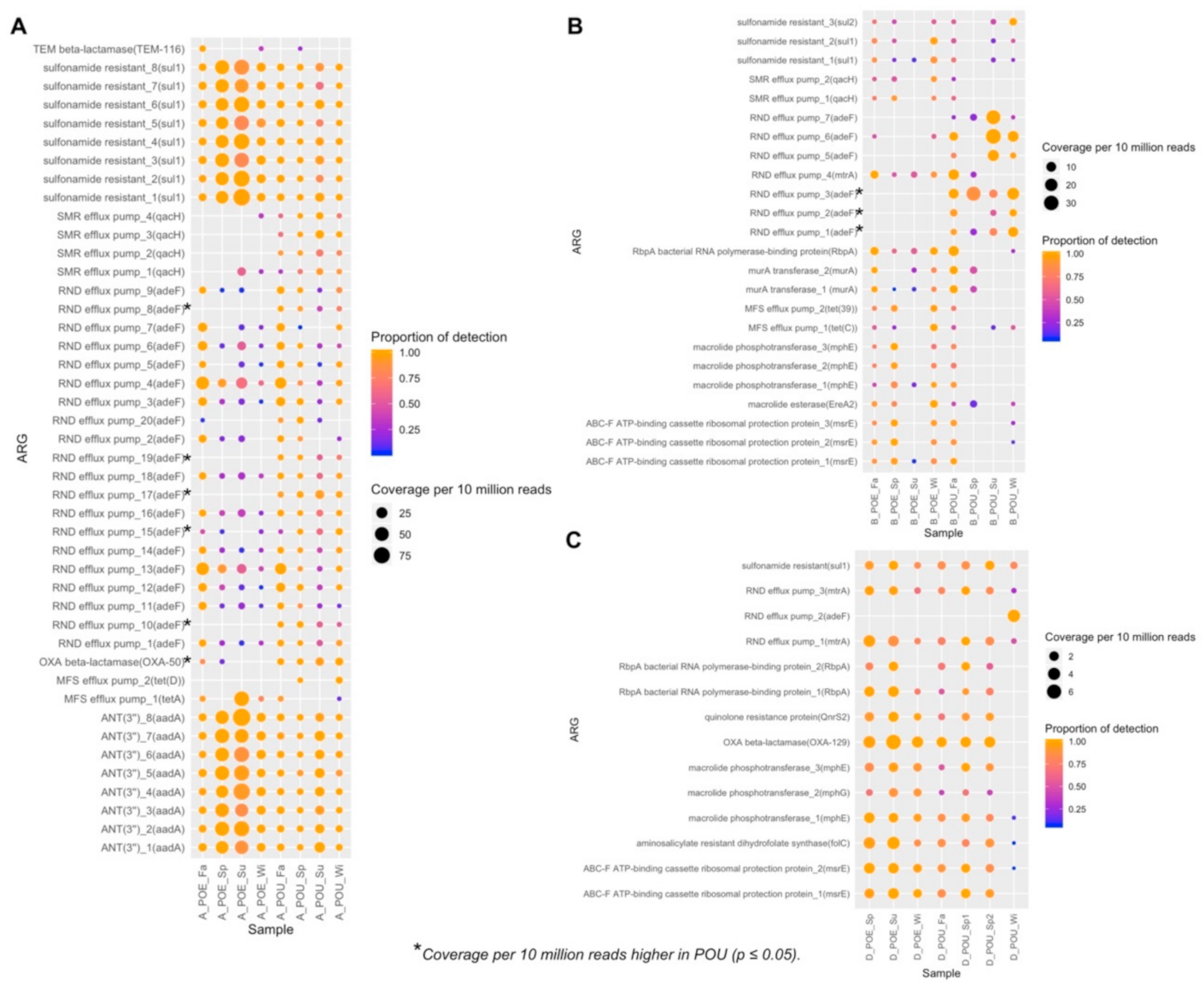
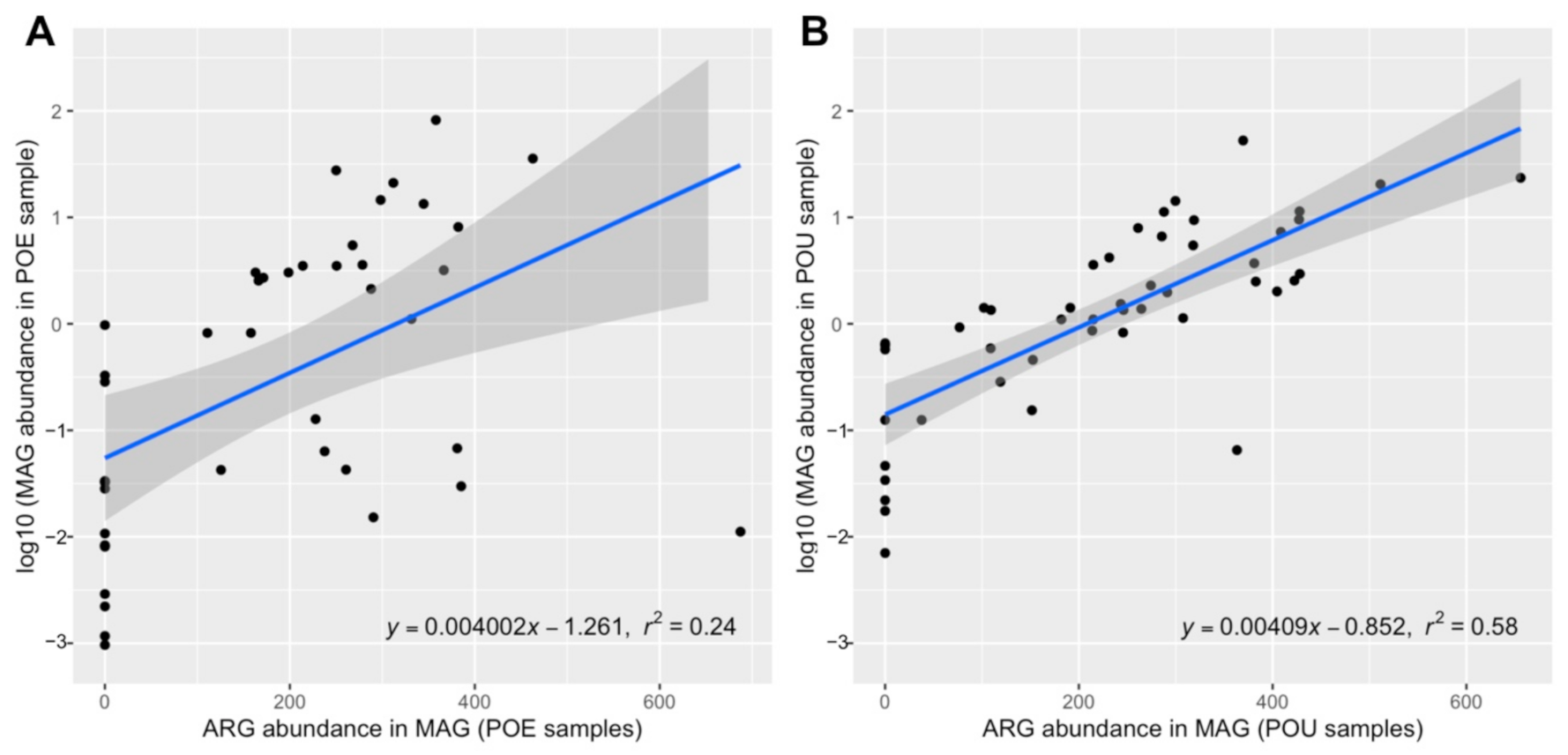
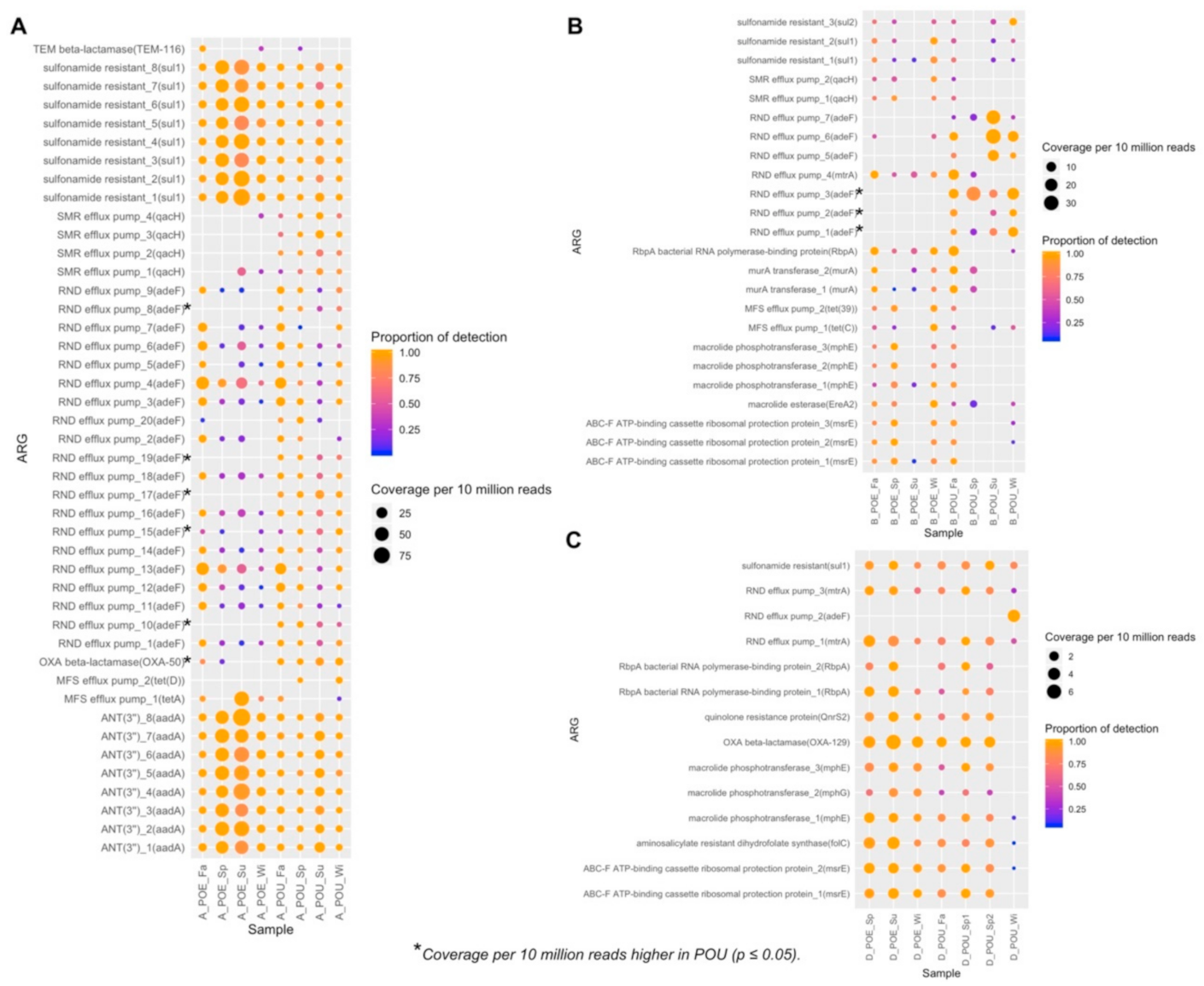
3.4. ARG Performance across Water Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boretti, A.; Rosa, L. Reassessing the projections of the World Water Development Report. NPJ Clean Water 2019, 2, 2. [Google Scholar] [CrossRef]
- Jimenez, B.; Asano, T. Water Reuse: An International Survey of current practice, issues and needs. Water Intell. Online 2015, 7. [Google Scholar] [CrossRef]
- Hong, P.-Y.; Wang, C.; Mantilla-Calderon, D. Mitigating Antimicrobial Resistance Risks When Using Reclaimed Municipal Wastewater for Agriculture. In Antibiotic Resistance in the Environment: A Worldwide Overview; Springer Nature: Cham, Switzerland, 2020; pp. 245–265. [Google Scholar]
- Hong, P.-Y.; Al-Jassim, N.; Ansari, M.I.; Mackie, R.I. Environmental and Public Health Implications of Water Reuse: Antibiotics, Antibiotic Resistant Bacteria, and Antibiotic Resistance Genes. Antibiotics 2013, 2, 367–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Zhang, X.-J. Biological stability in drinking water: A regression analysis of influencing factors. J. Environ. Sci. 2005, 17, 395–398. [Google Scholar]
- Urgun-Demirtas, M.; Sattayatewa, C.; Pagilla, K.R. Bioavailability of Dissolved Organic Nitrogen in Treated Effluents. Water Environ. Res. 2008, 80, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Courtois, S.; Rosikiewicz, M.; Piriou, P.; Aeby, S.; Robert, S.; Loret, J.-F.; Greub, G. Reduced Chlorine in Drinking Water Distribution Systems Impacts Bacterial Biodiversity in Biofilms. Front. Microbiol. 2018, 9, 2520. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Zhang, M.; Zhang, S.; Yu, X. Can chlorination co-select antibiotic-resistance genes? Chemosphere 2016, 156, 412–419. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, A.Z.; He, M.; Li, D.; Chen, J. Subinhibitory Concentrations of Disinfectants Promote the Horizontal Transfer of Multidrug Resistance Genes within and across Genera. Environ. Sci. Technol. 2017, 51, 570–580. [Google Scholar] [CrossRef]
- Aminov, R.I. Horizontal Gene Exchange in Environmental Microbiota. Front. Microbiol. 2011, 2, 158. [Google Scholar] [CrossRef] [Green Version]
- Hong, P.-Y.; Mantilla-Calderon, D.; Wang, C. Metagenomics as a Tool to Monitor Reclaimed-Water Quality. Appl. Environ. Microbiol. 2020, 86, 86. [Google Scholar] [CrossRef]
- Garner, E.; Chen, C.; Xia, K.; Bowers, J.; Engelthaler, D.M.; McLain, J.; Edwards, M.A.; Pruden, A. Metagenomic Characterization of Antibiotic Resistance Genes in Full-Scale Reclaimed Water Distribution Systems and Corresponding Potable Systems. Environ. Sci. Technol. 2018, 52, 6113–6125. [Google Scholar] [CrossRef]
- Garner, E.; McLain, J.; Bowers, J.; Engelthaler, D.M.; Edwards, M.A.; Pruden, A. Microbial Ecology and Water Chemistry Impact Regrowth of Opportunistic Pathogens in Full-Scale Reclaimed Water Distribution Systems. Environ. Sci. Technol. 2018, 52, 9056–9068. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; Souza, M.D.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.L.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community Ecol. Package Version 2013, 2, 1–295. [Google Scholar]
- Heberle, H.; Meirelles, G.V.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.W. Megahit: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Brown, C.T.; Olm, C.T.B.M.R.; Thomas, B.C.; Banfield, J.F. Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol. 2016, 34, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wilke, C.O.; Wickham, H.; Wilke, M.C.O. Package ‘Cowplot’. Streamlined Plot Theme and Plot Annotations for ‘ggplot2’. 2019. Available online: https://cran.r-project.org/web/packages/cowplot/index.html (accessed on 20 October 2020).
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Leplae, R.; Lima-Mendez, G.; Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010, 38, D57–D61. [Google Scholar] [CrossRef] [Green Version]
- Geck, C.E. Wiley StatsRef: Statistics Reference Online. Choice Rev. Online 2015, 52, 52. [Google Scholar] [CrossRef]
- Zaouri, N.; Jumat, M.R.; Cheema, T.; Hong, P.Y. Metagenomics-based evaluation of groundwater microbial profiles in response to treated wastewater discharge. Environ. Res. 2020, 180, 108835. [Google Scholar] [CrossRef]
- Weinrich, L.; Jjemba, P.K.; Giraldo, E.; Lechevallier, M. Implications of organic carbon in the deterioration of water quality in reclaimed water distribution systems. Water Res. 2010, 44, 5367–5375. [Google Scholar] [CrossRef]
- Kantor, R.S.; Miller, S.E.; Nelson, K.L. The Water Microbiome Through a Pilot Scale Advanced Treatment Facility for Direct Potable Reuse. Front. Microbiol. 2019, 10, 993. [Google Scholar] [CrossRef] [Green Version]
- Thayanukul, P.; Kurisu, F.; Kasuga, I.; Furumai, H. Characterization of bacterial isolates from water reclamation systems on the basis of substrate utilization patterns and regrowth potential in reclaimed water. Water Sci. Technol. 2013, 68, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guan, Y.; Zhao, R.; Feng, J.; Huang, J.; Ma, L.; Li, B. Metagenomic and network analyses decipher profiles and co-occurrence patterns of antibiotic resistome and bacterial taxa in the reclaimed wastewater distribution system. J. Hazard. Mater. 2020, 400, 123170. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, S.; Wang, Y.; Zhao, H.-P.; Song, L. Metagenomic analysis of antibiotic resistance genes (ARGs) during refuse decomposition. Sci. Total Environ. 2018, 634, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-H.; Qiao, M.; Su, J.-Q.; Chen, Z.; Zhou, X.; Zhu, Y.-G. High Throughput Profiling of Antibiotic Resistance Genes in Urban Park Soils with Reclaimed Water Irrigation. Environ. Sci. Technol. 2014, 48, 9079–9085. [Google Scholar] [CrossRef]
- Al-Jassim, N.; Ansari, M.I.; Harb, M.; Hong, P.-Y. Removal of bacterial contaminants and antibiotic resistance genes by conventional wastewater treatment processes in Saudi Arabia: Is the treated wastewater safe to reuse for agricultural irrigation? Water Res. 2015, 73, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Al-Jassim, N.; Mantilla-Calderon, D.; Scarascia, G.; Hong, P.-Y. Bacteriophages to Sensitize a Pathogenic New Delhi Metallo β-Lactamase-Positive Escherichia coli to Solar Disinfection. Environ. Sci. Technol. 2018, 52, 14331–14341. [Google Scholar] [CrossRef] [Green Version]
- AlJassim, N.I.; Mantilla-Calderon, D.; Wang, T.; Hong, P.-Y. Inactivation and Gene Expression of a Virulent Wastewater Escherichia coli Strain and the Nonvirulent Commensal Escherichia coli DSM1103 Strain upon Solar Irradiation. Environ. Sci. Technol. 2017, 51, 3649–3659. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Utility (Number of Samples) | Temperature (°C) | Total Cl (mg/L) | Free Cl (mg/L) | pH | Turbidity (NTU) | Conductivity (S/m) | Dissolved Oxygen (mg/L) | Total Organic Carbon (μg/L) | Dissolved Organic Carbon (μg/L) | Biodegradable Dissolved Organic Carbon (μg/L) |
|---|---|---|---|---|---|---|---|---|---|---|
| A (n = 20) | 26.7 ± 4.3 ↑ | 0.4 ± 0.4 ↓ | 0.2 ± 0.2 | 7.7 ± 0.3 ↑ | 5.4 ± 10.8 | 1354.4 ± 215.4 | 6.5 ± 1.3 | 5714 ± 2564 | 6351 ± 2761 | 2137 ± 2321 |
| B (n = 19) | 19.6 ± 2.9 | 2.3 ± 3.1 | 1.1 ± 2.1 ↑ | 7.3 ± 0.2 | 2.5 ± 2.6 | 736.9 ± 80.3 ↓ | 4 ± 1.4 ↓ | 10,123 ± 6173 ↑ | 11,087 ± 7014 ↑ | 6094 ± 8777 ↑ |
| D (n = 20) | 20 ± 1.8 | 2.7 ± 2.3 | 0.2 ± 0.2 | 7.2 ± 0.1 | 2 ± 1 | 1542.9 ± 283.1 | 6.8 ± 2.4 | 3961 ± 2120 | 4333 ± 2069 ↓ | 2621 ± 1238 |
| A-POE | A-POU | B-POE | B-POU | D-POE | D-POU | |
|---|---|---|---|---|---|---|
| Propionibacteriales | 49.1 ± 28.0 | 56.9 ± 29.8 | 46.7 ± 37.0 | 28.6 ± 45.8 | 10.0 ± 6.7 | 14.9 ± 10.9 |
| Rhizobiales | -- | 3.1 ± 2.1 | 0.2 ± 0.3 | 37.7 ± 41.5 | -- | 6.0 ± 11.0 |
| Sphingomonadales | -- | 2.7 ± 3.3 | -- | 8.9 ± 10.7 | 0.02 ± 0.04 | 6.8 ± 12.8 |
| Pseudomonadales | 2.2 ± 2.7 | 1.8 ± 1.2 | 3.8 ± 3.0 | 12.5 ± 15.0 | 8.4 ± 5.8 | 7.2 ± 6.9 |
| Flavobacteriales | 3.3 ± 4.3 | 0.8 ± 1.6 | 2.1 ± 3.0 | 8.2 ± 12.7 | 5.4 ± 0.1 | 3.2 ± 2.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Hong, P.-Y. Genome-Resolved Metagenomics and Antibiotic Resistance Genes Analysis in Reclaimed Water Distribution Systems. Water 2020, 12, 3477. https://doi.org/10.3390/w12123477
Wang C, Hong P-Y. Genome-Resolved Metagenomics and Antibiotic Resistance Genes Analysis in Reclaimed Water Distribution Systems. Water. 2020; 12(12):3477. https://doi.org/10.3390/w12123477
Chicago/Turabian StyleWang, Changzhi, and Pei-Ying Hong. 2020. "Genome-Resolved Metagenomics and Antibiotic Resistance Genes Analysis in Reclaimed Water Distribution Systems" Water 12, no. 12: 3477. https://doi.org/10.3390/w12123477
APA StyleWang, C., & Hong, P.-Y. (2020). Genome-Resolved Metagenomics and Antibiotic Resistance Genes Analysis in Reclaimed Water Distribution Systems. Water, 12(12), 3477. https://doi.org/10.3390/w12123477