Computational Study of the Dissociation Reactions of Secondary Ozonide

 ,
, 
 and
and
Abstract
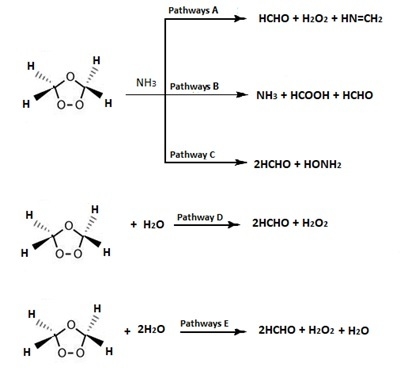
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. SOZ with Ammonia
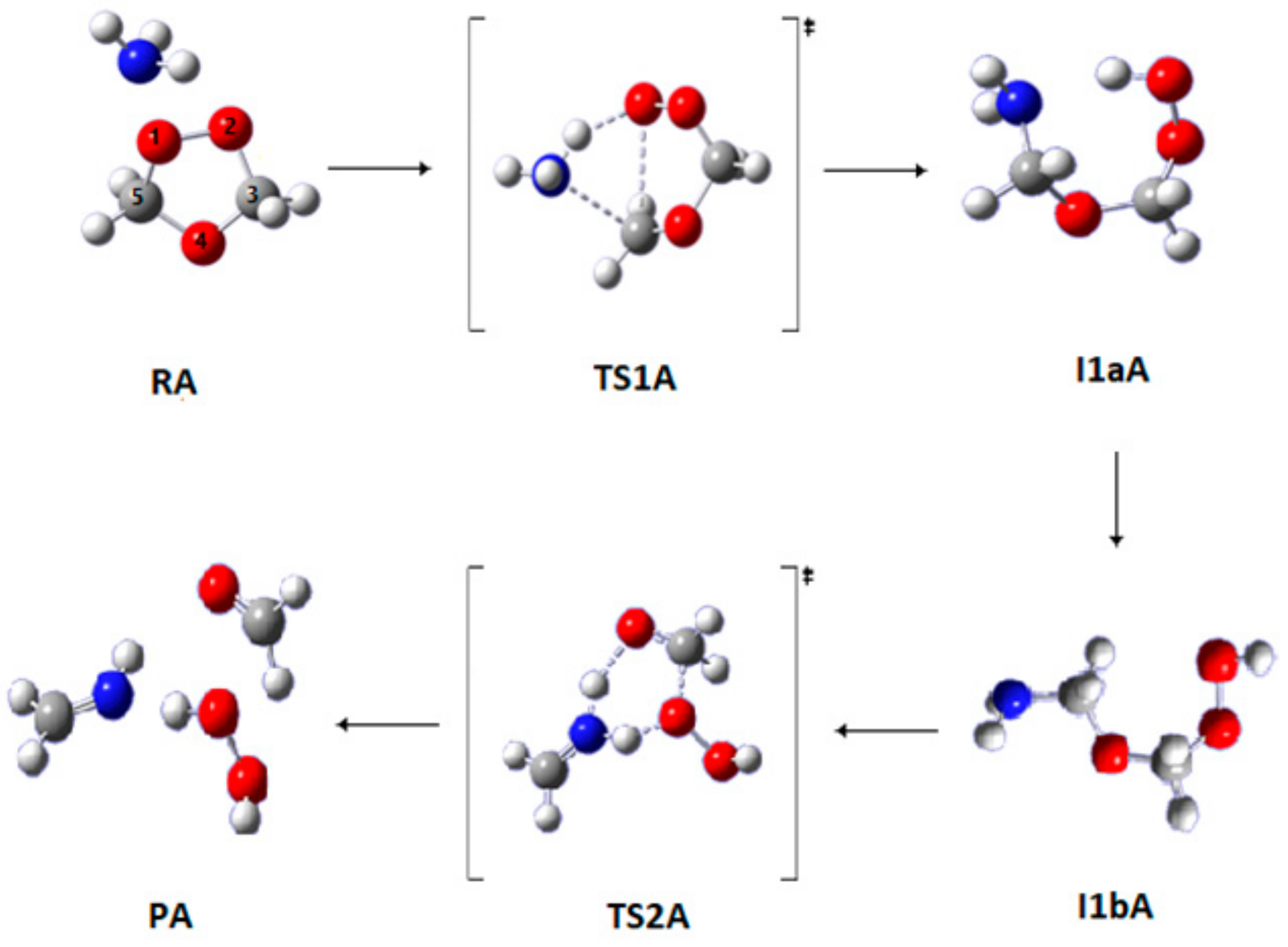
3.1.1. Pathway A
3.1.2. Pathway B
3.1.3. Pathway C
3.2. Reaction of SOZ with Water
3.2.1. Pathway D
3.2.2. Pathway E
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lamb, B.; Guenther, A.; Gay, D.; Westberg, H. A National Inventory of Biogenic Hydrocarbon Emissions. Atmos. Environ. 1987, 21, 1695–1705. [Google Scholar] [CrossRef]
- Almatarneh, M.H.; Elayan, I.A.; Poirier, R.A.; Altarawneh, M. The Ozonolysis of Cyclic Monoterpenes: A Computational Review. Can. J. Chem. 2017, 96, 281–292. [Google Scholar] [CrossRef]
- Aikin, A.C.; Herman, J.R.; Maier, E.J.; Mcquillan, C.J. Atmospheric Chemistry of Ethane and Ethylene. J. Geophys. Res. 1982, 87, 3105–3118. [Google Scholar] [CrossRef]
- Sawada, S.; Totsuka, T. Natural and Anthropogenic Sources and Fate of Atmospheric Ethylene. Atmos. Environ. 1967, 5, 821–832. [Google Scholar] [CrossRef]
- Bariseviciute, R.; Ceponkus, J.; Sablinskas, V. Matrix Isolation FTIR Spectroscopical Study of Ethene Secondary Ozonide. CEJC 2007, 5, 71–86. [Google Scholar] [CrossRef]
- Anglada, J.M.; Crehuet, R.; Bofill, M. The Ozonolysis of Ethylene: A Theoretical Study of the Gas-Phase Reaction Mechanism. Chem. Eur. J. 1999, 6, 1809–1822. [Google Scholar] [CrossRef]
- Almatarneh, M.H.; Alshamaileh, E.; Ahmad, Z.M.; Abu-Saleh, A.A.; Elayan, I.A. A Computational Study of the Ozonolysis of Phenanthrene. Acta Phys. Pol. A 2017, 132. [Google Scholar] [CrossRef]
- Neeb, N.; Horie, O.; Moortgat, G. Gas-phase Ozonolysis of Ethene in the Presence of Hydroxylic Compounds. Int. J. Chem. Kinet. 1996, 28, 721–730. [Google Scholar] [CrossRef]
- Aschmann, S.M.; Tuazon, E.C.; Arey, J.; Atkinson, R. Products of the Gas-phase Reaction of O3 with Cyclohexene. J. Phys. Chem. A 2003, 107, 2247–2255. [Google Scholar] [CrossRef]
- Vibenholt, A.; Nørgaard, A.W.; Clausen, P.A.; Wolkoff, P. Formation and Stability of Secondary Ozonides from Monoterpenes Studied by Mass Spectrometry. Chemosphere 2009, 76, 572–577. [Google Scholar] [CrossRef]
- Deng, J.; Chen, J.; Geng, C.; Liu, H.; Wang, W.; Bai, Z.; Xu, Y. The Overall Reaction Process of Ozone with Methacrolein and Isoprene in the Condensed Phase. J. Phys. Chem. A 2012, 116, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Naa, K.; Songa, C.; Cocker, D.R. Formation of Secondary Organic Aerosol from the Reaction of Styrene with Ozone in the Presence and Absence of Ammonia and Water. Atmos. Environ. 2006, 40, 1889–1900. [Google Scholar] [CrossRef]
- Behera, S.B.; Sharma, M.; Aneja, V.P.; Balasubramanian, R. Ammonia in the Atmosphere: A Review on Emission Sources, Atmospheric Chemistry and Deposition on Terrestrial Bodies. Environ. Sci. Pollut. Res. 2013, 20, 8092–8131. [Google Scholar] [CrossRef] [PubMed]
- Banu, T.; Sen, K.; Das, A.K. Atmospheric Fate of Criegee Intermediate Formed During Ozonolysis of Styrene in the Presence of H2O and NH3: The Crucial Role of Stereochemistry. J. Phys. Chem. A 2018, 122, 8377–8389. [Google Scholar] [CrossRef] [PubMed]
- Rousso, A.C.; Hansen, N.; Jasper, A.W.; Ju, Y. Identification of the Criegee Intermediate Reaction Network in Ethylene Ozonolysis: Impact on Energy Conversion Strategies and Atmospheric Chemistry. Phys. Chem. Chem. Phys. 2019, 21, 7341–7357. [Google Scholar] [CrossRef]
- Na, K.; Song, C.; Switzer, C.; Cocker, D.R. Effect of Ammonia on Secondary Organic Aerosol Formation from α-Pinene Ozonolysis in Dry and Humid Conditions. Environ. Sci. Technol. 2007, 41, 6096–6102. [Google Scholar] [CrossRef]
- Babar, Z.B.; Park, J.H.; Lim, H.J. Influence of NH3 on Secondary Organic Aerosols from the Ozonolysis and Photooxidation of α-Pinene in a Flow Reactor. Atmos. Environ. 2017, 146, 71–84. [Google Scholar] [CrossRef]
- Jørgensen, S.; Gross, A. Theoretical Investigation of Reactions Between Ammonia and Precursors from the Ozonolysis of Ethane. Chem. Phys. 2009, 362, 8–15. [Google Scholar] [CrossRef]
- Almatarneh, M.H.; Elayan, I.A.; Altarawneh, M.; Hollett, J.W. A computational study of the ozonolysis of sabinene. Theor. Chem. Acc. 2019, 138, 30. [Google Scholar] [CrossRef]
- Elayan, I.A.; Almatarneh, M.H.; Hollett, J.W. Reactivity of the anti-Criegee intermediate of β-pinene with prevalent atmospheric species. Struct. Chem. 2019, 30, 1353–1364. [Google Scholar] [CrossRef]
- Almatarneh, M.H.; Elayan, I.A.; Abu-Saleh, A.A.; Altarawneh, M.; Aryia, P.A. The Gas-Phase Ozonolysis Reaction of MethylButenol: A Mechanistic Study. Int. J. Quantum Chem. 2019, 119, e25888. [Google Scholar] [CrossRef]
- Elayan, I.A.; Almatarneh, M.H.; Hollett, J.W. The bimolecular catalytic transformation of methyl vinyl ketone oxide: A DFT study. Chem. Phys. 2020, 530, 110649. [Google Scholar] [CrossRef]
- Almatarneh, M.H.; Elayan, I.A.; Altarawneh, M.; Hollett, J.W. Hydration and Secondary Ozonide of the Criegee Intermediate of Sabinene. ACS-OMEGA 2018, 3, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; Revision a.02; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-energy Formula into A Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Mmodel Chemistry. I. The Total Energies of Closed-shell Atoms and Hydrides of the First-row Atoms. J. Chem. Phys 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 Theory Using Reduced Order Perturbation Theory. J. Chem. Phys 2007, 127, 124105. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A Complete Basis Set Model Chemistry. VI. Use of Density Functional Geometries and Frequencies. J. Chem. Phys 1999, 110, 2822–2827. [Google Scholar] [CrossRef]
- Wood, G.P.F.; Radom, L.; Petersson, G.A.; Barnes, E.C.; Frisch, M.J.; Montgomery, J.A. A Restricted-open-shell Complete-basis-set Model Chemistry. J. Chem. Phys 2006, 125, 094106. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Theory/Basis Set | TS1A | TS2A | TS1B | TS2B | ||||
|---|---|---|---|---|---|---|---|---|
| E‡ | ∆G‡ | E‡ | ∆G‡ | E‡ | ∆G‡ | E‡ | ∆G‡ | |
| B3LYP/6-31G(d) | 189 | 230 | 153 | 151 | 109 | 150 | 55 | 58 |
| B3LYP/6-31G(2df,p) | 191 | 232 | 148 | 146 | 114 | 155 | 57 | 60 |
| B3LYP/6-311++G(3df,3pd) | 189 | 228 | 140 | 137 | 112 | 151 | 58 | 61 |
| ωB97XD/6-311++G(3df,3pd) | 205 | 245 | 167 | 164 | 144 | 184 | 70 | 73 |
| APDF/6-311++G(3df,3pd) | 195 | 235 | 158 | 155 | 114 | 154 | 63 | 66 |
| M06-2X/6-311++G(3df,3pd) | 218 | 258 | 179 | 176 | 182 | 222 | 84 | 88 |
| M11/6-11++G(3df,3pd) | 204 | 243 | 161 | 157 | 171 | 212 | 73 | 76 |
| G3MP2 | 203 | 238 | 170 | 166 | 157 | 189 | 84 | 88 |
| SMD a | 158 | 169 | 136 | 129 | 96 | 102 | 26 | 29 |
| Theory/Basis Set | TS1C | TS2C | TS3C | |||
|---|---|---|---|---|---|---|
| E‡ | ∆G‡ | E‡ | ∆G‡ | E‡ | ∆G‡ | |
| B3LYP/6-31G(d) | 189 | 230 | 221 | 223 | 45 | 42 |
| B3LYP/6-31G(2df,p) | 191 | 232 | 225 | 228 | 45 | 42 |
| B3LYP/6-311++G(3df,3pd) | 189 | 228 | 213 | 215 | 48 | 45 |
| ωB97XD/6-311++G(3df,3pd) | 205 | 245 | 255 | 257 | 68 | 63 |
| APDF/6-311++G(3df,3pd) | 195 | 235 | 229 | 231 | 68 | 61 |
| M06-2X/6-311++G(3df,3pd) | 218 | 258 | 278 | 281 | 80 | 75 |
| M11/6-11++G(3df,3pd) | 204 | 243 | 273 | 276 | 70 | 75 |
| G3MP2 | 203 | 238 | 223 | 225 | 73 | 70 |
| SMD a | 158 | 169 | 179 | 181 | 77 | 67 |
| Theory/Basis Set | TS1D | TS2D | TS3D | TS1E | TS2E | TS3E | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E‡ | G‡ | E‡ | G‡ | E‡ | G‡ | E‡ | G‡ | E‡ | G‡ | E‡ | G‡ | |
| B3LYP/6-31G(d) | 174 | 214 | 164 | 166 | 183 | 179 | 128 | 205 | 82 | 89 | 99 | 105 |
| B3LYP/6-31G(2df,p) | 173 | 214 | 165 | 167 | 181 | 178 | 130 | 207 | 83 | 91 | 101 | 106 |
| B3LYP/6-311++G(3df,3pd) | 175 | 213 | 172 | 174 | 190 | 177 | 117 | 180 | 98 | 106 | 110 | 118 |
| ωB97XD/6-1++G(3df,3pd) | 193 | 232 | 178 | 181 | 194 | 192 | 159 | 233 | 104 | 112 | 122 | 130 |
| APDF/6-311++G(3df,3pd) | 180 | 220 | 169 | 172 | 186 | 183 | 147 | 224 | 82 | 90 | 104 | 110 |
| M06-2X/6-11++G(3df,3pd) | 201 | 240 | 191 | 193 | 200 | 196 | 168 | 241 | 97 | 105 | 120 | 126 |
| M11/6-++G(3df,3pd) | 187 | 224 | 187 | 189 | 196 | 191 | 155 | 225 | 95 | 102 | 112 | 120 |
| G3MP2 | 163 | 192 | 179 | 182 | 197 | 194 | 135 | 201 | 113 | 122 | 127 | 138 |
| SMD a | 147 | 153 | 173 | 174 | 197 | 182 | 75 | 80 | 98 | 106 | 43 | 42 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almatarneh, M.H.; Alrebei, S.F.; Altarawneh, M.; Zhao, Y.; Abu-Saleh, A.A.-A. Computational Study of the Dissociation Reactions of Secondary Ozonide. Atmosphere 2020, 11, 100. https://doi.org/10.3390/atmos11010100
Almatarneh MH, Alrebei SF, Altarawneh M, Zhao Y, Abu-Saleh AA-A. Computational Study of the Dissociation Reactions of Secondary Ozonide. Atmosphere. 2020; 11(1):100. https://doi.org/10.3390/atmos11010100
Chicago/Turabian StyleAlmatarneh, Mansour H., Shefa’ F. Alrebei, Mohammednoor Altarawneh, Yuming Zhao, and Abd Al-Aziz Abu-Saleh. 2020. "Computational Study of the Dissociation Reactions of Secondary Ozonide" Atmosphere 11, no. 1: 100. https://doi.org/10.3390/atmos11010100
APA StyleAlmatarneh, M. H., Alrebei, S. F., Altarawneh, M., Zhao, Y., & Abu-Saleh, A. A.-A. (2020). Computational Study of the Dissociation Reactions of Secondary Ozonide. Atmosphere, 11(1), 100. https://doi.org/10.3390/atmos11010100