Identification of Novel Candidate Markers of Type 2 Diabetes and Obesity in Russia by Exome Sequencing with a Limited Sample Size

 , , , ,
, , , ,  , and
, and
Abstract
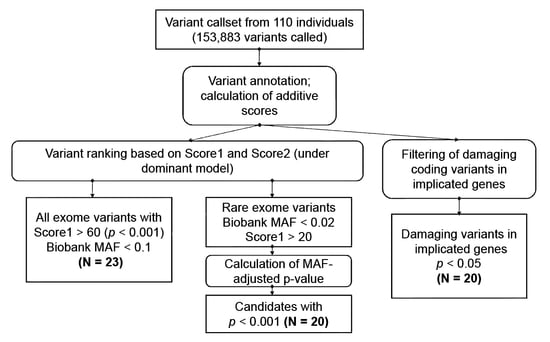
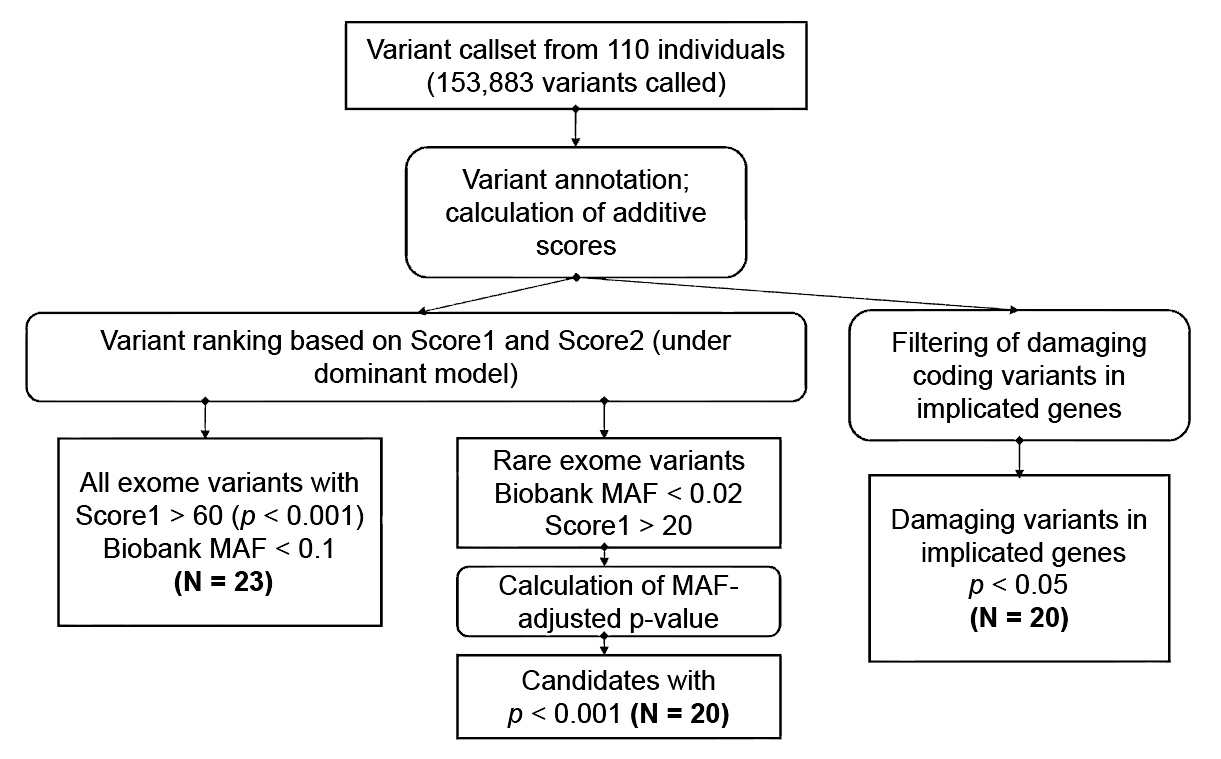
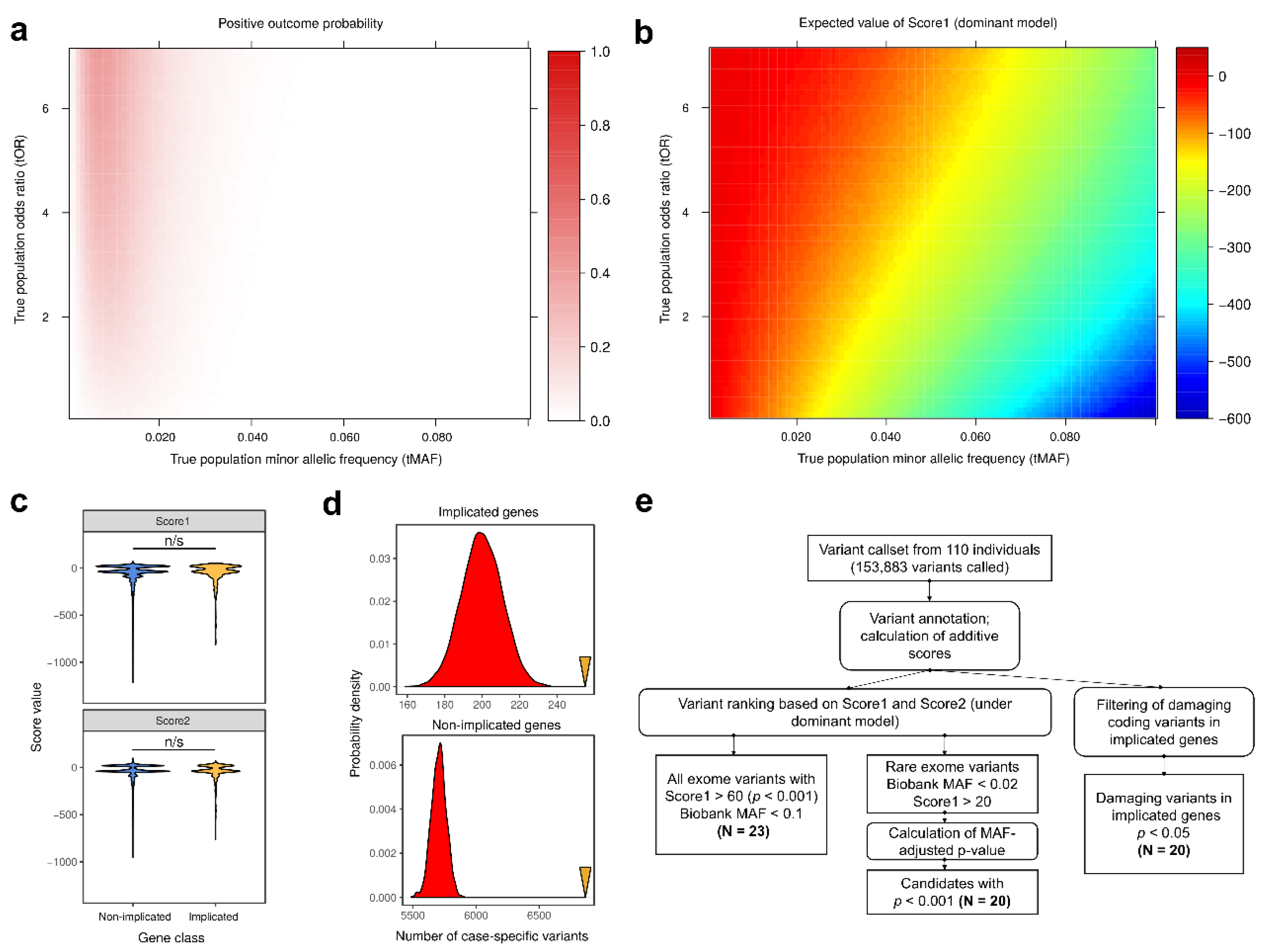
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA Isolation
2.3. Library Preparation and Exome Sequencing
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
- (i)
- variants in known genes implicated in T2D [10] and obesity [26] with significant effect on the corresponding protein (protein-altering variants) (based on the variant type (only non-synonymous substitutions and coding indels were selected) and pathogenicity prediction (for missense variants) by SIFT [27], PROVEAN [28] and Polyphen2 [29] packages); (significance level of 0.05 was applied for these variants)
- (ii)
- low-frequency variants (MAF between 1% and 10% according to SPBU Biobank data) that are highly specific to the case or control group (see Results for a more detailed analysis of such variants’ properties). (significance level of 0.001 was applied for case-specific variants).
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bougnères, P. Genetics of obesity and type 2 diabetes. Tracking pathogenic traits during the predisease period. Diabetes 2002, 51, S295–S303. [Google Scholar] [CrossRef] [PubMed]
- Langenberg, C.; Lotta, L.A. Genomic insights into the causes of type 2 diabetes. Lancet 2018, 391, 2463–2474. [Google Scholar] [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; Brussels International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Wang, X.; Strizich, G.; Hu, Y.; Wang, T.; Kaplan, R.C.; Qi, Q. Genetic markers of type 2 diabetes: Progress in genome-wide association studies and clinical application for risk prediction. J. Diabetes 2016, 8, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015, 6, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Hara, K.; Kubota, N.; Terauchi, Y.; Tobe, K.; Froguel, P.; Nagai, R.; Kadowaki, T. Dual roles of adiponectin/Acrp30 in vivo as an anti-diabetic and anti-atherogenic adipokine. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2003, 3, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.S.M.; Woo, J. Prevention of overweight and obesity: How effective is the current public health approach. Int. J. Environ. Res. Public Health 2010, 7, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, T. Type 2 diabetes mellitus in children and adolescents. World J. Diabetes 2013, 4, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Scott, L.J.; Mägi, R.; Marullo, L.; Gaulton, K.J.; Kaakinen, M.; Pervjakova, N.; Pers, T.H.; Johnson, A.D.; Eicher, J.D.; et al. An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes 2018, 66, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Yengo, L.; Sidorenko, J.; Kemper, K.E.; Zheng, Z.; Wood, A.R.; Weedon, M.N.; Frayling, T.M.; Hirschhorn, J.; Yang, J.; Visscher, P.M.; et al. Meta-analysis of genome-wide association studies for height and body mass index in ~700,000 individuals of European ancestry. bioRxiv 2018, 274654. [Google Scholar] [CrossRef]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Lohmueller, K.E.; Li, Q.; Andersson, E.; Korneliussen, T.; Albrechtsen, A.; Banasik, K.; Grarup, N.; Hallgrimsdottir, I.; Kiil, K.; Krarup, N.T.; et al. Whole-exome sequencing of 2000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am. J. Hum. Genet. 2013, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, A.; Grarup, N.; Li, Y.; Tian, G.; Cao, H.; Jiang, T.; Kim, S.Y.; Korneliussen, T.; Li, Q.; Nie, C.; et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia 2013, 56, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Ruden, D.M.; Lu, X. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E.; Jolla, L.; Genome, H. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human non-synonymous and splice site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; Birnbaum, D.P.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Bezdvornykh, I.V.; Polev, D.E.; Serebryakova, E.A.; Glotov, A.S.; Glotov, O.S.; Predeus, A.V. Catching hidden variation: Systematic correction of reference minor allele annotation in clinical variant calling. Genet. Med. 2018, 20, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Turcot, V.; Lu, Y.; Highland, H.M.; Schurmann, C.; Justice, A.E.; Fine, R.S.; Bradfield, J.P.; Esko, T.; Giri, A.; Graff, M.; et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat. Genet. 2018, 50, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Jagannadham, J.; Jaiswal, H.K.; Agrawal, S.; Rawal, K. Comprehensive map of molecules implicated in obesity. PLoS ONE 2016, 11, e0146759. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Adzhubey, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Glotov, A.S.; Kazakov, S.V.; Zhukova, E.A.; Alexandrov, A.V.; Glotov, O.S.; Pakin, V.S.; Danilova, M.M.; Tarkovskaya, I.V.; Niyazova, S.V.; Chakova, N.N.; et al. Targeted next-generation sequencing (NGS) of nine candidate genes with custom AmpliSeq in patients and a cardiomyopathy risk group. Clin. Chim. Acta 2015, 15, 132–140. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org/ (accessed on 10 February 2018).
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015, 518, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.W.; et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes article. Nat. Genet. 2018, 50, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Zabetian, C.P.; Romero, R.; Robertson, D.; Sharma, S.; Padbury, J.F.; Kuivaniemi, H.; Kim, K.S.; Kim, C.H.; Kohnke, M.D.; Kranzler, H.R.; et al. A revised allele frequency estimate and haplotype analysis of the DBH deficiency mutation IVS1+2T->C in African- and European-Americans. Am. J. Med. Genet. Part A 2003, 123, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Ehret, G.B.; Ferreira, T.; Chasman, D.I.; Jackson, A.U.; Schmidt, E.M.; Johnson, T.; Thorleifsson, G.; Luan, J.; Donnelly, L.A.; Kanoni, S.; et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat. Genet. 2016, 48, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, A.; De Bustros, A.; Borges, M.; Jasti, R.; Compton, D.; Diamond, L.; Mabry, M.; Ball, D.W.; Baylin, S.B.; Nelkin, B.D. RREB-1, a novel zinc finger protein, is involved in the differentiation response to Ras in human medullary thyroid carcinomas. Mol. Cell. Biol. 1996, 16, 5335–5345. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto-Nishiyama, A.; Ishii, S.; Matsuda, S.; Inoue, J.; Yamamoto, T. A novel zinc finger protein, Finb, is a transcriptional activator and localized in nuclear bodies. Gene 1997, 195, 267–275. [Google Scholar] [CrossRef]
- Ray, S.K.; Nishitani, J.; Petry, M.W.; Fessing, M.Y.; Leiter, A.B. Novel transcriptional potentiation of BETA2/NeuroD on the secretin gene promoter by the DNA-binding protein Finb/RREB-1. Mol. Cell. Biol. 2003, 23, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Flajollet, S.; Poras, I.; Carosella, E.D.; Moreau, P. RREB-1 is a transcriptional repressor of HLA-G. J. Immunol. 2009, 183, 6948–6959. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.T.; Monda, K.L.; Taylor, K.C.; Lange, L.; Demerath, E.W.; Palmas, W.; Wojczynski, M.K.; Ellis, J.C.; Vitolins, M.Z.; Liu, S.; et al. Genome-wide association of body fat distribution in African ancestry populations suggests new loci. PLoS Genet. 2013, 9, e1003681. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Lagou, V.; Welch, R.P.; Wheeler, W.; Montasser, M.E.; Luan, J.; Magi, R.; Strawbridge, R.J.; Rehnberg, E.; Gustafsson, S.; et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012, 44, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.H.; Kyong, S.P. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48, e220. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, T.; Komaki, S.; Hasegawa, Y.; Ohmomo, H.; Tanno, K.; Hozawa, A.; Tamiya, G.; Yamamoto, M.; Ogasawara, K.; Nakamura, M.; et al. Genome-wide meta-analysis in Japanese populations identifies novel variants at the TMC6-TMC8 and SIX3-SIX2 loci associated with HbA1c. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, A.M.; Hamilton-Williams, E.E.; Cristino, A.; Ziegler, A.; Bonifacio, E.; Le Cao, K.-A.; Harris, M.; Thomas, R. A peripheral blood transcriptomic signature predicts autoantibody development in infants at risk of type 1 diabetes. JCI Insight 2018, 3, e98212. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, M.; Taniuchi, K.; Furihata, M.; Seiji, N.; Masashi, K.; Watanabe, R.; Shimizu, T.; Saito, M.; Dabanaka, K.; Kazuhiro, H.; et al. Vav3 is linked to poor prognosis of pancreatic cancers and promotes the motility and invasiveness of pancreatic cancer cells. Pancreatology 2016, 16, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Stehouwer, C.D.; Fischer, H.R.; Van Kuijk, A.W.; Polak, B.C.; Donker, A.J. Endothelial dysfunction precedes development of microalbuminuria in IDDM. Diabetes 1995, 44, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Zeldenrust, G.C.; Hackeng, W.H.; Donker, A.J.; Den Ottolander, G.J. Urinary albumin excretion, cardiovascular disease, and endothelial dysfunction in noninsulin-dependent diabetes mellitus. Lancet 1992, 340, 319–323. [Google Scholar]
- Skeppholm, M.; Kallner, A.; Kalani, M.; Jörneskog, G.; Blombäck, M.; Wallén, H.N. ADAMTS13 and von Willebrand factor concentrations in patients with diabetes mellitus. Blood Coagul. Fibrinol. 2009, 20, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Stehouwer, C.D.; Gall, M.A.; Twisk, J.W.; Knudsen, E.; Emeis, J.J.; Parving, H.H. Increased urinary albumin excretion, endothelial dysfunction, and chronic low-grade inflammation in type 2 diabetes: Progressive, interrelated, and independently associated with risk of death. Diabetes 2002, 51, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Standl, E.; Balletshofer, B.; Dahl, B.; Weichenhain, B.; Stiegler, H.; Hormann, A.; Holle, R. Predictors of 10-year macrovascular and overall mortality in patients with NIDDM: The Munich General Practitioner Project. Diabetologia 1996, 39, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Nair, A.K.; Muller, Y.L.; Piaggi, P.; Bian, L.; Knowler, W.C.; Kobes, S.; Hanson, R.L.; Bogardus, C.; Baier, L.J. Whole exome sequencing identifies variation in CYB5A and RNF10 associated with adiposity and type 2 diabetes. Obesity 2014, 22, 984–988. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Characteristic | Control Group (n = 40) | Obesity Group (n = 21) | T2D (with Obesity) (n = 49) |
|---|---|---|---|
| Age (years) | 56.44 ± 10.8 | 55.76 ± 8.60 | 50.21 ± 10.82 |
| Male | 52.5% | 61.9% | 34.7% |
| Ethnicity | Russian | Russian | Russian |
| BMI (kg/m2) | 22.56 ± 1.85 | 43.05 ± 5.70 | 32.10 ± 8.92 |
| Family History (based on the results of the questionnaire) | |||
| Family history of obesity | no (40/40) | yes (21/21) | yes (49/49) |
| Family history of T2D | no (40/40) | yes (21/21) | yes (49/49) |
| Family history of other endocrine pathologies | no (40/40) | yes (21/21) | yes (49/49) |
| Test | Position | rsID | Gene | Effect | p | MAF | padj * | OR/β | Comment |
|---|---|---|---|---|---|---|---|---|---|
| 1. Replication of Association (SNP-Level) with Binary/Quantitative Traits | |||||||||
| BMI vs. all variants | chr2:272203:C>T | rs11553746 | ACP1 | Missense variant | 0.012 | 0.352 | - | 3.92 | |
| chr14:60928201:G>A | rs1956549 | C14orf39 | Intron variant | 0.013 | 0.073 | - | 2.60 | ||
| chr16:24578458:T>C | rs7195386 | RBBP6 | Splice variant | 0.029 | 0.473 | - | −1.24 | [11] | |
| OB vs. control | chr5:180166677:G>A | rs11960429 | OR2Y1 | Missense variant | 0.043 | 0.014 | - | 13.6 | |
| T2D vs. C, T2D vs. OBC | chr6:7231843:G>A | rs9379084 | RREB1 | Missense variant | 0.013, 0.042 | 0.097 | - | 0.26, 0.35 | [10,34] |
| chr12:49399132:G>C | rs1126930 | PRKAG1 | Missense variant | 0.041, 0.037 | 0.032 | 0.09,0.09 | [9,25] | ||
| 2. Association with Quantitative Traits ** | |||||||||
| BMI vs. all variants | chr16:69752464:C>G | rs689452 | NQO1 | Intron variant | 9 × 10−6 | 0.129 | - | 2.04 | Associated with height [33] |
| 3. Association with Protein-Altering Variants in Known Genes (0.01 < SPBU MAF < 0.1) | |||||||||
| OB + T2D vs. control | chr8:19819724:C>G | rs328 | LPL | Stop gained | 0.023 | 0.065 | - | 0.26 | Recently associated [34] |
| OB vs. control | chr9:136522274:C>T | rs6271 | DBH | Missense variant | 0.043 | 0.041 | - | 13.5 | Associated with BP *** [36] |
| chr11:32956:C>T | rs62618693 | QSER1 | Missense variant | 0.021 | 0.038 | - | 10.1 | Recently associated [34] | |
| T2D vs. control | chr6:7231843:G>A | rs9379084 | RREB1 | Missense variant | 0.013 | 0.097 | - | 0.26 | Recently associated [34] |
| chr6:31079264:C>T | rs2233984 | C6orf15 | Missense variant | 0.030 | 0.067 | - | 0.31 | Associated with height [33] | |
| T2D vs. OBC | chr2:160994293:C>T | rs61737764 | ITGB6 | Missense variant | 0.035 | 0.007 | - | 12.1 | Linked withrs7593730 |
| chr6:7231843:G>A | rs9379084 | RREB1 | Missense variant | 0.042 | 0.097 | - | 0.35 | ||
| 4. Association with Case-Specific Exome Variants (SPBU MAF < 0.1) | |||||||||
| OB vs. control | chr5:140168291:T>C | rs34042554 | PCDHA1 | Missense variant | 0.003 | 0.017 | 1.0 × 10−4 | 25.1 | |
| chr14:69061228:G>A | rs61758785 | RAD51B | Missense variant | 0.035 | 0.007 | 1.7 × 10−4 | 12.6 | ||
| chr12:19408017:G>A | rs144183813 | PLEKHA5 | Missense variant | 0.015 | 0.007 | 1.7 × 10−4 | 17.9 | Gene expression affects T1D † [45] | |
| T2D vs. OBC | chr12:48376869:T>C | rs17801742 | COL2A1 | Splice variant | 3.1 × 10−4 | 0.089 | 8.5 × 10−5 | 10.4 | Associated with height [33] |
| OB + T2D vs. control | chr16:230578:A>G | rs11863726 | HBQ1 | Splice variant | 3.9 × 10−4 | 0.094 | 8 × 10−5 | 22.8 | |
| chr1:108119007:G>C | rs112984085 | VAV3 | Intron variant | 7.1 × 10−4 | 0.063 | 4.8 × 10−4 | 21.3 | ||
| T2D vs. C, T2D vs. OBC | chr9:136310908:C>T | rs685523 | ADAMTS13 | Missense variant | 2.7 × 10−4, 1.6 × 10−5 | 0.083 | 7 × 10−5, 1 × 10−6 | 26.0, 38.9 | |
| chr17:76136994:C>T | rs139972217 | TMC8 | Missense variant | 0.032, 0.006 | 0.009 | 1.4 × 10−4, 9.9 × 10−5 | 11.6, 17.9 | Gene associated with HbA1c [44] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbitoff, Y.A.; Serebryakova, E.A.; Nasykhova, Y.A.; Predeus, A.V.; Polev, D.E.; Shuvalova, A.R.; Vasiliev, E.V.; Urazov, S.P.; Sarana, A.M.; Scherbak, S.G.; et al. Identification of Novel Candidate Markers of Type 2 Diabetes and Obesity in Russia by Exome Sequencing with a Limited Sample Size. Genes 2018, 9, 415. https://doi.org/10.3390/genes9080415
Barbitoff YA, Serebryakova EA, Nasykhova YA, Predeus AV, Polev DE, Shuvalova AR, Vasiliev EV, Urazov SP, Sarana AM, Scherbak SG, et al. Identification of Novel Candidate Markers of Type 2 Diabetes and Obesity in Russia by Exome Sequencing with a Limited Sample Size. Genes. 2018; 9(8):415. https://doi.org/10.3390/genes9080415
Chicago/Turabian StyleBarbitoff, Yury A., Elena A. Serebryakova, Yulia A. Nasykhova, Alexander V. Predeus, Dmitrii E. Polev, Anna R. Shuvalova, Evgenii V. Vasiliev, Stanislav P. Urazov, Andrey M. Sarana, Sergey G. Scherbak, and et al. 2018. "Identification of Novel Candidate Markers of Type 2 Diabetes and Obesity in Russia by Exome Sequencing with a Limited Sample Size" Genes 9, no. 8: 415. https://doi.org/10.3390/genes9080415
APA StyleBarbitoff, Y. A., Serebryakova, E. A., Nasykhova, Y. A., Predeus, A. V., Polev, D. E., Shuvalova, A. R., Vasiliev, E. V., Urazov, S. P., Sarana, A. M., Scherbak, S. G., Gladyshev, D. V., Pokrovskaya, M. S., Sivakova, O. V., Meshkov, A. N., Drapkina, O. M., Glotov, O. S., & Glotov, A. S. (2018). Identification of Novel Candidate Markers of Type 2 Diabetes and Obesity in Russia by Exome Sequencing with a Limited Sample Size. Genes, 9(8), 415. https://doi.org/10.3390/genes9080415