
Removal of Atmospheric Methane by Increasing Hydroxyl Radicals via a Water Vapor Enhancement Strategy

 ,
,  , and
, and
Abstract
1. Introduction
1.1. Importance of Methane Mitigation for Climate
1.2. Relationship between Methane, Atmospheric OH, and Water Vapor
1.2.1. Relationship between Methane and Atmospheric OH
1.2.2. Relationship between Atmospheric OH and Water Vapor
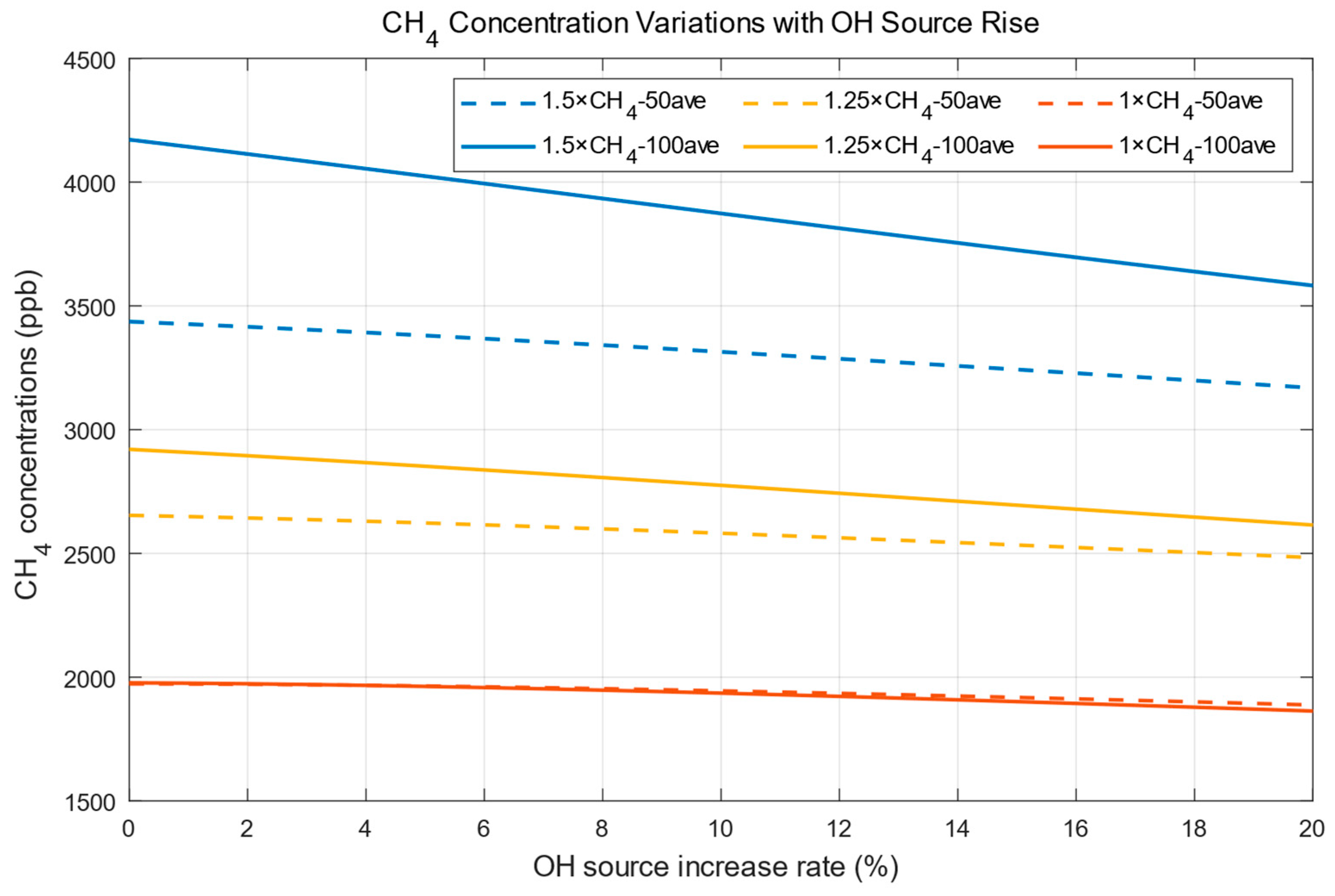
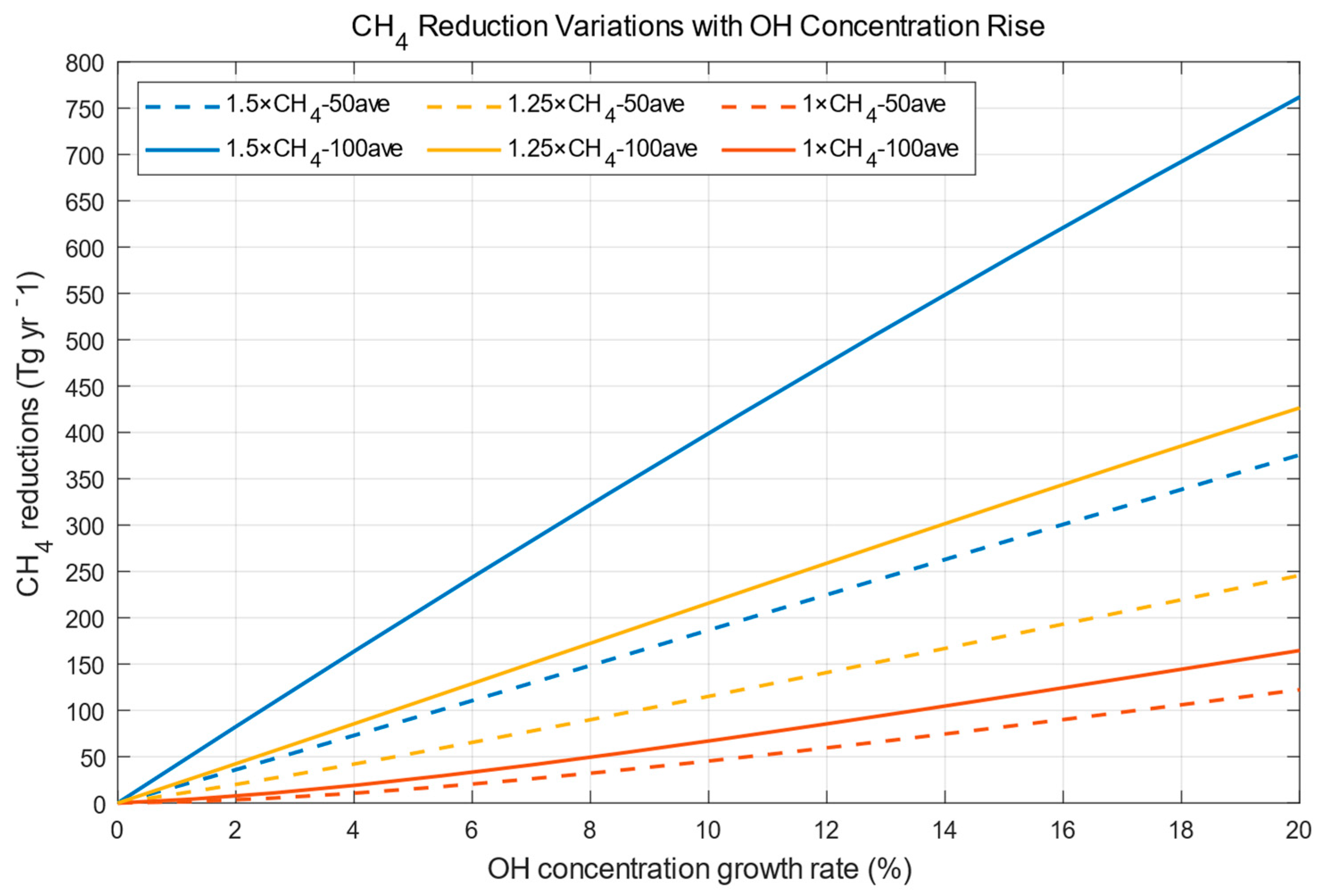
2. Effects of OH on Methane Concentration
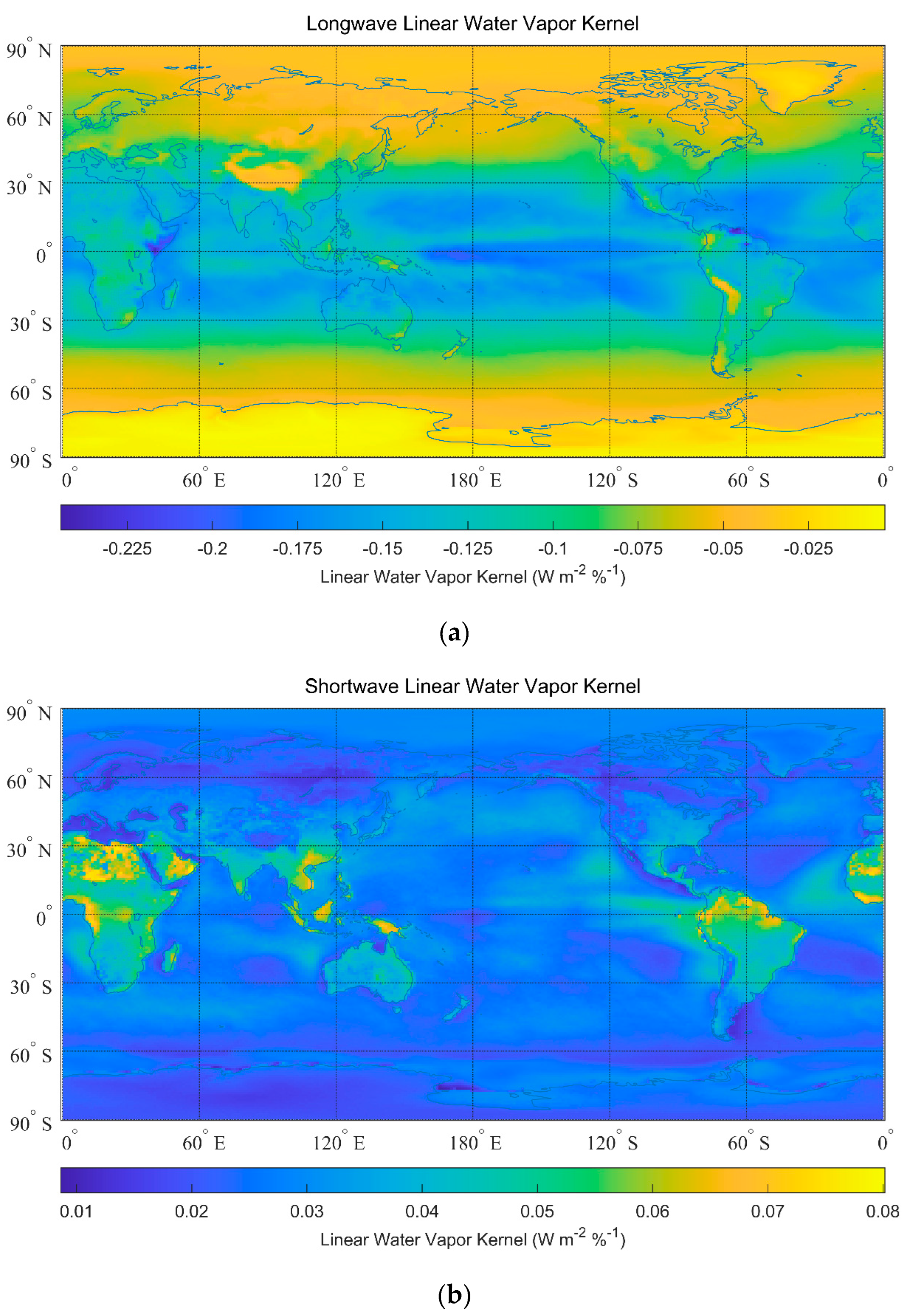
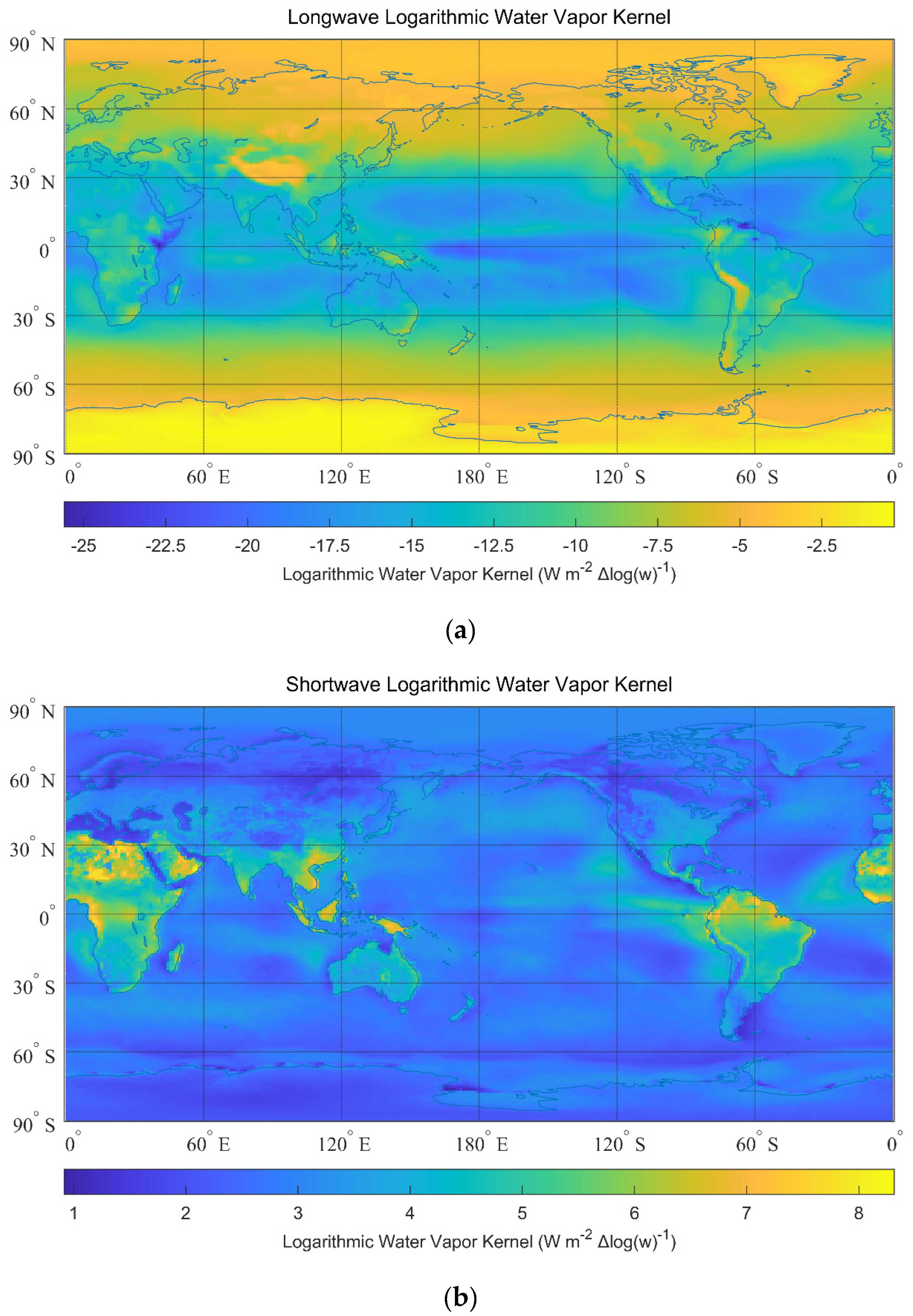
3. Greenhouse Effects of Water Vapor
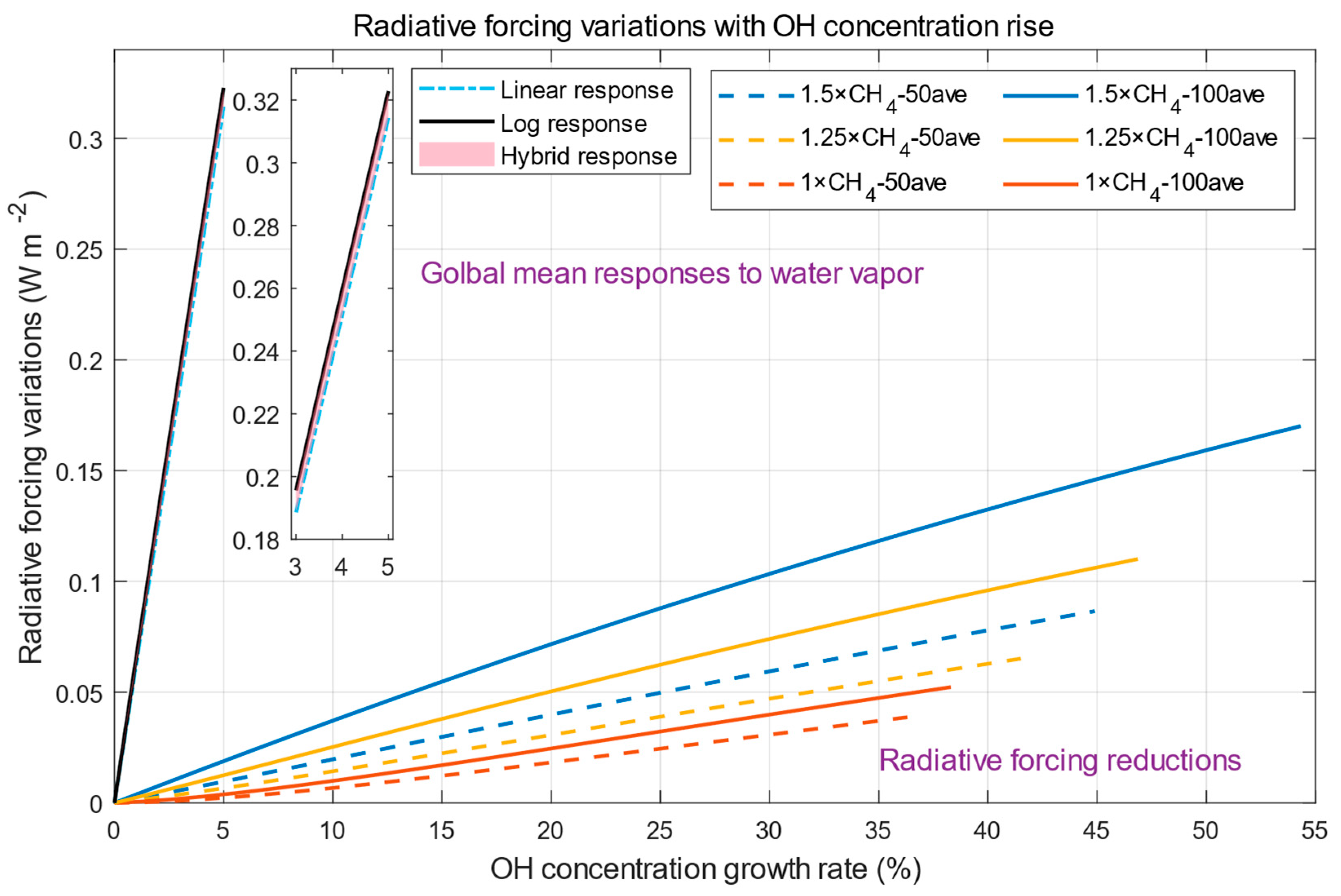
4. Comparison of Radiative Forcing Due to Methane and Water Vapor Variations
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abernethy, S.; O’Connor, F.M.; Jones, C.D.; Jackson, R.B. Methane removal and the proportional reductions in surface temperature and ozone. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2021, 379, 20210104. [Google Scholar] [CrossRef]
- Shahabadi, M.B.; Huang, Y. Logarithmic radiative effect of water vapor and spectral kernels. J. Geophys. Res. Atmos. 2014, 119, 6000–6008. [Google Scholar] [CrossRef]
- Collins, W.J.; Webber, C.P.; Cox, P.M.; Huntingford, C.; Lowe, J.; Sitch, S.; E Chadburn, S.; Comyn-Platt, E.; Harper, A.B.; Hayman, G.; et al. Increased importance of methane reduction for a 1.5 degree target. Environ. Res. Lett. 2018, 13, 054003. [Google Scholar] [CrossRef]
- Collins, W.J.; Frame, D.J.; Fuglestvedt, J.S.; Shine, K.P. Stable climate metrics for emissions of short and long-lived species—Combining steps and pulses. Environ. Res. Lett. 2020, 15, 024018. [Google Scholar] [CrossRef]
- Dang, R.; Liao, H. Radiative Forcing and Health Impact of Aerosols and Ozone in China as the Consequence of Clean Air Actions over 2012–2017. Geophys. Res. Lett. 2019, 46, 12511–12519. [Google Scholar] [CrossRef]
- Dean, J.F.; Middelburg, J.J.; Röckmann, T.; Aerts, R.; Blauw, L.G.; Egger, M.; Jetten, M.S.M.; de Jong, A.E.E.; Meisel, O.H.; Rasigraf, O.; et al. Methane Feedbacks to the Global Climate System in a Warmer World. Rev. Geophys. 2018, 56, 207–250. [Google Scholar] [CrossRef]
- Dentener, F.; Stevenson, D.; Cofala, J.; Mechler, R.; Amann, M.; Bergamaschi, P.; Raes, F.; Derwent, R. The impact of air pollutant and methane emission controls on tropospheric ozone and radiative forcing: CTM calculations for the period 1990–2030. Atmos. Chem. Phys. 2005, 5, 1731–1755. [Google Scholar] [CrossRef]
- Dessler, A.E.; Zhang, Z.; Yang, P. Water-vapor climate feedback inferred from climate fluctuations, 2003–2008. Geophys. Res. Lett. 2008, 35, L20704. [Google Scholar] [CrossRef]
- Etminan, M.; Myhre, G.; Highwood, E.J.; Shine, K.P. Radiative forcing of carbon dioxide, methane, and nitrous oxide: A significant revision of the methane radiative forcing. Geophys. Res. Lett. 2016, 43, 12614–12623. [Google Scholar] [CrossRef]
- Feldl, N.; Bordoni, S.; Merlis, T.M. Coupled High-Latitude Climate Feedbacks and Their Impact on Atmospheric Heat Transport. J. Clim. 2017, 30, 189–201. [Google Scholar] [CrossRef]
- Gedney, N.; Huntingford, C.; Comyn-Platt, E.; Wiltshire, A. Significant feedbacks of wetland methane release on climate change and the causes of their uncertainty. Environ. Res. Lett. 2019, 14, 084027. [Google Scholar] [CrossRef]
- Gordon, N.D.; Jonko, A.K.; Forster, P.M.; Shell, K.M. An observationally based constraint on the water-vapor feedback. J. Geophys. Res. Atmos. 2013, 118, 12435–12443. [Google Scholar] [CrossRef]
- Haines, A.; Amann, M.; Borgford-Parnell, N.; Leonard, S.; Kuylenstierna, J.; Shindell, D. Short-lived climate pollutant mitigation and the Sustainable Development Goals. Nat. Clim. Chang. 2017, 7, 863–869. [Google Scholar] [CrossRef]
- Harmsen, M.; Fricko, O.; Hilaire, J.; van Vuuren, D.P.; Drouet, L.; Durand-Lasserve, O.; Fujimori, S.; Keramidas, K.; Klimont, Z.; Luderer, G.; et al. Taking some heat off the NDCs? The limited potential of additional short-lived climate forcers’ mitigation. Clim. Chang. 2020, 163, 1443–1461. [Google Scholar] [CrossRef]
- Heimann, I.; Griffiths, P.T.; Warwick, N.J.; Abraham, N.L.; Archibald, A.T.; Pyle, J.A. Methane Emissions in a Chemistry-Climate Model: Feedbacks and Climate Response. J. Adv. Model. Earth Syst. 2020, 12, e2019MS002019. [Google Scholar] [CrossRef]
- Heinze, C.; Eyring, V.; Friedlingstein, P.; Jones, C.; Balkanski, Y.; Collins, W.; Fichefet, T.; Gao, S.; Hall, A.; Ivanova, D.; et al. ESD Reviews: Climate feedbacks in the Earth system and prospects for their evaluation. Earth Syst. Dyn. 2019, 10, 379–452. [Google Scholar] [CrossRef]
- Held, I.M.; Soden, B.J. Water Vapor Feedback and Global Warming. Annu. Rev. Energy Environ. 2000, 25, 441–475. [Google Scholar] [CrossRef]
- Holmes, C.D.; Prather, M.J.; Søvde, O.A.; Myhre, G. Future methane, hydroxyl, and their uncertainties: Key climate and emission parameters for future predictions. Atmos. Chem. Phys. 2013, 13, 285–302. [Google Scholar] [CrossRef]
- Holmes, C.D. Methane Feedback on Atmospheric Chemistry: Methods, Models, and Mechanisms. J. Adv. Model. Earth Syst. 2018, 10, 1087–1099. [Google Scholar] [CrossRef]
- Huang, Y.; Ramaswamy, V.; Soden, B. An investigation of the sensitivity of the clear-sky outgoing longwave radiation to atmospheric temperature and water vapor. J. Geophys. Res. Atmos. 2007, 112, D05104. [Google Scholar] [CrossRef]
- Huang, Y.; Xia, Y.; Tan, X. On the pattern of CO2 radiative forcing and poleward energy transport. J. Geophys. Res. Atmos. 2017, 122, 10578–10593. [Google Scholar] [CrossRef]
- IPCC. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- IPCC. Summary for Policymakers. In Global Warming of 1.5 °C. An IPCC Special Report on the Impacts of Global Warming of 1.5 °C above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty; Masson-Delmotte, V., Zhai, P., Pörtner, H.-O., Roberts, D., Skea, J., Shukla, P.R., Pirani, A., Moufouma-Okia, W., Péan, C., Pidcock, R., et al., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2018; pp. 3–24. [Google Scholar]
- IPCC. Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S.L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M.I., et al., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2021; p. 2391. [Google Scholar]
- Isaksen, I.S.A.; Gauss, M.; Myhre, G.; Anthony, K.M.W.; Ruppel, C. Strong atmospheric chemistry feedback to climate warming from Arctic methane emissions. Glob. Biogeochem. Cycles 2011, 25, GB2002. [Google Scholar] [CrossRef]
- Burkholder, J.B.; Sander, S.P.; Abbatt, J.; Barker, J.R.; Cappa, C.; Crounse, J.D.; Dibble, T.S.; Huie, R.E.; Kolb, C.E.; Kurylo, M.J.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19; Jet Propulsion Laboratory: Pasadena, CA, USA, 2019. [Google Scholar]
- Jackson, R.B.; Solomon, E.I.; Canadell, J.G.; Cargnello, M.; Field, C.B. Methane removal and atmospheric restoration. Nat. Sustain. 2019, 2, 436–438. [Google Scholar] [CrossRef]
- Koffi, E.N.; Bergamaschi, P.; Alkama, R.; Cescatti, A. An observation-constrained assessment of the climate sensitivity and future trajectories of wetland methane emissions. Sci. Adv. 2020, 6, eaay4444. [Google Scholar] [CrossRef]
- Kramer, R.J.; Matus, A.V.; Soden, B.J.; L’Ecuyer, T.S. Observation-Based Radiative Kernels From CloudSat/CALIPSO. J. Geophys. Res. Atmos. 2019, 124, 5431–5444. [Google Scholar] [CrossRef]
- Lan, X.; Thoning, K.W.; Dlugokencky, E.J. Trends in Globally-Averaged CH4, N2O, and SF6 Determined from NOAA Global Monitoring Laboratory Measurements (No. Version 2023-04). 2023. Available online: https://gml.noaa.gov/ccgg/trends_doi.html (accessed on 1 June 2023).
- Lelieveld, J.; Gromov, S.; Pozzer, A.; Taraborrelli, D. Global tropospheric hydroxyl distribution, budget and reactivity. Atmos. Chem. Phys. 2016, 16, 12477–12493. [Google Scholar] [CrossRef]
- Li, M.; Karu, E.; Brenninkmeijer, C.; Fischer, H.; Lelieveld, J.; Williams, J. Tropospheric OH and stratospheric OH and Cl concentrations determined from CH4, CH3Cl, and SF6 measurements. NPJ Clim. Atmospheric Sci. 2018, 1, 29. [Google Scholar] [CrossRef]
- Liu, R.; Su, H.; Liou, K.-N.; Jiang, J.H.; Gu, Y.; Liu, S.C.; Shiu, C. An Assessment of Tropospheric Water Vapor Feedback Using Radiative Kernels. J. Geophys. Res. Atmos. 2018, 123, 1499–1509. [Google Scholar] [CrossRef]
- López-Comí, L.; Morgenstern, O.; Zeng, G.; Masters, S.L.; Querel, R.R.; Nedoluha, G.E. Assessing the sensitivity of the hydroxyl radical to model biases in composition and temperature using a single-column photochemical model for Lauder, New Zealand. Atmos. Chem. Phys. 2016, 16, 14599–14619. [Google Scholar] [CrossRef]
- Lund, M.T.; Aamaas, B.; Stjern, C.W.; Klimont, Z.; Berntsen, T.K.; Samset, B.H. A continued role of short-lived climate forcers under the Shared Socioeconomic Pathways. Earth Syst. Dyn. 2020, 11, 977–993. [Google Scholar] [CrossRef]
- Maycock, A.C.; Smith, C.J.; Rap, A.; Rutherford, O. On the Structure of Instantaneous Radiative Forcing Kernels for Greenhouse Gases. J. Atmospheric Sci. 2021, 78, 949–965. [Google Scholar] [CrossRef]
- Meinshausen, M.; Nicholls, Z.R.J.; Lewis, J.; Gidden, M.J.; Vogel, E.; Freund, M.; Beyerle, U.; Gessner, C.; Nauels, A.; Bauer, N.; et al. The shared socio-economic pathway (SSP) greenhouse gas concentrations and their extensions to 2500. Geosci. Model Dev. 2020, 13, 3571–3605. [Google Scholar] [CrossRef]
- Miner, K.R.; Turetsky, M.R.; Malina, E.; Bartsch, A.; Tamminen, J.; McGuire, A.D.; Fix, A.; Sweeney, C.; Elder, C.D.; Miller, C.E. Permafrost carbon emissions in a changing Arctic. Nat. Rev. Earth Environ. 2022, 3, 55–67. [Google Scholar] [CrossRef]
- Monks, S.A.; Arnold, S.R.; Emmons, L.K.; Law, K.S.; Turquety, S.; Duncan, B.N.; Flemming, J.; Huijnen, V.; Tilmes, S.; Langner, J.; et al. Multi-model study of chemical and physical controls on transport of anthropogenic and biomass burning pollution to the Arctic. Atmos. Chem. Phys. 2015, 15, 3575–3603. [Google Scholar] [CrossRef]
- Montzka, S.A.; Spivakovsky, C.M.; Butler, J.H.; Elkins, J.W.; Lock, L.T.; Mondeel, D.J. New Observational Constraints for Atmospheric Hydroxyl on Global and Hemispheric Scales. Science 2000, 288, 500–503. [Google Scholar] [CrossRef]
- Naik, V.; Voulgarakis, A.; Fiore, A.M.; Horowitz, L.W.; Lamarque, J.-F.; Lin, M.; Prather, M.J.; Young, P.J.; Bergmann, D.; Cameron-Smith, P.J.; et al. Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmos. Chem. Phys. 2013, 13, 5277–5298. [Google Scholar] [CrossRef]
- Naus, S.; Montzka, S.A.; Pandey, S.; Basu, S.; Dlugokencky, E.J.; Krol, M. Constraints and biases in a tropospheric two-box model of OH. Atmos. Chem. Phys. 2019, 19, 407–424. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Turner, A.J.; Yin, Y.; Prather, M.J.; Frankenberg, C. Effects of Chemical Feedbacks on Decadal Methane Emissions Estimates. Geophys. Res. Lett. 2020, 47, e2019GL085706. [Google Scholar] [CrossRef]
- Nicely, J.M.; Salawitch, R.J.; Canty, T.; Anderson, D.C.; Arnold, S.R.; Chipperfield, M.P.; Emmons, L.K.; Flemming, J.; Huijnen, V.; Kinnison, D.E.; et al. Quantifying the causes of differences in tropospheric OH within global models. J. Geophys. Res. Atmos. 2017, 122, 1983–2007. [Google Scholar] [CrossRef]
- Nicely, J.M.; Canty, T.P.; Manyin, M.; Oman, L.D.; Salawitch, R.J.; Steenrod, S.D.; Strahan, S.E.; Strode, S.A. Changes in Global Tropospheric OH Expected as a Result of Climate Change Over the Last Several Decades. J. Geophys. Res. Atmos. 2018, 123, 10774–10795. [Google Scholar] [CrossRef]
- Nisbet, E.G.; Fisher, R.E.; Lowry, D.; France, J.L.; Allen, G.; Bakkaloglu, S.; Broderick, T.J.; Cain, M.; Coleman, M.; Fernandez, J.; et al. Methane Mitigation: Methods to Reduce Emissions, on the Path to the Paris Agreement. Rev. Geophys. 2020, 58, e2019RG000675. [Google Scholar] [CrossRef]
- Ocko, I.B.; Sun, T.; Shindell, D.; Oppenheimer, M.; Hristov, A.N.; Pacala, S.W.; Mauzerall, D.L.; Xu, Y.; Hamburg, S.P. Acting rapidly to deploy readily available methane mitigation measures by sector can immediately slow global warming. Environ. Res. Lett. 2021, 16, 054042. [Google Scholar] [CrossRef]
- Patra, P.K.; Krol, M.C.; Montzka, S.A.; Arnold, T.; Atlas, E.L.; Lintner, B.R.; Stephens, B.B.; Xiang, B.; Elkins, J.W.; Fraser, P.J.; et al. Observational evidence for interhemispheric hydroxyl-radical parity. Nature 2014, 513, 219–223. [Google Scholar] [CrossRef]
- Pendergrass, A.G.; Conley, A.; Vitt, F.M. Surface and top-of-atmosphere radiative feedback kernels for CESM-CAM5. Earth Syst. Sci. Data 2018, 10, 317–324. [Google Scholar] [CrossRef]
- Peng, S.; Lin, X.; Thompson, R.L.; Xi, Y.; Liu, G.; Hauglustaine, D.; Lan, X.; Poulter, B.; Ramonet, M.; Saunois, M.; et al. Wetland emission and atmospheric sink changes explain methane growth in 2020. Nature 2022, 612, 477–482. [Google Scholar] [CrossRef]
- Prather, M.J. Time scales in atmospheric chemistry: Theory, GWPs for CH4 and CO, and runaway growth. Geophys. Res. Lett. 1996, 23, 2597–2600. [Google Scholar] [CrossRef]
- Prather, M.J. Lifetimes and time scales in atmospheric chemistry. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2007, 365, 1705–1726. [Google Scholar] [CrossRef]
- Prather, M.J.; Holmes, C.D.; Hsu, J. Reactive greenhouse gas scenarios: Systematic exploration of uncertainties and the role of atmospheric chemistry. Geophys. Res. Lett. 2012, 39, L09803. [Google Scholar] [CrossRef]
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; et al. The Global Methane Budget 2000–2017. Earth Syst. Sci. Data 2020, 12, 1561–1623. [Google Scholar] [CrossRef]
- Shell, K.M.; Kiehl, J.T.; Shields, C.A. Using the Radiative Kernel Technique to Calculate Climate Feedbacks in NCAR’s Community Atmospheric Model. J. Clim. 2008, 21, 2269–2282. [Google Scholar] [CrossRef]
- Sherwood, S.C.; Dixit, V.; Salomez, C. The global warming potential of near-surface emitted water vapour. Environ. Res. Lett. 2018, 13, 104006. [Google Scholar] [CrossRef]
- Smith, C.J.; Kramer, R.J.; Myhre, G.; Forster, P.M.; Soden, B.J.; Andrews, T.; Boucher, O.; Faluvegi, G.; Fläschner, D.; Hodnebrog, Ø.; et al. Understanding Rapid Adjustments to Diverse Forcing Agents. Geophys. Res. Lett. 2018, 45, 12023–12031. [Google Scholar] [CrossRef] [PubMed]
- Soden, B.J.; Held, I.M.; Colman, R.; Shell, K.M.; Kiehl, J.T.; Shields, C.A. Quantifying Climate Feedbacks Using Radiative Kernels. J. Clim. 2008, 21, 3504–3520. [Google Scholar] [CrossRef]
- Stevenson, D.S.; Zhao, A.; Naik, V.; O’Connor, F.M.; Tilmes, S.; Zeng, G.; Murray, L.T.; Collins, W.J.; Griffiths, P.T.; Shim, S.; et al. Trends in global tropospheric hydroxyl radical and methane lifetime since 1850 from AerChemMIP. Atmos. Chem. Phys. 2020, 20, 12905–12920. [Google Scholar] [CrossRef]
- Sun, T.; Ocko, I.B.; Hamburg, S.P. The value of early methane mitigation in preserving Arctic summer sea ice. Environ. Res. Lett. 2022, 17, 044001. [Google Scholar] [CrossRef]
- Thorsen, T.J.; Kato, S.; Loeb, N.G.; Rose, F.G. Observation-Based Decomposition of Radiative Perturbations and Radiative Kernels. J. Clim. 2018, 31, 10039–10058. [Google Scholar] [CrossRef]
- Tian, H.Q.; Xu, R.T.; Canadell, J.G.; Thompson, R.L.; Winiwarter, W.; Suntharalingam, P.; Davidson, E.A.; Ciais, P.; Jackson, R.B.; Janssens-Maenhout, G.; et al. A comprehensive quantification of global nitrous oxide sources and sinks. Nature 2020, 586, 248–256. [Google Scholar] [CrossRef]
- Turetsky, M.R.; Abbott, B.W.; Jones, M.C.; Anthony, K.W.; Olefeldt, D.; Schuur, E.A.G.; Grosse, G.; Kuhry, P.; Hugelius, G.; Koven, C.; et al. Carbon release through abrupt permafrost thaw. Nat. Geosci. 2020, 13, 138–143. [Google Scholar] [CrossRef]
- Turner, A.J.; Frankenberg, C.; Wennberg, P.O.; Jacob, D.J. Ambiguity in the causes for decadal trends in atmospheric methane and hydroxyl. Proc. Natl. Acad. Sci. USA 2017, 114, 5367–5372. [Google Scholar] [CrossRef]
- Turner, A.J.; Frankenberg, C.; Kort, E.A. Interpreting contemporary trends in atmospheric methane. Proc. Natl. Acad. Sci. USA 2019, 116, 2805–2813. [Google Scholar] [CrossRef]
- Anthony, K.W.; von Deimling, T.S.; Nitze, I.; Frolking, S.; Emond, A.; Daanen, R.; Anthony, P.; Lindgren, P.; Jones, B.; Grosse, G. 21st-century modeled permafrost carbon emissions accelerated by abrupt thaw beneath lakes. Nat. Commun. 2018, 9, 3262. [Google Scholar] [CrossRef]
- Wang, X.; Jacob, D.J.; Eastham, S.D.; Sulprizio, M.P.; Zhu, L.; Chen, Q.; Alexander, B.; Sherwen, T.; Evans, M.J.; Lee, B.H.; et al. The role of chlorine in global tropospheric chemistry. Atmos. Chem. Phys. 2019, 19, 3981–4003. [Google Scholar] [CrossRef]
- Wang, Y.; Ming, T.; Li, W.; Yuan, Q.; de Richter, R.; Davies, P.; Caillol, S. Atmospheric removal of methane by enhancing the natural hydroxyl radical sink. Greenh. Gases Sci. Technol. 2022, 12, 784–795. [Google Scholar] [CrossRef]
- Wuebbles, D.J.; Tamaresis, J.S. The Role of Methane in the Global Environment. In Atmospheric Methane: Sources, Sinks, and Role in Global Change; Khalil, M.A.K., Ed.; Springer: Berlin/Heidelberg, Germany, 1993; pp. 469–513. [Google Scholar]
- Xiong, H.; Ming, T.; Wu, Y.; Li, W.; Mu, L.; de Richter, R.; Yan, S.; Yuan, Y.; Peng, C. Numerical analysis of a negative emission technology of methane to mitigate climate change. Sol. Energy 2023, 255, 416–424. [Google Scholar] [CrossRef]
- Yin, Y.; Chevallier, F.; Ciais, P.; Broquet, G.; Fortems-Cheiney, A.; Pison, I.; Saunois, M. Decadal trends in global CO emissions as seen by MOPITT. Atmos. Chem. Phys. 2015, 15, 13433–13451. [Google Scholar] [CrossRef]
- Yin, Y.; Chevallier, F.; Ciais, P.; Bousquet, P.; Saunois, M.; Zheng, B.; Worden, J.; Bloom, A.A.; Parker, R.J.; Jacob, D.J.; et al. Accelerating methane growth rate from 2010 to 2017: Leading contributions from the tropics and East Asia. Atmos. Chem. Phys. 2021, 21, 12631–12647. [Google Scholar] [CrossRef]
- Yuan, W.; Zheng, Y.; Piao, S.; Ciais, P.; Lombardozzi, D.; Wang, Y.; Ryu, Y.; Chen, G.; Dong, W.; Hu, Z.; et al. Increased atmospheric vapor pressure deficit reduces global vegetation growth. Sci. Adv. 2019, 5, eaax1396. [Google Scholar] [CrossRef] [PubMed]
- Zelinka, M.D.; Hartmann, D.L. Climate Feedbacks and Their Implications for Poleward Energy Flux Changes in a Warming Climate. J. Clim. 2012, 25, 608–624. [Google Scholar] [CrossRef]
- Zhang, Z.; Zimmermann, N.E.; Stenke, A.; Li, X.; Hodson, E.L.; Zhu, G.; Huang, C.; Poulter, B. Emerging role of wetland methane emissions in driving 21st century climate change. Proc. Natl. Acad. Sci. USA 2017, 114, 9647–9652. [Google Scholar] [CrossRef]
- Zhao, Y.; Saunois, M.; Bousquet, P.; Lin, X.; Berchet, A.; Hegglin, M.I.; Canadell, J.G.; Jackson, R.B.; Dlugokencky, E.J.; Langenfelds, R.L.; et al. Influences of hydroxyl radicals (OH) on top-down estimates of the global and regional methane budgets. Atmos. Chem. Phys. 2020, 20, 9525–9546. [Google Scholar] [CrossRef]
- Zhao, Y.; Saunois, M.; Bousquet, P.; Lin, X.; Berchet, A.; Hegglin, M.I.; Canadell, J.G.; Jackson, R.B.; Deushi, M.; Jöckel, P.; et al. On the role of trend and variability in the hydroxyl radical (OH) in the global methane budget. Atmos. Chem. Phys. 2020, 20, 13011–13022. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Concentrations | Initial Emission Rates | Reaction Rate Coefficients | |
|---|---|---|---|
| CH4 (N) | 1975 ppb | 395 Tg yr−1 | |
| CH4 (S) | 1875 ppb | 167 Tg yr−1 | |
| CO (N) | 120 ppb | 910 Tg yr−1 | |
| CO (S) | 60 ppb | 420 Tg yr−1 | |
| OH (Global mean) | 1.09 × 106 molecules cm−3 | 251.2 Tmol yr−1 | = 0.432 s−1 |
| 60° N to 90° N | 60° S to 60° N | 60° S to 90° S | ||||
|---|---|---|---|---|---|---|
| Longwave | Shortwave | Longwave | Shortwave | Longwave | Shortwave | |
| Linear kernel | −0.0032 | 0.0016 | −0.1047 | 0.0272 | −0.0024 | 0.0016 |
| Logarithmic kernel | −0.3370 | 0.1694 | −11.0373 | 2.8357 | −0.2593 | 0.1645 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Yao, X.; Zhou, L.; Ming, T.; Li, W.; de Richter, R. Removal of Atmospheric Methane by Increasing Hydroxyl Radicals via a Water Vapor Enhancement Strategy. Atmosphere 2024, 15, 1046. https://doi.org/10.3390/atmos15091046
Liu Y, Yao X, Zhou L, Ming T, Li W, de Richter R. Removal of Atmospheric Methane by Increasing Hydroxyl Radicals via a Water Vapor Enhancement Strategy. Atmosphere. 2024; 15(9):1046. https://doi.org/10.3390/atmos15091046
Chicago/Turabian StyleLiu, Yang, Xiaokun Yao, Li Zhou, Tingzhen Ming, Wei Li, and Renaud de Richter. 2024. "Removal of Atmospheric Methane by Increasing Hydroxyl Radicals via a Water Vapor Enhancement Strategy" Atmosphere 15, no. 9: 1046. https://doi.org/10.3390/atmos15091046
APA StyleLiu, Y., Yao, X., Zhou, L., Ming, T., Li, W., & de Richter, R. (2024). Removal of Atmospheric Methane by Increasing Hydroxyl Radicals via a Water Vapor Enhancement Strategy. Atmosphere, 15(9), 1046. https://doi.org/10.3390/atmos15091046