Palmitic Acid on Salt Subphases and in Mixed Monolayers of Cerebrosides: Application to Atmospheric Aerosol Chemistry

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
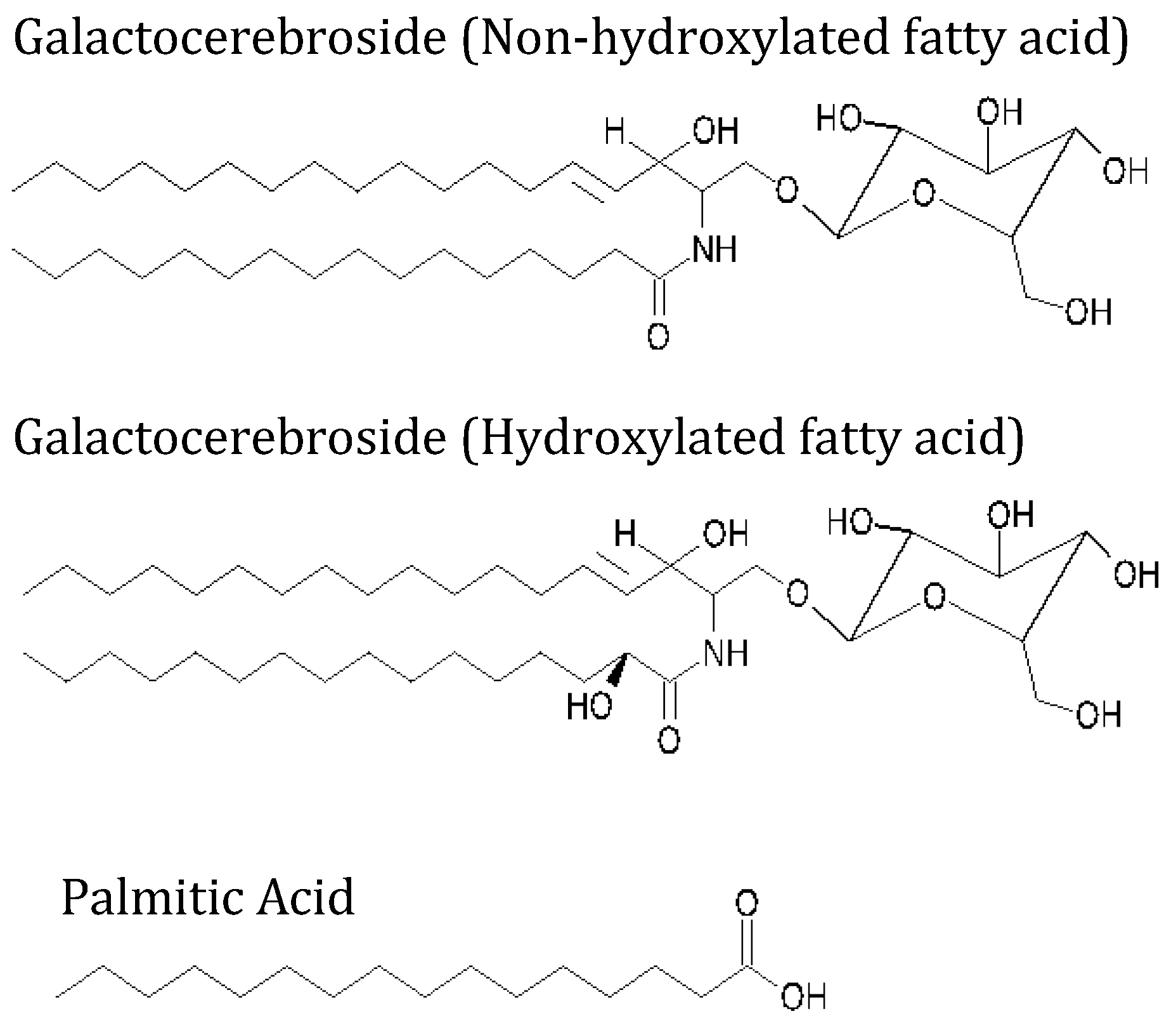
2.2. Methods
2.2.1. Langmuir Film Balance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | Concentration (g ion/kg Water) | Concentration (mol/L) |
|---|---|---|
| Na+ | 10.780 | 0.4521 |
| K+ | 0.420 | 0.0104 |
| Mg2+ | 1.320 | 0.0530 |
| Ca2+ | 0.400 | 0.0096 |
| Sr2+ | 0.0088 | 9.64 × 10−5 |
| Cl− | 19.290 | 0.5315 |
| SO42− | 2.660 | 0.0267 |
| HCO3− | 0.200 | 0.0032 |
| Br− | 0.056 | 6.75 × 10−4 |
| F− | 0.001 | 5.11 × 10−5 |
2.2.2. Brewster Angle Microscopy
2.2.3. Vibrational Sum Frequency Generation Spectroscopy
3. Results and Discussion
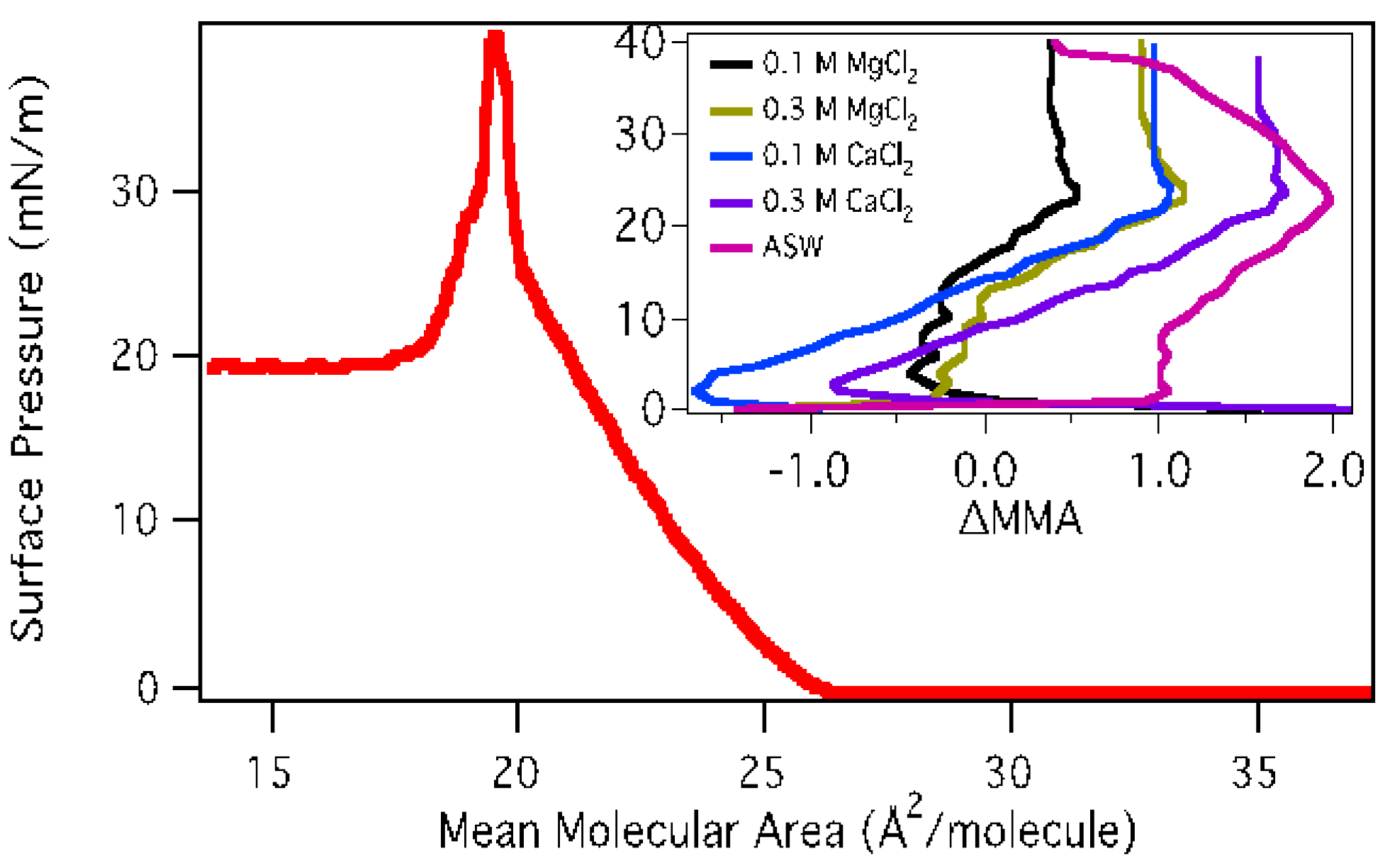
3.1. Effect of Subphase Composition on Surface Morphology of Palmitic Acid
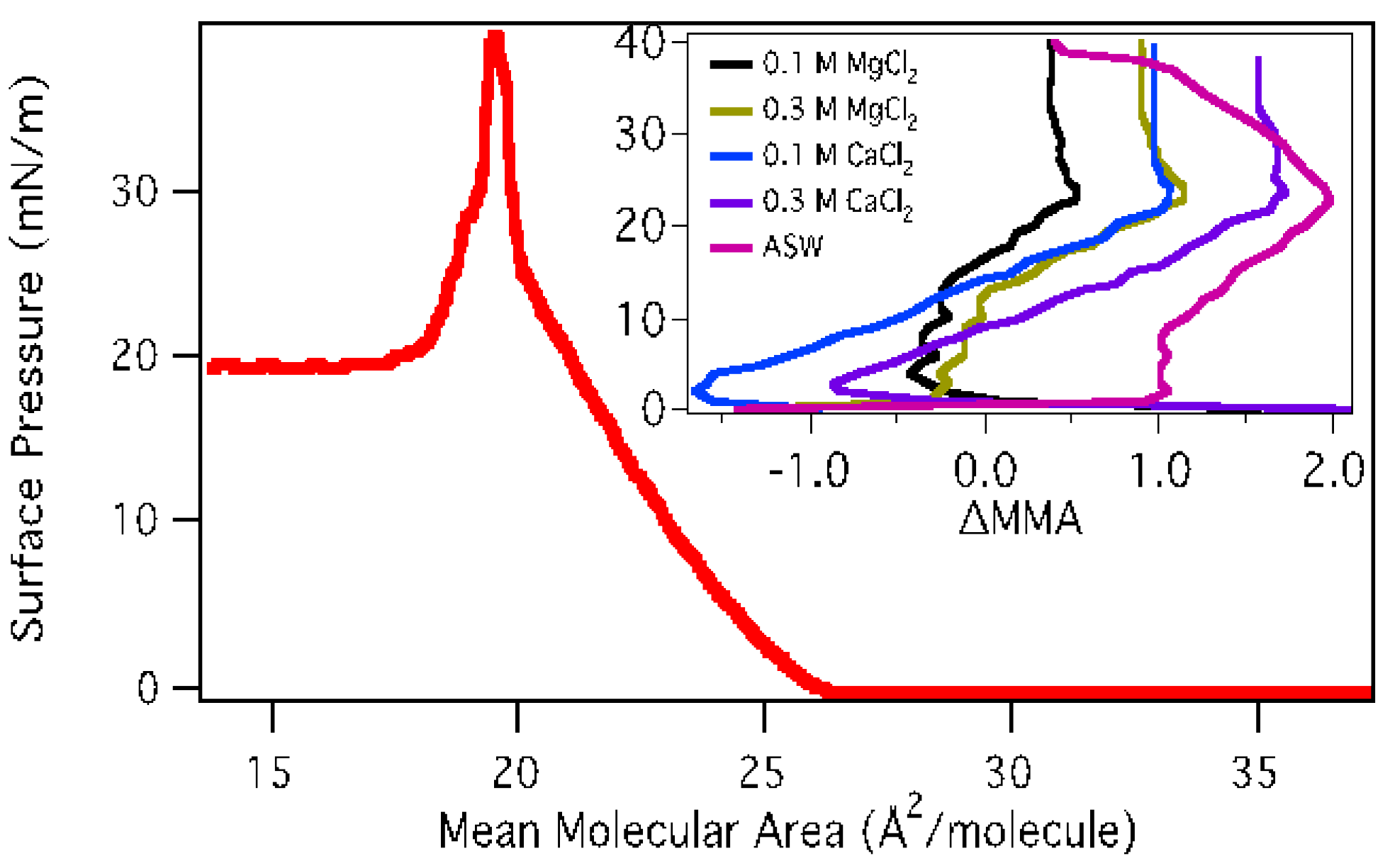
3.1.1. Surface Pressure-Area Isotherms
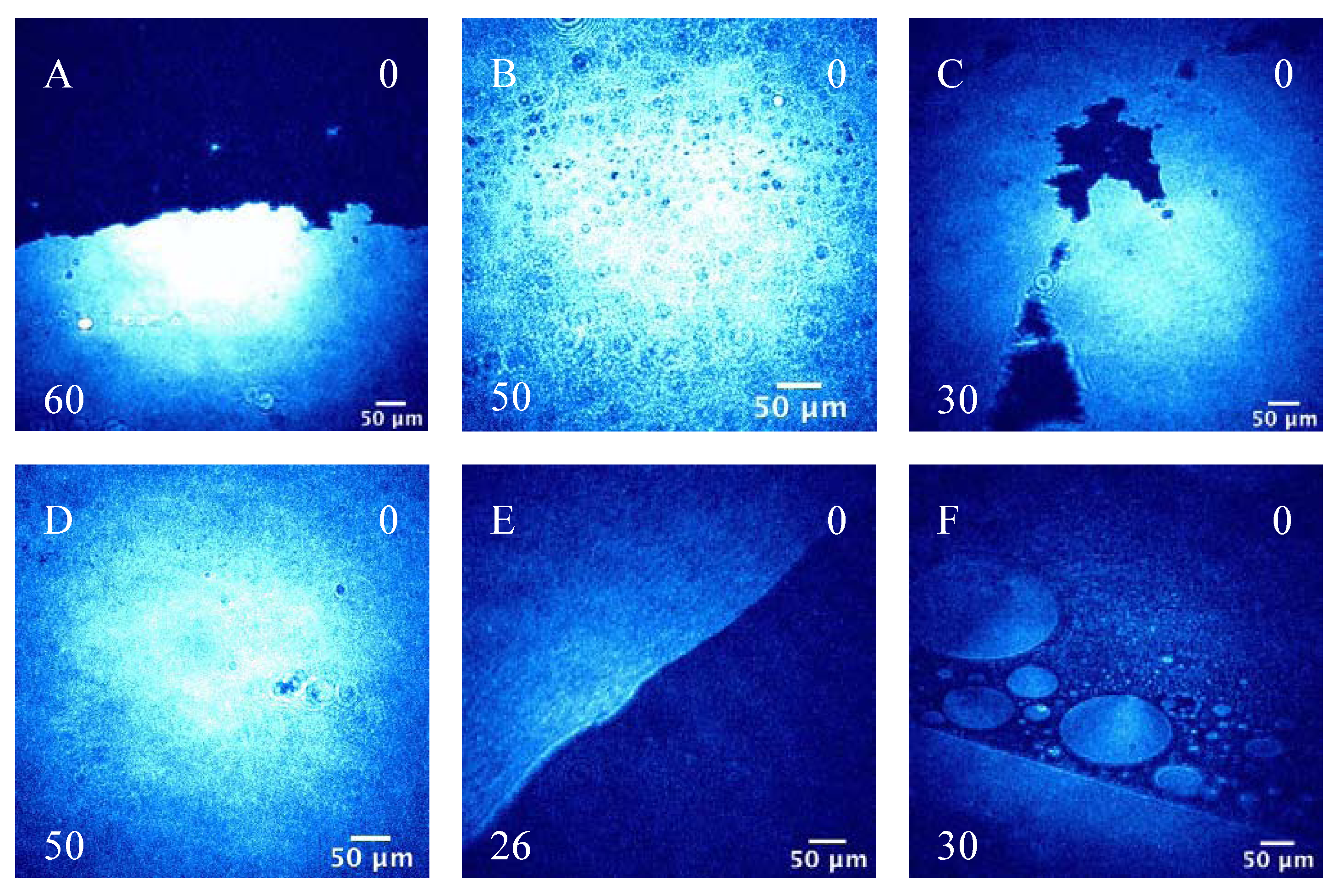
3.1.2. BAM Images
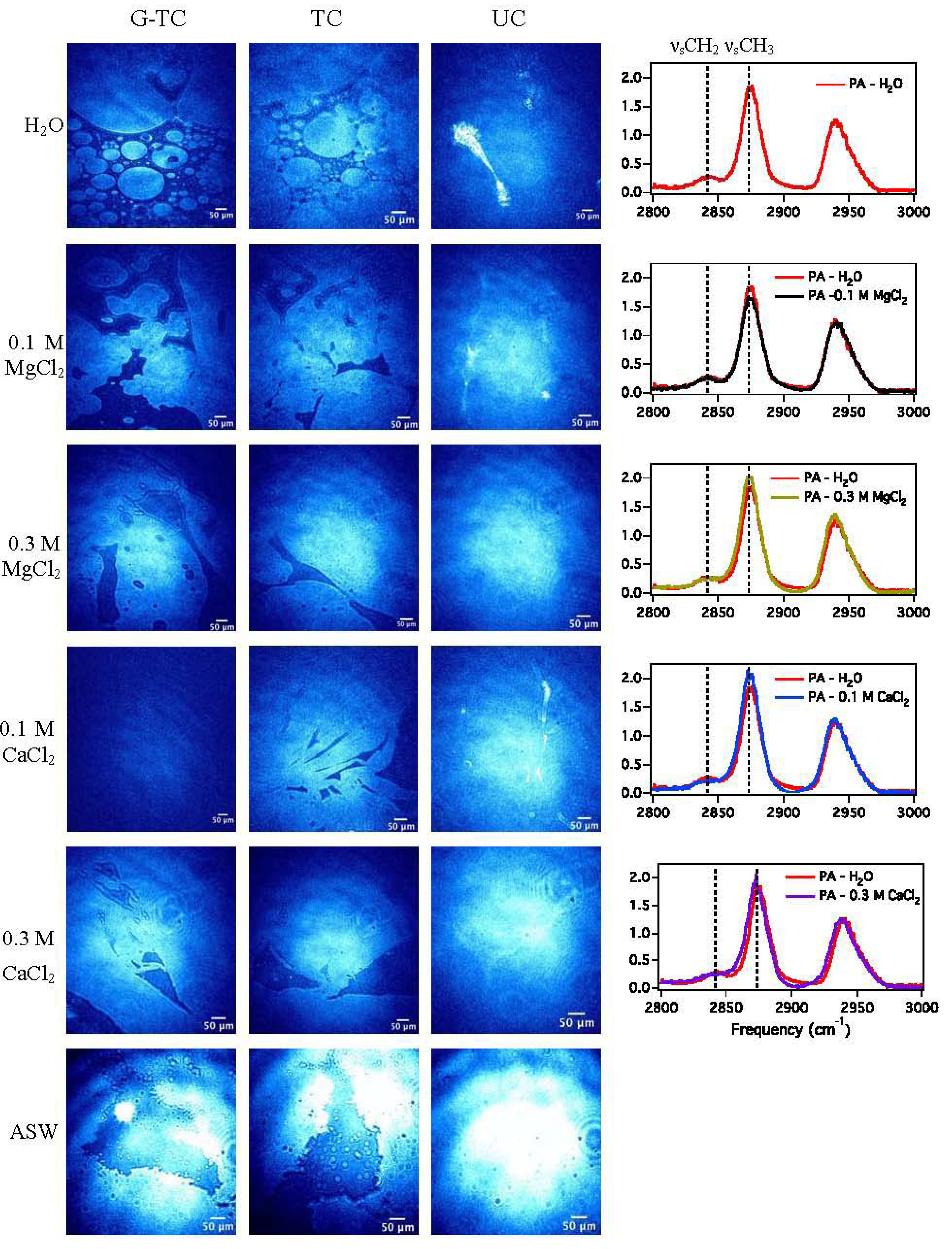
3.1.3. Sum Frequency Generation
| Subphase | νsCH3/νsCH2 |
|---|---|
| H2O | 6.7 |
| 0.1 M MgCl2 | 6.8 |
| 0.3 M MgCl2 | 7.9 |
| 0.1 M CaCl2 | 11.3 |
| 0.3 M CaCl2 | 8.1 |
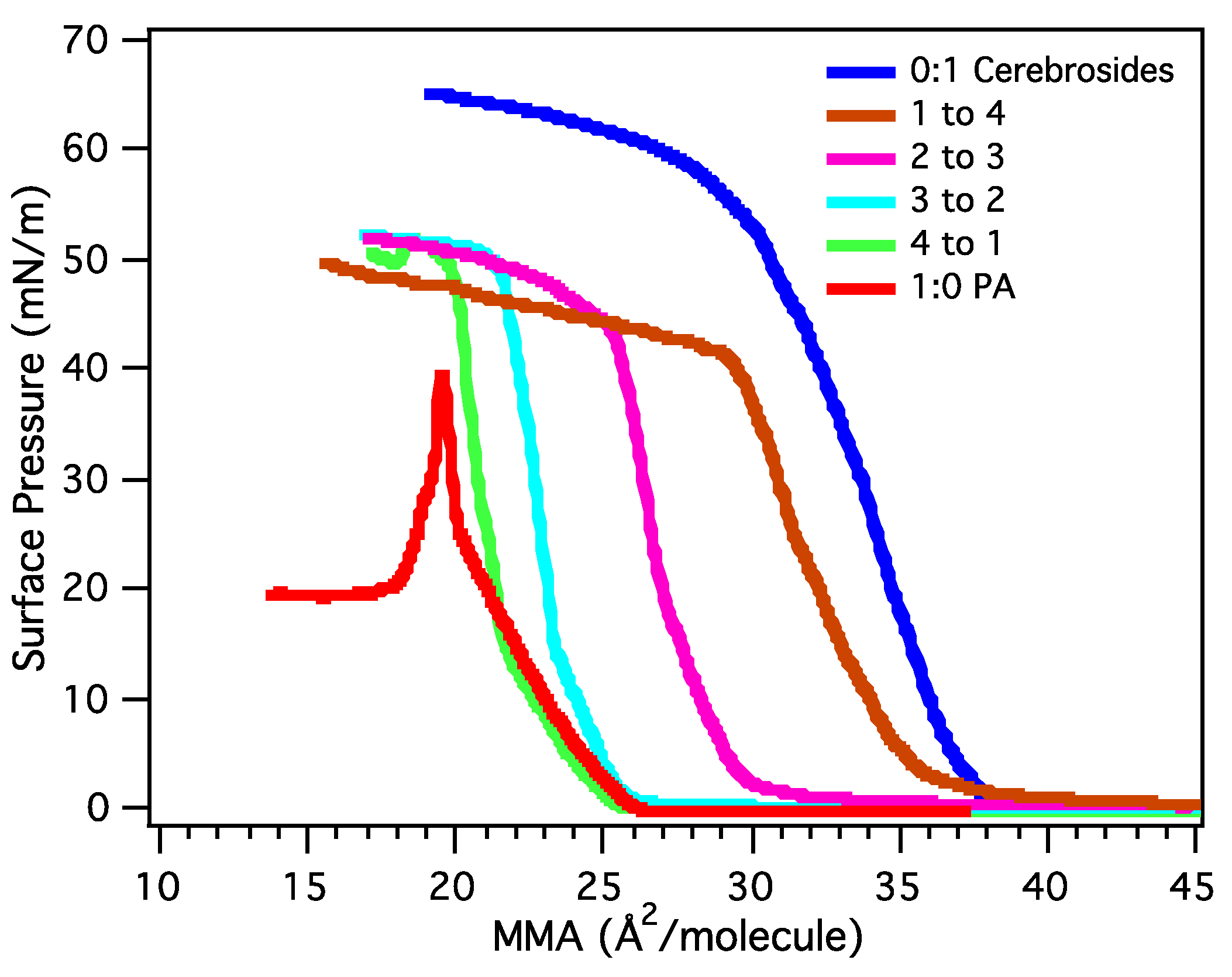
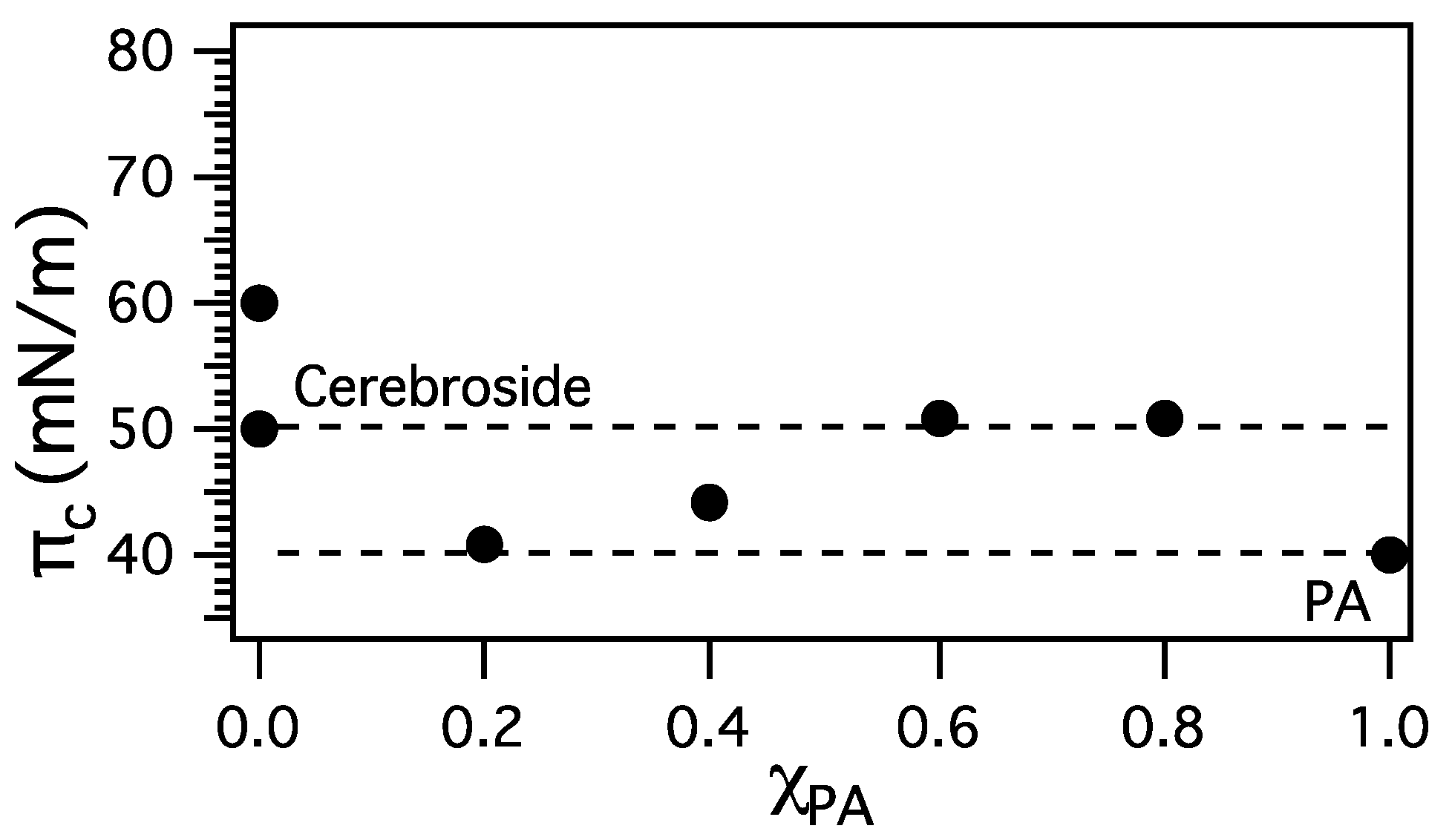
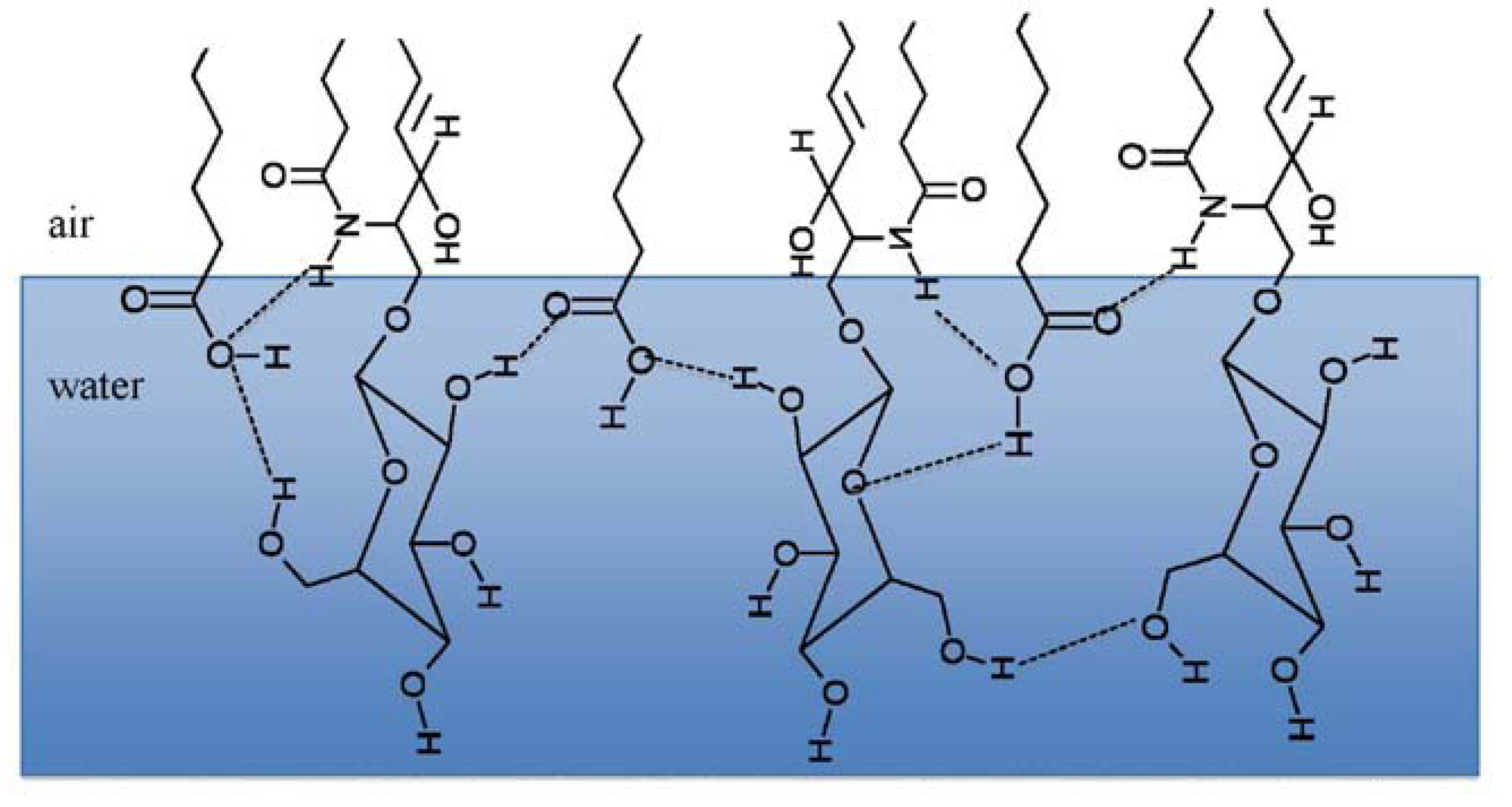
3.2. Miscibility and Surface Morphology of Mixed Monolayers of PA and Cerebrosides
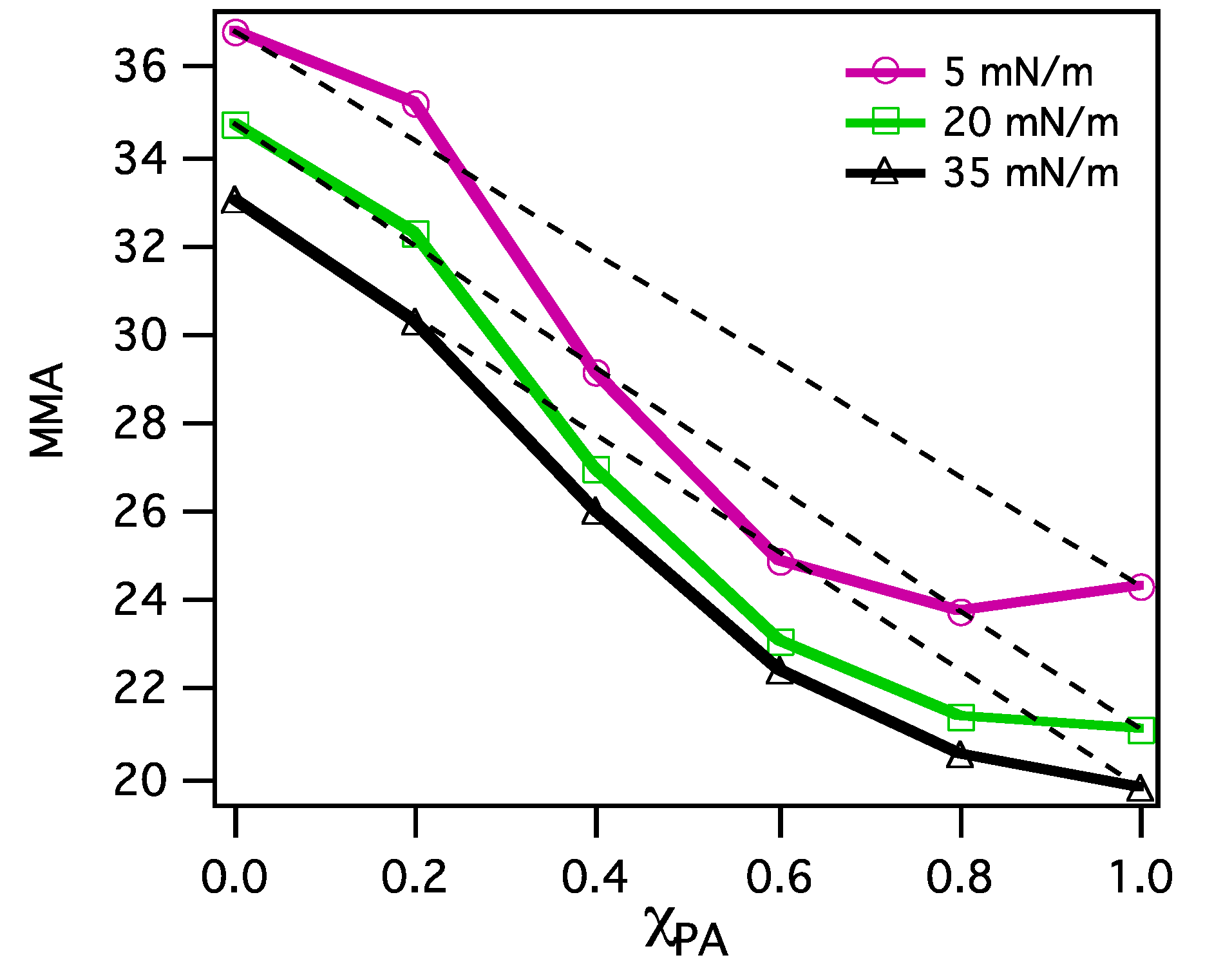
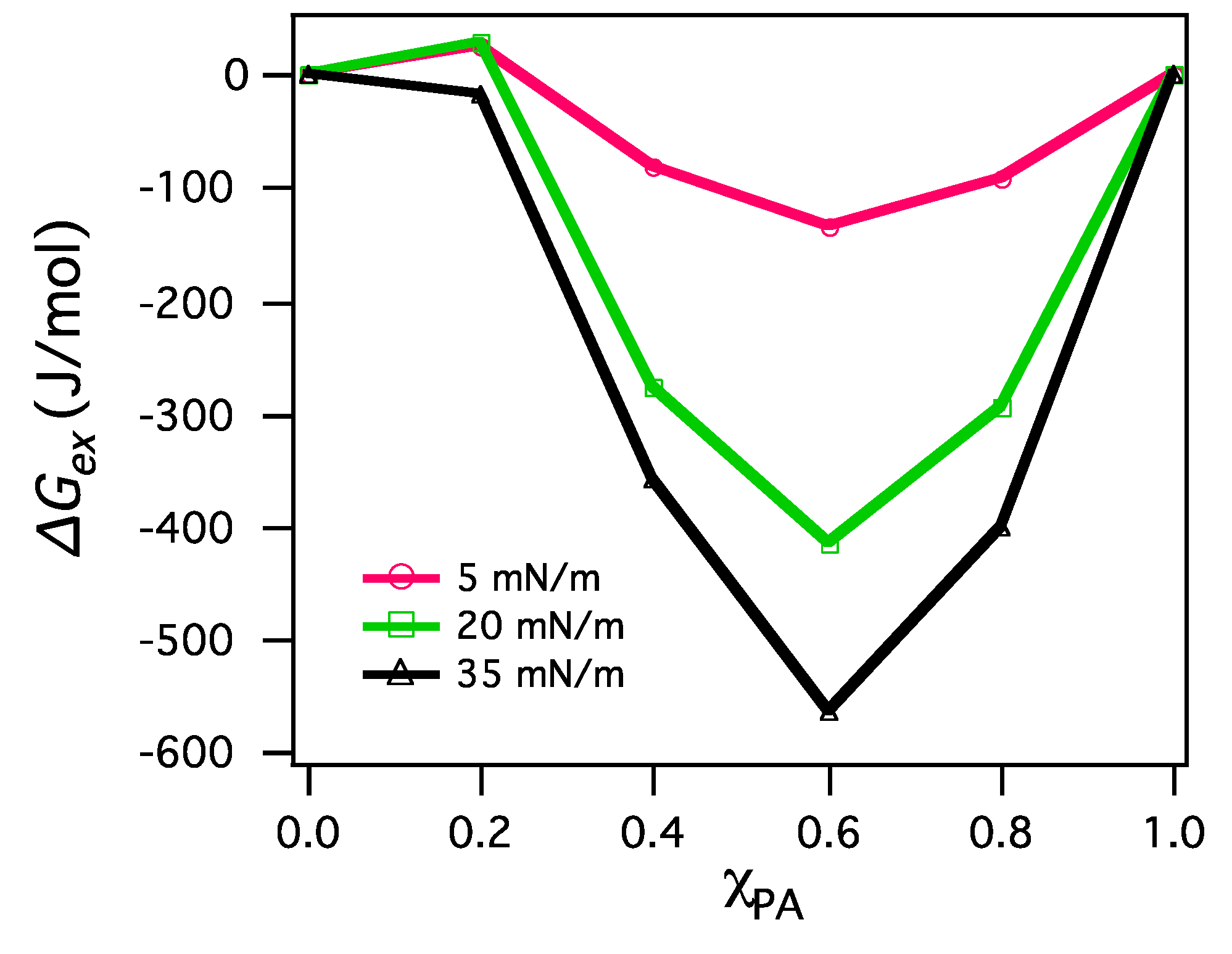
3.2.1. Surface Pressure-Area Isotherms
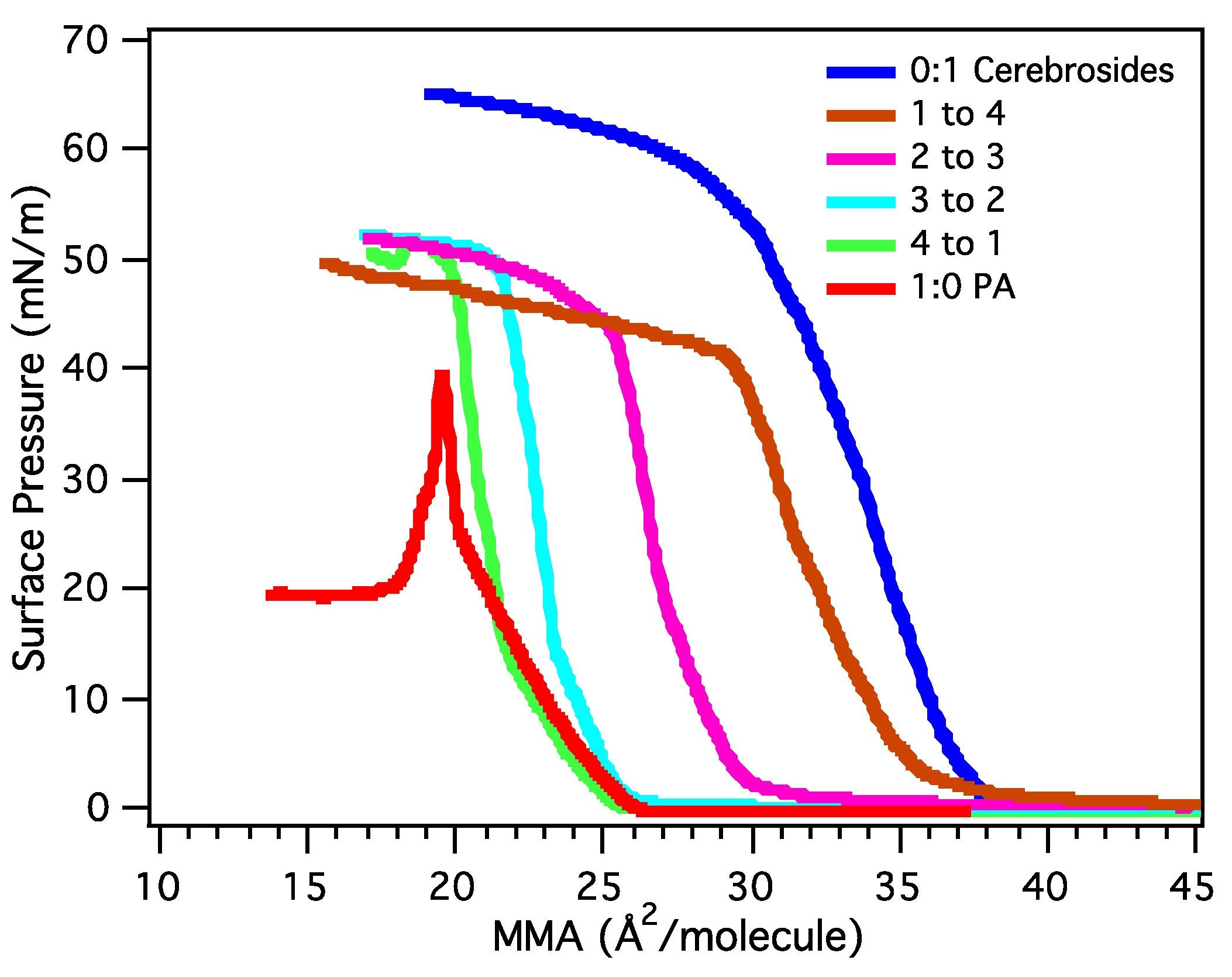
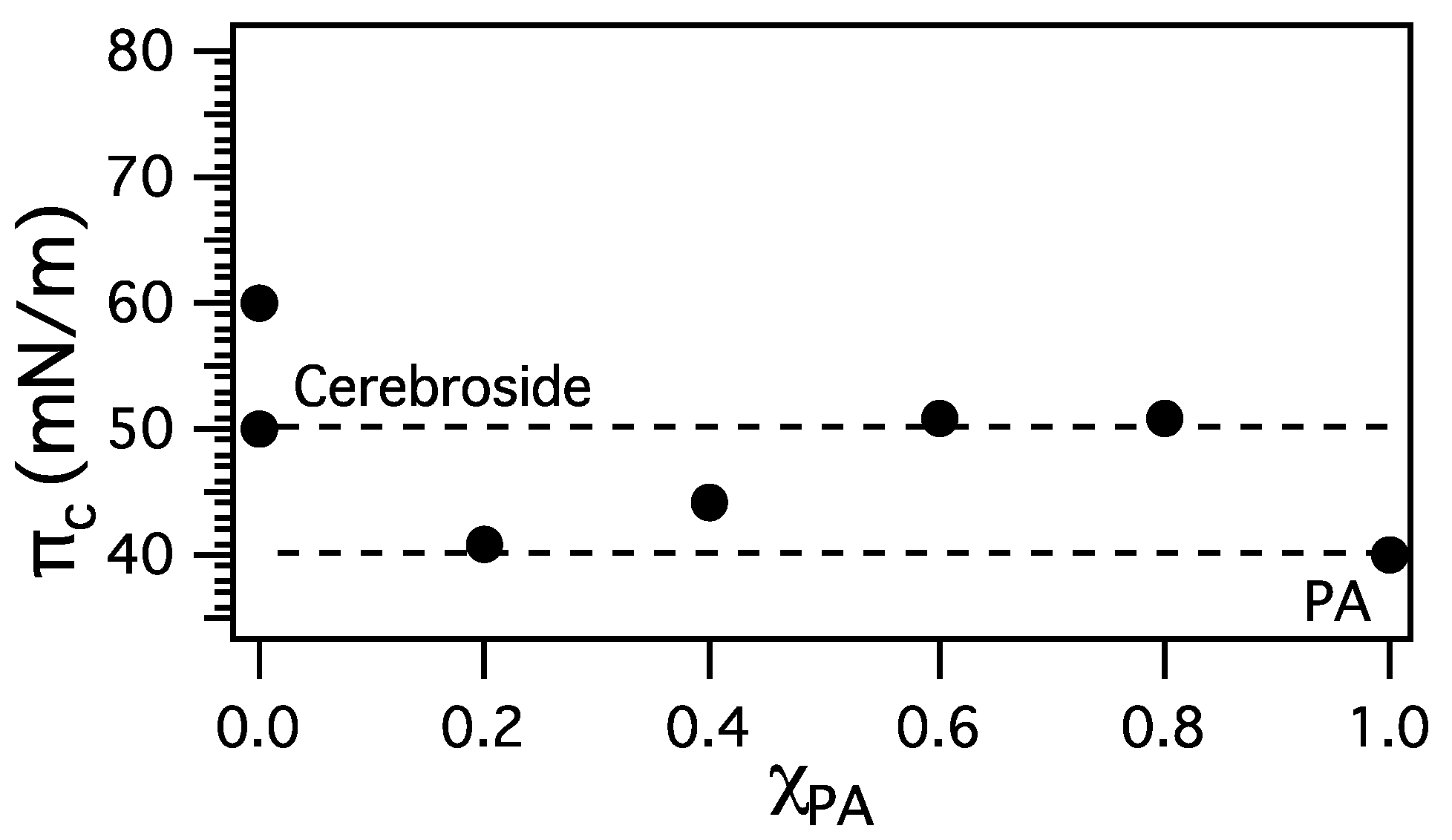
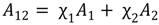
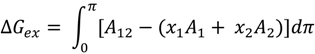
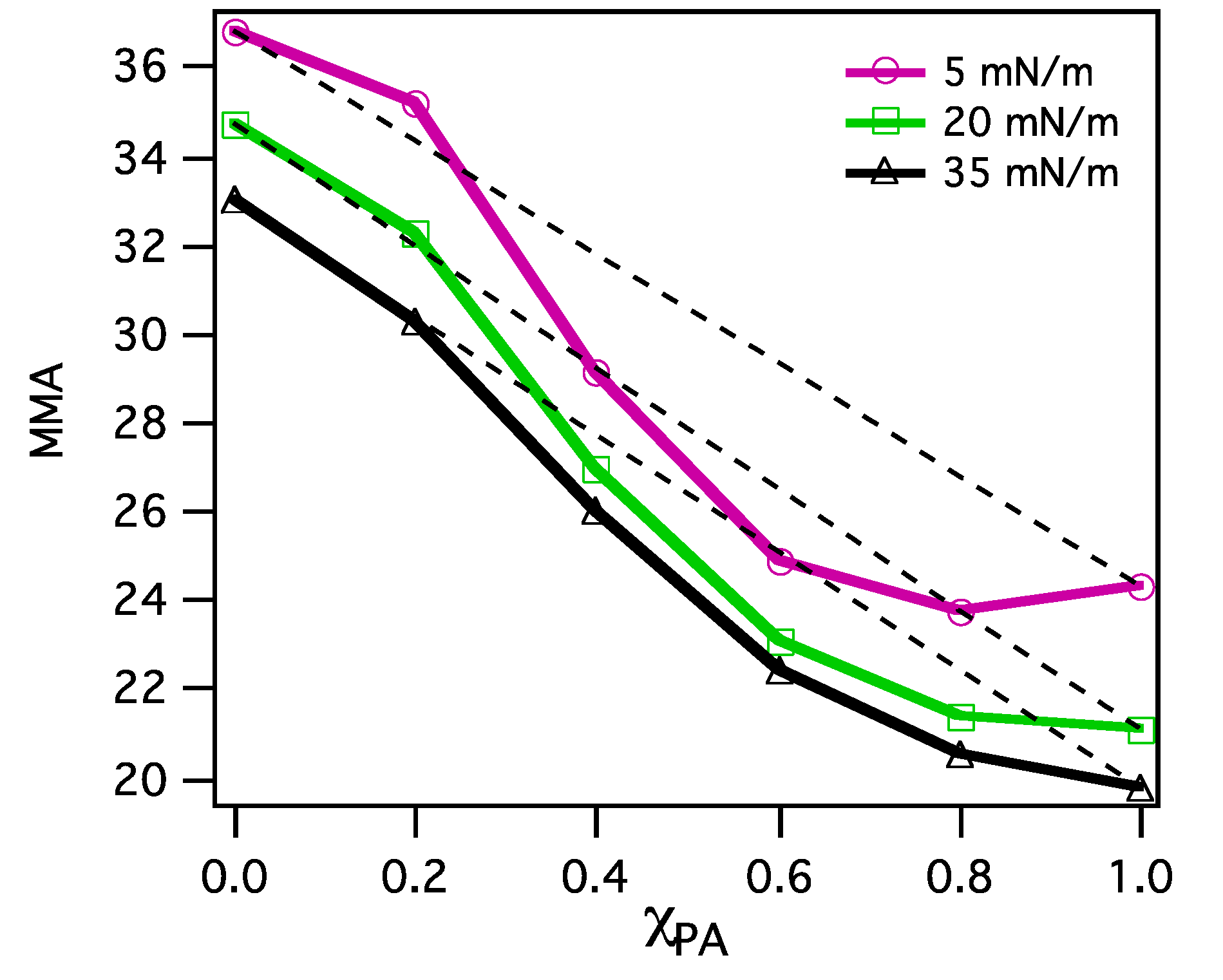
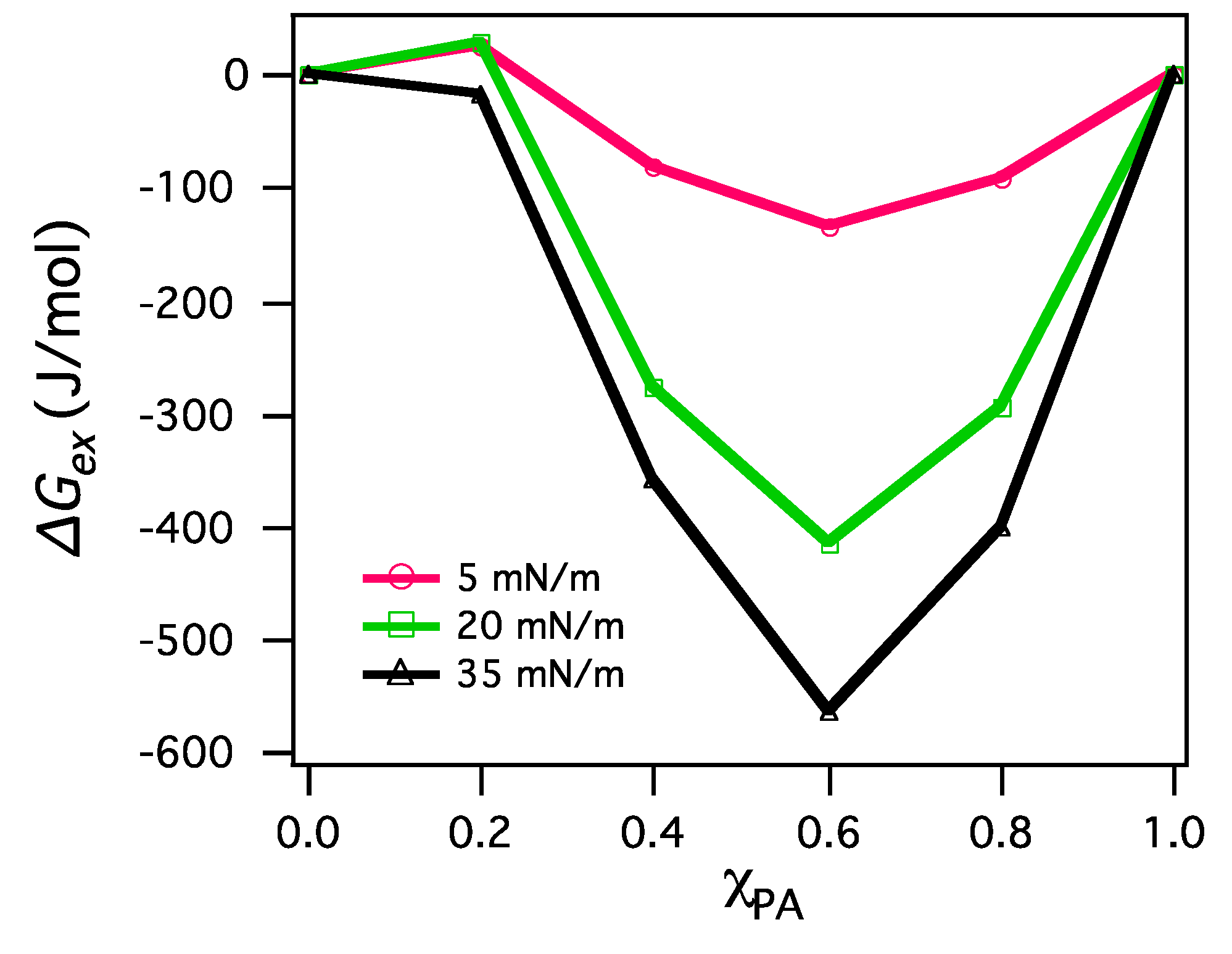
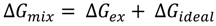
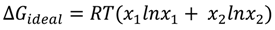
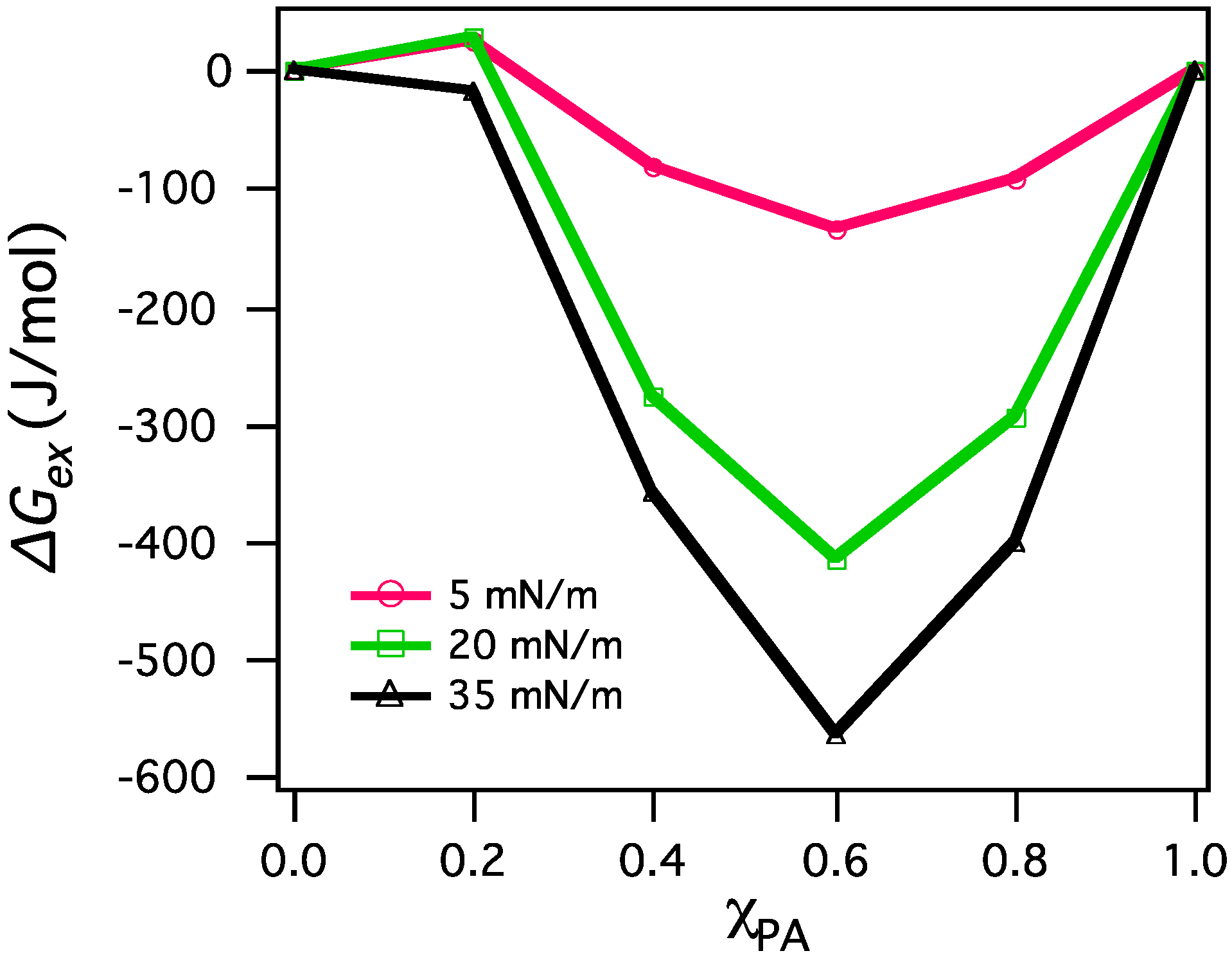
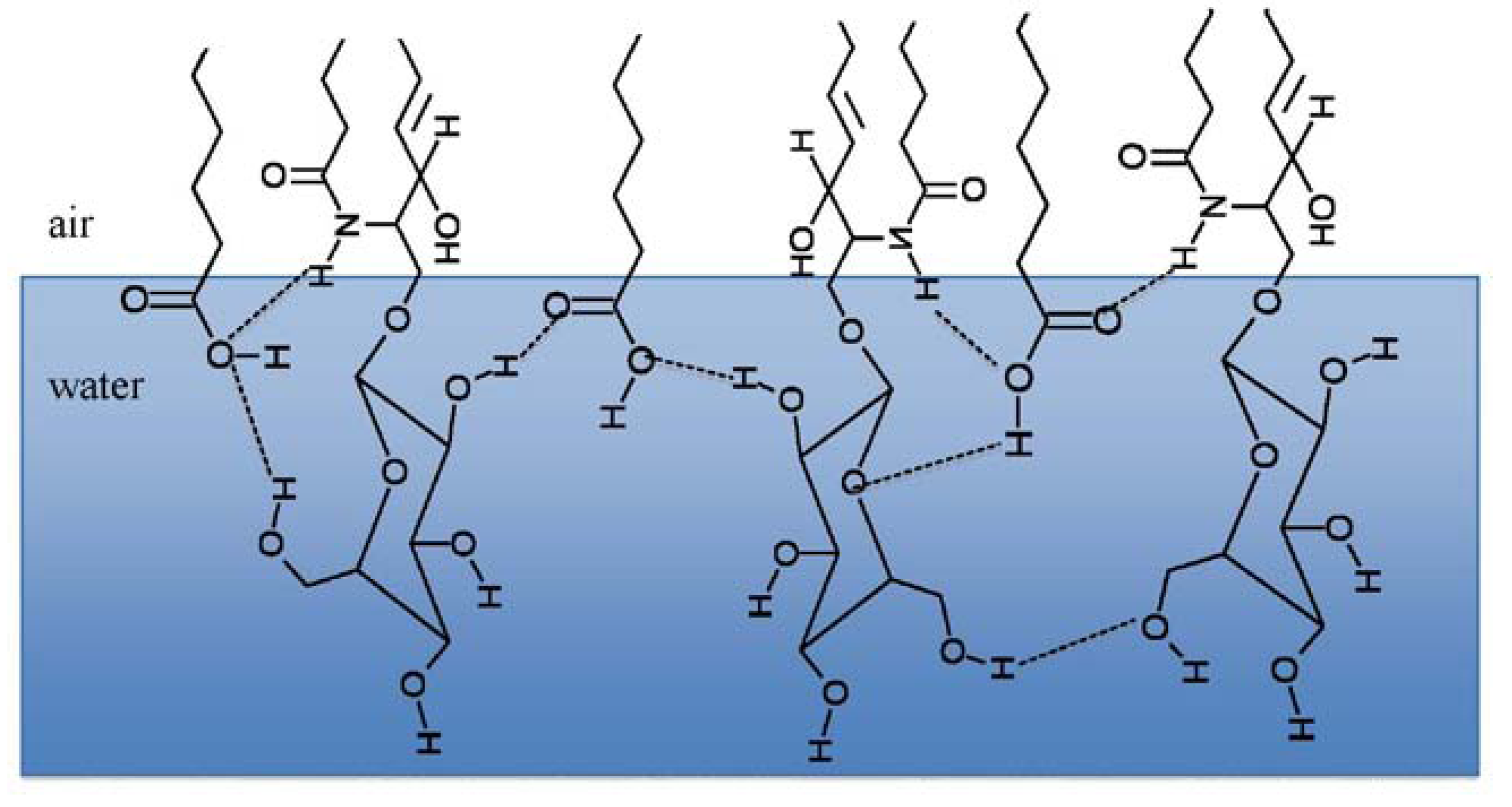
3.2.2. BAM Images
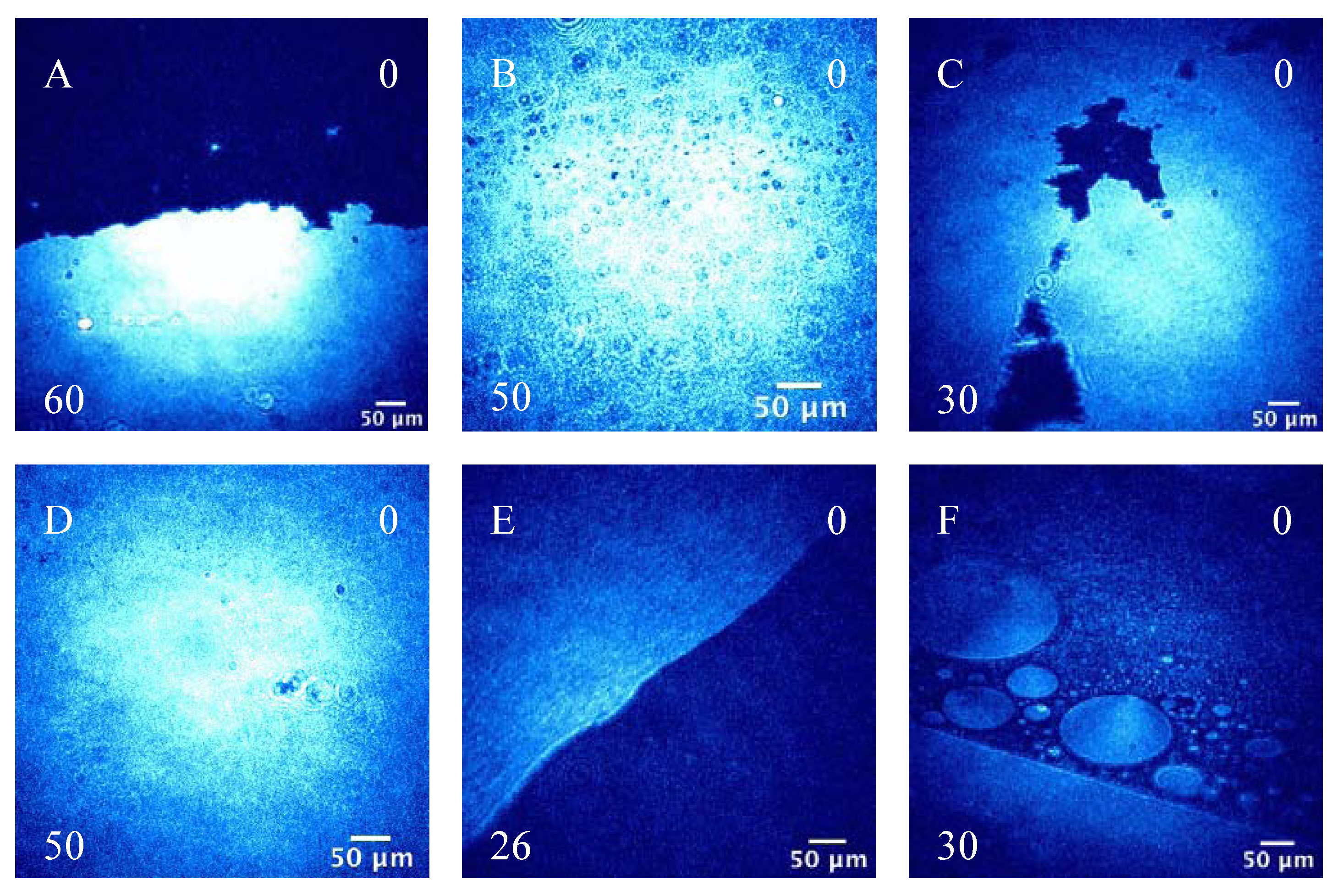

4. Conclusions
5. Atmospheric Implications
Acknowledgments
Conflicts of Interest
References
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Ellison, G.B.; Tuck, A.F.; Vaida, V. Atmospheric processing of organic aerosols. J. Geophys. Res. 1999, 104, 11633–11641. [Google Scholar] [CrossRef]
- Jimenez, J.L.; Canagaratna, M.R.; Donahue, N.M.; Prevot, A.S.H.; Zhang, Q.; Kroll, J.H.; DeCarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N.L.; et al. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef]
- Rinaldi, M.; Decesari, S.; Finessi, E.; Giulianelli, L.; Carbone, C.; Fuzzi, S.; O’Dowd, C.D.; Ceburnis, D.; Facchini, M.C. Primary and secondary organic marine aerosol and oceanic biological activity: recent results and new perspectives for future studies. Adv. Meterol. 2010. [Google Scholar] [CrossRef]
- O’Dowd, C.D.; Facchini, M.C.; Cavalli, F.; Ceburnis, D.; Mircea, M.; Decesari, S.; Fuzzi, S.; Yoon, Y.J.; Putaud, J.P. Biogenically driven organic contribution to marine aerosol. Nature 2004, 431, 676–680. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Ceburnis, D.; Cavalli, F.; Jourdan, O.; Putaud, J.P.; Facchini, M.C.; Decesari, S.; Fuzzi, S.; Sellegri, K.; Jennings, S.G.; et al. Seasonal characteristics of the physiochemical properties of North Atlantic marine atmospheric aerosols. J. Geophys. Res. 2007, 112. [Google Scholar] [CrossRef]
- Laurain, A.M.; Reid, J.P. Characterizing internally mixed insoluble organic inclusions in aqueous aerosol droplets and their influence on light absorption. J. Phys. Chem. 2009, 113, 7039–7047. [Google Scholar] [CrossRef]
- Sierra-Hernandez, M.R.; Allen, H.C. Incorporation and exclusion of long chain alkyl halides in fatty acid monolayers at the air-water interface. Langmuir 2010, 26, 18806–18816. [Google Scholar] [CrossRef]
- Quinn, P.K.; Kapustin, V.N.; Bates, T.S.; Covert, D.S. Chemical and optical properties of marine boundary layer aerosol particles of the mid-Pacific in relation to sources and meteorological transport. J. Geophys. Res. 1996, 101, 6931–6951. [Google Scholar] [CrossRef]
- Beaver, M.R.; Freedman, M.A.; Hasenkopf, C.A.; Tolbert, M.A. Cooling enhancement of aerosol particles due to surfactant precipitation. J. Phys. Chem. 2010, 114, 7070–7076. [Google Scholar] [CrossRef]
- Novakov, T.; Corrigan, C.E.; Penner, J.E.; Chuang, C.C. Organic aerosols in the Caribbean trade winds: A natural source? J. Geophys. Res. 1997, 102, 21307–21313. [Google Scholar]
- Garland, R.M.; Wise, M.E.; Beaver, M.R.; Dewitt, H.L.; Aiken, A.C.; Jimenez, J.L.; Tolbert, M.A. Impact of palmitic acid coating on the water uptake and loss of ammonium sulfate particles. Atmos. Chem. Phys. 2005, 5, 1951–1961. [Google Scholar] [CrossRef]
- Abbatt, J.P.D.; Broekhuizen, K.; Kumar, P.P. Cloud condensation nucleus activity of internally mixed ammonium sulfate/organic acid aerosol particles. Atmos. Envrion. 2005, 39, 4767–4778. [Google Scholar] [CrossRef]
- Fuzzi, S.; Andreae, M.O.; Huebert, B.J.; Kulmala, M.; Bond, T.C.; Boy, M.; Doherty, S.J.; Guenther, A.; Kanakidou, M.; Kawamura, K.; et al. Critical assessment of the current state of scientific knowledge, terminology, and research needs concerning the role of organic aerosols in the atmosphere, climate, and global change. Atmos. Chem. Phys. 2006, 6, 2017–2038. [Google Scholar] [CrossRef]
- Rudlich, Y. Laboratory perspectives on the chemical transformations of organic matter in atmospheric particles. Chem. Rev. 2003, 103, 5097–5124. [Google Scholar] [CrossRef]
- Rogge, W.F.; Mazurek, M.A.; Hildenmann, L.M.; Cass, G.R. Qualification of urban organic aerosols at a molecular level: Identification, abundance, and seasonal variation. Atmos. Environ. 1993, 27A, 1309–1330. [Google Scholar]
- Simoneit, B.R.T. Application of molecular marker analysis to reconcile sources of carbonaceous particulates in tropospheric aerosols. Sci. Total Environ. 1984, 36, 61–72. [Google Scholar] [CrossRef]
- Sicre, M.A.; Marty, J.C.; Saliot, A. n-Alkanes, fatty acid esters, and fatty acid salts in size fractioned aerosols collected over the Mediterranean Sea. J. Geophys. Res. 1990, 95, 3649–3657. [Google Scholar] [CrossRef]
- Gogou, A.I.; Apostolaki, M.; Stephanou, E.G. Determination of organic molecular markers in marine aerosols and sediments: one-step flash chromatography compound class fractionation and capillary gas chromatographic anaylsis. J. Chromatogr. 1998, 799, 215–231. [Google Scholar] [CrossRef]
- Gogou, A.; Stratigakis, N.; Kanakidou, M.; Stephanou, E.G. Organic aerosols in Eastern Mediterranean: Components source reconciliation by using molecular markers and atmospheric back trajectories. Org. Geochem. 1996, 25, 79–96. [Google Scholar] [CrossRef]
- Barbier, M.; Tusseau, D.; Marty, J.C.; Saliot, A. Sterols in aerosols, surface microlayer, and subsurface water in the North-Eastern tropical Atlantic. Ocean. Acta 1981, 4, 77–84. [Google Scholar]
- Barger, W.R.; Garrett, W.D. Surface active organic material in the marine atmosphere. J. Geophys. Res. 1970, 75, 4561. [Google Scholar] [CrossRef]
- Barger, W.R.; Garrett, W.D. Surface active organic material in air over the mediterranean and over the Eastern Equatorial Pacific. J. Geophys. Res. 1976, 81, 3151. [Google Scholar] [CrossRef]
- Duce, R.A.; Mohnen, V.A.; Zimmerman, P.R.; Grosjean, D.; Cautreels, W.; Chatfield, R. Organic material in the global troposphere. Rev. Geophys. Space Phys. 1983, 21, 921–952. [Google Scholar] [CrossRef]
- Crahan, K.K.; Hegg, D.A.; Covert, D.S.; Jonsson, H.; Reid, J.S.; Khelif, D.; Brooks, B.J. Speciation of organic aerosols in the tropical mid-pacific and their relationshipe to light scattering. J. Atmos. Sci. 2004, 61, 2544–2558. [Google Scholar] [CrossRef]
- Tervahattu, H.; Hartonen, K.; Kerminen, V.; Kupiainen, K.; Aarnio, P.; Koskentalo, T.; Tuck, A.F.; Vaida, V. New evidence of an organic layer on marine aerosols. J. Geophys. Res. 2002, 107. [Google Scholar] [CrossRef]
- Tervahattu, H.; Juhanoja, J.; Kupiainen, K. Identification of an organic coating on marine aerosol particles by TOF-SIMS. J. Geophys. Res. 2002, 107. [Google Scholar] [CrossRef]
- Dobson, C.M.; Ellison, G.B.; Tuck, A.F.; Vaida, V. Atmospheric aerosols as prebiotic chemical reactors. Proc. Natl. Acad. Sci. USA 2000, 97, 11864–11868. [Google Scholar] [CrossRef]
- Donaldson, D.J.; Vaida, V. The influence of organic films at the air-aqueous boundary on atmospheric processes. Chem. Rev. 2006, 106, 1445–1461. [Google Scholar] [CrossRef]
- Lawrence, J.R.; Glass, S.V.; Nathanson, G.M. Evaporation of water through butanol films at the surface of supercooled sulfuric acid. J. Phys. Chem. 2005, 109, 7449–7457. [Google Scholar] [CrossRef]
- Langmuir, I.; Schaefer, J. The effect of dissolved salts on insoluble monolayers. J. Am. Chem. Soc. 1937, 59, 2400–2414. [Google Scholar] [CrossRef]
- Deamer, D.W.; Cornwell, D.G. Calcium action on fatty acid and phospholipid monolayers and its relation to the cell membrane. Biochim. Biophys. Acta 1966, 116, 555–562. [Google Scholar] [CrossRef]
- MacDonald, R.C.; Simon, S.A.; Baer, E. Ionic influences on the phase transitions of dipalmitoylphosphatidylserine. Biochemistry 1976, 15, 885–891. [Google Scholar] [CrossRef]
- Seimiya, T.; Ishiwata, S.; Watanabe, N.; Miyasaka, H.; Iwahashi, M.; Tajima, K.; Hayashi, M.; Muramatsu, T. Effect of structural separation between phosphate and ammonium groups of phospholipids on calcium binding properties as studied by pressure-area analysis of the monolayer. J. Colloid Interface Sci. 1984, 101, 267–276. [Google Scholar] [CrossRef]
- Virden, J.W.; Berg, J.C. NaCl-induced aggregation of dipalmitoylphosphatidylglycerol small unilamellar vesicles with varying amounts of incorporated cholesterol. Langmuir 1992, 8, 1532–1537. [Google Scholar] [CrossRef]
- Tang, C.Y.; Allen, H.C. Ionic binding of Na+ versus K+ to the carboxylic acid headgroup of palmitic acid monolayers studied by vibrational sum frequency generation spectroscopy. J. Phys. Chem. 2009, 113, 7383–7393. [Google Scholar] [CrossRef]
- Casillas-Ituarte, N.N.; Chen, X.; Castada, H.; Allen, H.C. Na+ and Ca2+ effect on the hydration and orientation of the phosphate group of DPPC at air-water and air-hydrated silica surface. J. Phys. Chem. B 2010, 114, 9485–9495. [Google Scholar] [CrossRef]
- Quinn, P.J.; Sherman, W.R. Monolayer characteristics and calcium adsorption to cerebroside and cerebroside sulphate oriented at the air-water interface. Biochim. Biophys. Acta 1971, 233, 734–752. [Google Scholar] [CrossRef]
- Colacicco, G.; Buckelew, A.R., Jr.; Scarpelli, E.M. Lipid monolayers: surface viscosity of dipalmitoyl lecithin in relation to surface potential and ion binding. J. Colloid Interface Sci. 1974, 46, 147–151. [Google Scholar] [CrossRef]
- Tang, C.Y.; Huang, Z.; Allen, H.C. Binding of Mg2+ and Ca2+ to palmitic acid and deprotonation of the COOH headgroup studied by vibrational sum frequency generation spectroscopy. J. Phys. Chem. B 2010, 114, 17068–17076. [Google Scholar] [CrossRef]
- Tang, C.Y.; Huang, Z.; Allen, H.C. Interfacial water structure and effects of Mg2+ and Ca2+ binding to the COOH headgroup of a palmitic acid monolayer studied by sum frequency spectroscopy. J. Phys. Chem. B 2011, 115, 34–40. [Google Scholar] [CrossRef]
- Lipp, M.M.; Lee, K.Y.C.; Waring, A.; Zasadzinski, J.A. Fluorescence, polarized fluoresence, and brewster angle microscopy of palmitic acid and lung surfactant protein B monolayers. Biophys. J. 1997, 72, 2783–2804. [Google Scholar] [CrossRef]
- Cuvillier, N.; Bernon, R.; Doux, J.-C.; Merzeau, P.; Mingotaud, C.; Delhaes, P. Adsorption of Inorganic poylanions along a langmuir film: A brewster angle microscopy study. Langmuir 1998, 14, 5573–5580. [Google Scholar] [CrossRef]
- Aller, J.Y.; Kuznetsova, M.R.; Jahns, C.J.; Kemp, P.F. The sea surface microlayer as a source of viral and bacterial enrichment in marine aerosols. Aerosol Sci. 2005, 36, 801–812. [Google Scholar] [CrossRef]
- Muralidhar, P.; Radhika, P.; Krishna, N.; Rao, D.V.; Rao, C.B. Sphingolipids from marine organisms: A Review. Nat. Prod. Sci. 2003, 9, 117–142. [Google Scholar]
- Lass, K.; Friedrichs, G. Revealing structural properties of the marine nanolayer from vibrational sum frequency generation spectra. J. Geophys. Res. 2011, 116, C08042. [Google Scholar] [CrossRef]
- Hawkins, L.N.; Russell, L.M. Polysaccharides, proteins, and phytoplankton fragments: Four chemically distinct types of marine primary organic aerosol classified by single particle spectromicroscopy. Adv. Meterol. 2010. [Google Scholar] [CrossRef]
- Russell, L.M.; Hawkins, L.N.; Frossard, A.A.; Quinn, P.K.; Bates, T.S. Carbohydrate-like composition of submicron atmosphere particles and their production from ocean bubble bursting. Proc. Natl. Acad. Sci. USA 2010, 107, 6652–6657. [Google Scholar]
- Facchini, M.C.; Rinaldi, M.; Decesari, S.; Carbone, C.; Finessi, E.; Mircea, M.; Fuzzi, S.; Ceburnis, D.; Flanagan, R.; Nilsson, E.D.; et al. Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates. Geophys. Res. Lett. 2008, 35, L17814. [Google Scholar] [CrossRef]
- Pagarete, A.; Allen, M.J.; Wilson, W.H.; Kimmance, S.A.; de Vargas, C. Host-virus shift of the sphingolipid pathway along an Emiliania huxleyi bloom: Survival of the fattest. Env. Microbiol. 2009, 11, 2840–2848. [Google Scholar] [CrossRef]
- Michaelson, L.V.; Dunn, T.M.; Napier, J.A. Viral trans-dominant manipulation of algal sphingolipids. Trends Plant Sci. 2010, 15, 651–655. [Google Scholar] [CrossRef]
- Vardi, A.; Haramaty, L.; Van Mooy, B.A.S.; Fredricks, H.F.; Kimmance, S.A.; Larsen, A.; Bidle, K.D. Host-virus dyanmics and subcellular controls of cell fate in a natural coccolithophore population. Proc. Natl. Acad. Sci. USA 2012, 109, 19327–19332. [Google Scholar]
- Kester, D.R.; Duedall, I.W.; Connors, D.N.; Pytkowicz, R.M. Preparation of artificial seawater. Limnol. Oceanogr. 1967, 12, 176–179. [Google Scholar] [CrossRef]
- Millero, F.J.; Feistal, R.; Wright, D.G.; McDougall, T.J. The composition of Standard Seawater and the definition of the References-Composition Salinity Scale. Deep Sea Res. Part I Oceanogr. Res. Pap. 2008, 55, 50–72. [Google Scholar] [CrossRef]
- Finlayson, A.C. The pH range of the mohr titration for chloride ion can be usefully extended to 4-10.5. J. Chem. Educ. 1992, 69, 559. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Z.; Hua, W.; Castada, H.; Allen, H.C. Reorganization and caging of DPPC, DPPE, DPPG, and DPPS monolayers caused by dimethylsulfoxide observed using brewster angle microscopy. Langmuir 2010, 26, 18902–18908. [Google Scholar] [CrossRef]
- Hommel, E.L.; Allen, H.C. Broadband sum frequency generation with two regenerative amplifiers: Temporal overlap of femtosecond and picosecond light pulses. Anal. Sci. 2001, 17, 137–139. [Google Scholar] [CrossRef]
- Ma, G.; Allen, H.C. Surface studies of aqueous methanol solutions by vibrational broad bandwidth sum frequency generation spectroscopy. J. Phys. Chem. B 2003, 107, 6343–6349. [Google Scholar] [CrossRef]
- Gaines, G.L., Jr. Insoluble Monolayers at Liquid-Gas Interfaces; Interscience Publishers: New York, NY, USA, 1966. [Google Scholar]
- Nutting, G.C.; Harkins, W.D. Pressure-area relations of fatty acid and alcohol monolayers. J. Am. Chem. Soc. 1939, 61, 1180–1187. [Google Scholar] [CrossRef]
- Fischer, A.; Sackmann, E. Electron microscopy and electron diffraction study of coexisting phases of pure and mixed monolayers transferred onto solid substrates. J. Colloid Interface Sci. 1986, 112, 1–14. [Google Scholar] [CrossRef]
- Ma, G.; Allen, H.C. Condensing effect of palmitic Acid on DPPC in mixed langmuir monolayers. Langmuir 2007, 23, 589–597. [Google Scholar] [CrossRef]
- Kaganer, V.M.; Moehwald, H.; Dutta, P. Structure and Phase transitions in langmuir monolayers. Rev. Mod. Phys. 1999, 71, 779–819. [Google Scholar] [CrossRef]
- Flach, C.R.; Mendelsohn, R.; Rerek, M.E.; Moore, D.J. Biophysical studies of model stratum corneum lipid monolayers by infrared reflection-absorption spectroscopy and brewster angle microscopy. J. Phys. Chem. B 2000, 104, 2159–2165. [Google Scholar] [CrossRef]
- Qi, S.; Roser, S.J.; Deutsch, D.; Barker, S.A.; Craig, D.Q.M. A laser imaging and neutron reflection investigation into the monolayer behavior of fatty acids used for taste making microspheres. J. Pharm. Sci. 2008, 97, 1864–1877. [Google Scholar] [CrossRef]
- Kundu, S.; Langevin, D. Fatty acid monolayer dissociation and collapse: Effect of pH and cations. Colloids Surf. Physiochem. Eng. Asp. 2008, 325, 81–85. [Google Scholar] [CrossRef]
- Johnston, D.S.; Coppard, E.; Chapman, D. A langmuir film balance study of the interactions of ionic and polar solutes with glycolipid monolayers. Biochim. Biophys. Acta 1985, 815, 325–333. [Google Scholar] [CrossRef]
- Johnston, D.S.; Chapman, D. The properties of brain galactocerebroside monolayers. Biochim. Biophys. Acta 1988, 937, 10–22. [Google Scholar] [CrossRef]
- Chou, T.; Chang, C. Thermodynamic behavior and relaxation processes of mixed DPPC/cholesterol monolayers at the air/water interface. Colloid Surf. B 2000, 17, 71–79. [Google Scholar] [CrossRef]
- Gomez-Fernandez, J.C.; Villalain, J. The use of FT-IR for quantitative studies of the apparent pKa of lipid carboxyl groups and dehydration degree of the phosphate group of phospholipids. Chem. Phys. Lipids 1998, 96, 41–52. [Google Scholar] [CrossRef]
- Gershevitz, O.; Sukenik, C.N. In situ FTIR-ATR analysis and titration of carboxylic acid-terminated SAMs. J. Am. Chem. Soc. 2004, 126, 482–483. [Google Scholar] [CrossRef]
- Rosetti, C.M.; Oliveira, R.G.; Maggio, B. Reflectance and topography of glycosphingolipid monolayers at the air-water interface. Langmuir 2003, 19, 377–384. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Adams, E.M.; Allen, H.C. Palmitic Acid on Salt Subphases and in Mixed Monolayers of Cerebrosides: Application to Atmospheric Aerosol Chemistry. Atmosphere 2013, 4, 315-336. https://doi.org/10.3390/atmos4040315
Adams EM, Allen HC. Palmitic Acid on Salt Subphases and in Mixed Monolayers of Cerebrosides: Application to Atmospheric Aerosol Chemistry. Atmosphere. 2013; 4(4):315-336. https://doi.org/10.3390/atmos4040315
Chicago/Turabian StyleAdams, Ellen M., and Heather C. Allen. 2013. "Palmitic Acid on Salt Subphases and in Mixed Monolayers of Cerebrosides: Application to Atmospheric Aerosol Chemistry" Atmosphere 4, no. 4: 315-336. https://doi.org/10.3390/atmos4040315