Decoding the Heart through Next Generation Sequencing Approaches


Abstract
1. Introduction
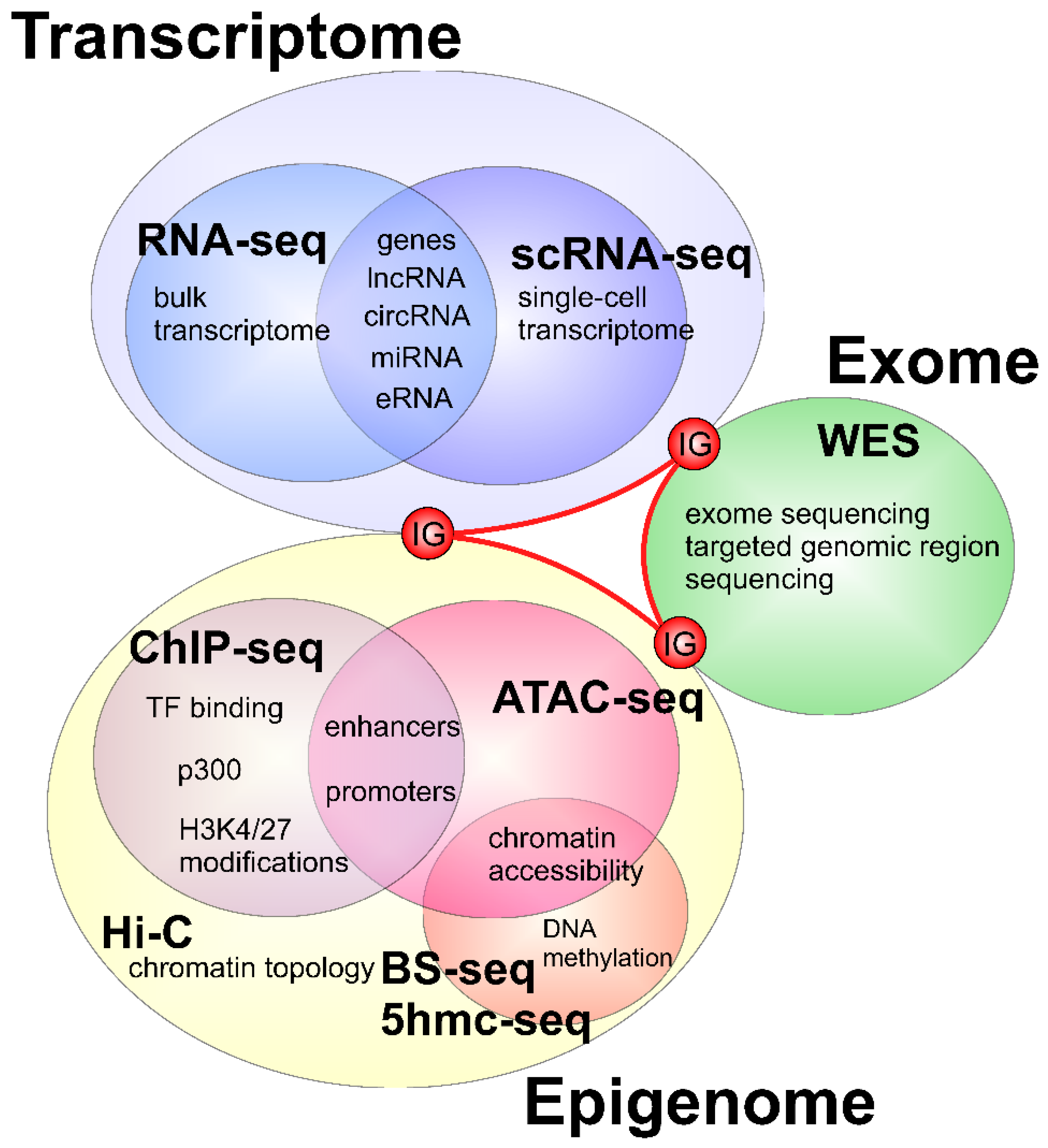
2. Applications of Next Generation Sequencing to Study Heart Development
2.1. Cardiac Cell Specification and Differentiation
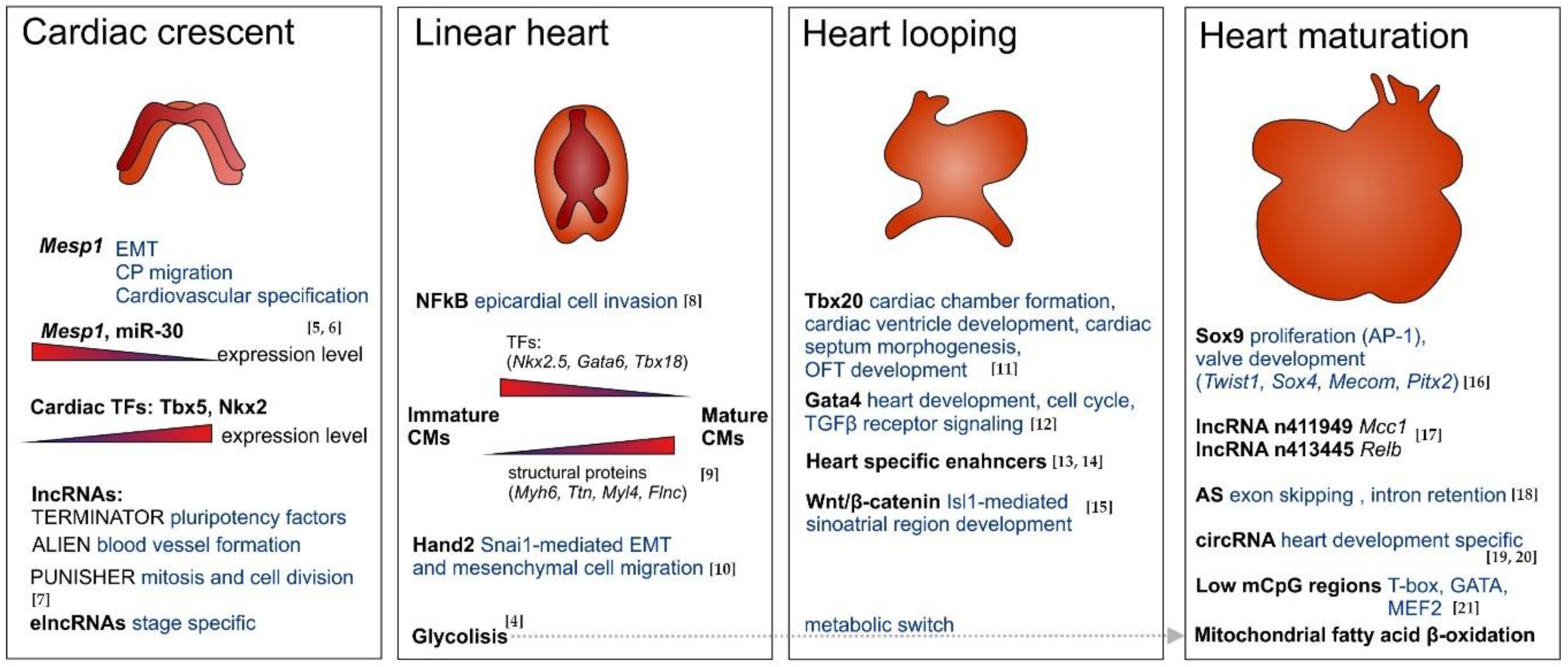
2.2. Insights into Heart Morphogenesis
2.2.1. Early Morphogenesis
2.2.2. Late Morphogenesis
2.3. Lessons from Next Generation Sequencing Application in Clinical Studies of Congenital Heart Disease
3. Next Generation Sequencing in Heart Regeneration
3.1. Heart Regeneration—Decades of Intensive Studies
3.2. Molecular Mechanism of Cardiac Regeneration
4. Conclusions and Future Perspectives for the Application of Emerging Next Generation Sequencing -Based Assays on Heart Biology
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.L. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Vecoli, C.; Pulignani, S.; Foffa, I.; Andreassi, M.G. Congenital heart disease: The crossroads of genetics, epigenetics and environment. Curr. Genom. 2014, 15, 390–399. [Google Scholar] [CrossRef] [PubMed]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-cell resolution of temporal gene expression during heart development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sanchez-Danes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Rustagi, Y.; Jaiswal, H.K.; Rawal, K.; Kundu, G.C.; Rani, V. Comparative characterization of cardiac development specific microRNAs: Fetal regulators for future. PLoS ONE 2015, 10, e0139359. [Google Scholar] [CrossRef] [PubMed]
- Kurian, L.; Aguirre, A.; Sancho-Martinez, I.; Benner, C.; Hishida, T.; Nguyen, T.B.; Reddy, P.; Nivet, E.; Krause, M.N.; Nelles, D.A.; et al. Identification of novel long non-coding RNAs underlying vertebrate cardiovascular development. Circulation 2015, 131, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- DeLaughter, D.M.; Clark, C.R.; Christodoulou, D.C.; Seidman, C.E.; Baldwin, H.S.; Seidman, J.G.; Barnett, J.V. Transcriptional profiling of cultured, embryonic epicardial cells identifies novel genes and signaling pathways regulated by TGFβR3 in vitro. PLoS ONE 2016, 11, e0159710. [Google Scholar] [CrossRef] [PubMed]
- Sereti, K.I.; Nguyen, N.B.; Kamran, P.; Zhao, P.; Ranjbarvaziri, S.; Park, S.; Sabri, S.; Engel, J.L.; Sung, K.; Kulkarni, R.P.; et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat. Commun. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Laurent, F.; Girdziusaite, A.; Gamart, J.; Barozzi, I.; Osterwalder, M.; Akiyama, J.A.; Lincoln, J.; Lopez-Rios, J.; Visel, A.; Zuniga, A.; et al. HAND2 target gene regulatory networks control atrioventricular canal and cardiac valve development. Cell Rep. 2017, 19, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Boogerd, C.J.; Aneas, I.; Sakabe, N.; Dirschinger, R.J.; Cheng, Q.J.; Zhou, B.; Chen, J.; Nobrega, M.A.; Evans, S.M. Probing chromatin landscape reveals roles of endocardial TBX20 in septation. J. Clin. Investig. 2016, 126, 3023–3035. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; McCulley, D.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 2010, 42, 806–810. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2011, 44, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, S.B.; Bakkers, J. Spatially resolved RNA-sequencing of the embryonic heart identifies a role for Wnt/β-catenin signaling in autonomic control of heart rate. elife 2018, 7, e31515. [Google Scholar] [CrossRef] [PubMed]
- Garside, V.C.; Cullum, R.; Alder, O.; Lu, D.Y.; Vander Werff, R.; Bilenky, M.; Zhao, Y.; Jones, S.J.; Marra, M.A.; Underhill, T.M.; et al. SOX9 modulates the expression of key transcription factors required for heart valve development. Development 2015, 142, 4340–4350. [Google Scholar] [CrossRef] [PubMed]
- Matkovich, S.J.; Edwards, J.R.; Grossenheider, T.C.; de Guzman Strong, C.; Dorn, G.W., 2nd. Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc. Natl. Acad. Sci. USA 2014, 111, 12264–12269. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Y.; Li, X.; Chen, G.; Zhong, L.; Chen, G.; Liao, Y.; Liao, W.; Bin, J. Genome-wide analysis of alternative splicing during human heart development. Sci. Rep. 2016, 6, 35520. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wu, J.; Han, P.; Zhao, Z.; Song, X. Circular RNA expression profiles and features in human tissues: A study using RNA-seq data. BMC Genom. 2017, 18, 680. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.L.; Lim, B.T.; Anene-Nzelu, C.G.; Ackers-Johnson, M.; Dashi, A.; See, K.; Tiang, Z.; Lee, D.P.; Chua, W.W.; Luu, T.D.; et al. A landscape of circular RNA expression in the human heart. Cardiovasc. Res. 2017, 113, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Gruning, B.A.; Kranzhofer, D.; Schneider, P.; Nuhrenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.W.; Sinha, R.; Barkal, A.A.; Morganti, R.M.; Chen, A.; Weissman, I.L.; Ang, L.T.; Kundaje, A.; Loh, K.M. An atlas of transcriptional, chromatin accessibility, and surface marker changes in human mesoderm development. Sci. Data 2016, 3, 160109. [Google Scholar] [CrossRef] [PubMed]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Ounzain, S.; Pezzuto, I.; Micheletti, R.; Burdet, F.; Sheta, R.; Nemir, M.; Gonzales, C.; Sarre, A.; Alexanian, M.; Blow, M.J.; et al. Functional importance of cardiac enhancer-associated noncoding RNAs in heart development and disease. J. Mol. Cell. Cardiol. 2014, 76, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [PubMed]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Fields, P.A.; Ramani, V.; Bonora, G.; Yardimci, G.G.; Bertero, A.; Reinecke, H.; Pabon, L.; Noble, W.S.; Shendure, J.; Murry, C. Dynamic reorganization of nuclear architecture during human cardiogenesis. bioRxiv 2017. [Google Scholar] [CrossRef]
- Hoffmann, A.D.; Yang, X.H.; Burnicka-Turek, O.; Bosman, J.D.; Ren, X.; Steimle, J.D.; Vokes, S.A.; McMahon, A.P.; Kalinichenko, V.V.; Moskowitz, I.P. Foxf genes integrate Tbx5 and Hedgehog pathways in the second heart field for cardiac septation. PLoS Genet. 2014, 10, e1004604. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Kruse, F.; Junker, J.P.; van Oudenaarden, A.; Bakkers, J. Tomo-seq: A method to obtain genome-wide expression data with spatial resolution. Methods Cell Biol. 2016, 135, 299–307. [Google Scholar] [PubMed]
- Hill, J.T.; Demarest, B.; Gorsi, B.; Smith, M.; Yost, H.J. Heart morphogenesis gene regulatory networks revealed by temporal expression analysis. Development 2017, 144, 3487–3498. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Samtani, R.; Dhanantwari, P.; Lee, E.; Yamada, S.; Shiota, K.; Donofrio, M.T.; Leatherbury, L.; Lo, C.W. A detailed comparison of mouse and human cardiac development. Pediatr. Res. 2014, 76, 500–507. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Gu, F.; Hu, Y.; Ma, Q.; Ye, L.Y.; Akiyama, J.A.; Visel, A.; Pennacchio, L.A.; Pu, W.T. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat. Commun. 2014, 5, 4907. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Kong, S.W.; Ma, Q.; Pu, W.T. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. USA 2011, 108, 5632–5637. [Google Scholar] [CrossRef] [PubMed]
- Tayal, U.; Prasad, S.; Cook, S.A. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Blue, G.M.; Kirk, E.P.; Giannoulatou, E.; Sholler, G.F.; Dunwoodie, S.L.; Harvey, R.P.; Winlaw, D.S. Advances in the genetics of congenital heart disease: A clinician’s guide. J. Am. Coll. Cardiol. 2017, 69, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Bonachea, E.M.; Zender, G.; White, P.; Corsmeier, D.; Newsom, D.; Fitzgerald-Butt, S.; Garg, V.; McBride, K.L. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Med. Genom. 2014, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, X.; Wang, L.; Jiang, J.; Sun, Y.; Zhu, Q.; Chen, Z.; He, Y.; Hu, P.; Xu, Q.; et al. Targeted next-generation sequencing identified ADAMTS5 as novel genetic substrate in patients with bicuspid aortic valve. Int. J. Cardiol. 2018, 252, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Pulignani, S.; Vecoli, C.; Borghini, A.; Foffa, I.; Ait-Ali, L.; Andreassi, M.G. Targeted next-generation sequencing in patients with non-syndromic congenital heart disease. Pediatr. Cardiol. 2018, 39, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Preuss, C.; Capredon, M.; Wunnemann, F.; Chetaille, P.; Prince, A.; Godard, B.; Leclerc, S.; Sobreira, N.; Ling, H.; Awadalla, P.; et al. Family based whole exome sequencing reveals the multifaceted role of Notch signaling in congenital heart disease. PLoS Genet. 2016, 12, e1006335. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Abaci, N.; Arikan, M.; Tansel, T.; Sahin, N.; Cakiris, A.; Pacal, F.; Sirma Ekmekci, S.; Gok, E.; Ustek, D. Mitochondrial mutations in patients with congenital heart defects by next generation sequencing technology. Cardiol. Young 2015, 25, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.D.; Shen, P.; Fung, E.; Karakikes, I.; Zhang, A.; InanlooRahatloo, K.; Odegaard, J.; Sallam, K.; Davis, R.W.; Lui, G.K.; et al. A rapid, high-quality, cost-effective, comprehensive and expandable targeted next-generation sequencing assay for inherited heart diseases. Circ. Res. 2015, 117, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Manheimer, K.B.; Richter, F.; Edelmann, L.J.; D’Souza, S.L.; Shi, L.; Shen, Y.; Homsy, J.; Boskovski, M.T.; Tai, A.C.; Gorham, J.; et al. Robust identification of mosaic variants in congenital heart disease. Hum. Genet. 2018, 137, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Bu, H.; Yang, Y.; Tan, Z.; Zhang, F.; Hu, S.; Zhao, T. A targeted, next-generation genetic sequencing study on tetralogy of fallot, combined with cleft lip and palate. J. Craniofac. Surg. 2017, 28, e351–e355. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, H.D.; Cui, C.Y.; Qin, Y.Y.; Fan, T.B.; Peng, B.T.; Zhang, L.Z.; Wang, C.Z. Whole exome sequencing identifies novel mutation in eight Chinese children with isolated tetralogy of Fallot. Oncotarget 2017, 8, 106976–106988. [Google Scholar] [CrossRef] [PubMed]
- LaHaye, S.; Corsmeier, D.; Basu, M.; Bowman, J.L.; Fitzgerald-Butt, S.; Zender, G.; Bosse, K.; McBride, K.L.; White, P.; Garg, V. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circ. Cardiovasc. Genet. 2016, 9, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qi, B.; Zhao, J.; Liu, W.; Duan, R.; Zhang, M. A novel mutation of GATA4 (K300T) associated with familial atrial septal defect. Gene 2016, 575, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Hanchard, N.A.; Furthner, D.; Fernbach, S.; Azamian, M.; Nicosia, A.; Rosenfeld, J.; Muzny, D.; D’Alessandro, L.C.A.; Morris, S.; et al. Whole exome sequencing in 342 congenital cardiac left sided lesion cases reveals extensive genetic heterogeneity and complex inheritance patterns. Genome Med. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Guimier, A.; Gabriel, G.C.; Bajolle, F.; Tsang, M.; Liu, H.; Noll, A.; Schwartz, M.; El Malti, R.; Smith, L.D.; Klena, N.T.; et al. MMP21 is mutated in human heterotaxy and is required for normal left-right asymmetry in vertebrates. Nat. Genet. 2015, 47, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Sifrim, A.; Hitz, M.P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Priest, J.R.; Osoegawa, K.; Mohammed, N.; Nanda, V.; Kundu, R.; Schultz, K.; Lammer, E.J.; Girirajan, S.; Scheetz, T.; Waggott, D.; et al. De novo and rare variants at multiple loci support the oligogenic origins of atrioventricular septal heart defects. PLoS Genet. 2016, 12, e1005963. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, M.; Wang, Y.; Hou, X.; Wu, H.; Wang, D.; Shen, H.; Hu, Z.; Zou, J. Whole-exome sequencing identify a new mutation of MYH7 in a chinese family with left ventricular noncompaction. Gene 2015, 558, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Zdunek, S.; Frisen, J.; Bernard, S.; Druid, H.; Jovinge, S. Cardiomyocyte renewal in humans. Circ. Res. 2012, 110, e17–e18. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of cell generation and turnover in the human heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Poss, K.D. Cardiac regenerative capacity and mechanisms. Annu. Rev. Cell Dev. Biol. 2012, 28, 719–741. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics-2018 update: A report from the American heart association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Atlas Writing, G.; Timmis, A.; Townsend, N.; Gale, C.; Grobbee, R.; Maniadakis, N.; Flather, M.; Wilkins, E.; Wright, L.; Vos, R.; et al. European society of cardiology: Cardiovascular disease statistics 2017. Eur. Heart J. 2018, 39, 508–579. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Raya, A.; Koth, C.M.; Buscher, D.; Kawakami, Y.; Itoh, T.; Raya, R.M.; Sternik, G.; Tsai, H.J.; Rodriguez-Esteban, C.; Izpisua-Belmonte, J.C. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. Proc. Natl. Acad. Sci. USA 2003, 100 (Suppl. 1), 11889–11895. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Panakova, D.; Kikuchi, K.; Holdway, J.E.; Gemberling, M.; Burris, J.S.; Singh, S.P.; Dickson, A.L.; Lin, Y.F.; Sabeh, M.K.; et al. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 2011, 138, 3421–3430. [Google Scholar] [CrossRef] [PubMed]
- Chablais, F.; Veit, J.; Rainer, G.; Jazwinska, A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev. Biol. 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rosa, J.M.; Martin, V.; Peralta, M.; Torres, M.; Mercader, N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development 2011, 138, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, K.; Wu, C.C.; Kurth, T.; Weidinger, G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS ONE 2011, 6, e18503. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Sleep, E.; Raya, M.; Marti, M.; Raya, A.; Izpisua Belmonte, J.C. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Holdway, J.E.; Werdich, A.A.; Anderson, R.M.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; Macrae, C.A.; Stainier, D.Y.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4+ cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Hu, J.; Karra, R.; Dickson, A.L.; Tornini, V.A.; Nachtrab, G.; Gemberling, M.; Goldman, J.A.; Black, B.L.; Poss, K.D. Modulation of tissue repair by regeneration enhancer elements. Nature 2016, 532, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Kruse, F.; Vasudevarao, M.D.; Junker, J.P.; Zebrowski, D.C.; Fischer, K.; Noel, E.S.; Grun, D.; Berezikov, E.; Engel, F.B.; et al. Spatially resolved genome-wide transcriptional profiling identifies BMP signaling as essential regulator of zebrafish cardiomyocyte regeneration. Dev. Cell 2016, 36, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Iranzo, H.; Galardi-Castilla, M.; Minguillon, C.; Sanz-Morejon, A.; Gonzalez-Rosa, J.M.; Felker, A.; Ernst, A.; Guzman-Martinez, G.; Mosimann, C.; Mercader, N. Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nat. Commun. 2018, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, D.; Gonzalez-Rosa, J.M.; Guzman-Martinez, G.; Gutierrez-Gutierrez, O.; Aguado, T.; Sanchez-Ferrer, C.; Marques, I.J.; Galardi-Castilla, M.; de Diego, I.; Gomez, M.J.; et al. Telomerase is essential for zebrafish heart regeneration. Cell Rep. 2015, 12, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Marin-Juez, R.; Moura, P.L.; Kuenne, C.; Lai, J.K.H.; Tsedeke, A.T.; Guenther, S.; Looso, M.; Stainier, D.Y. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. elife 2017, 6, e25605. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Feng, Y.; Liang, J.; Yu, H.; Wang, C.; Wang, B.; Wang, M.; Jiang, L.; Meng, W.; Cai, W.; et al. Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat. Commun. 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.I.; O’Meara, C.C.; Gemberling, M.; Zhao, L.; Bryant, D.M.; Zheng, R.; Gannon, J.B.; Cai, L.; Choi, W.Y.; Egnaczyk, G.F.; et al. Nerves regulate cardiomyocyte proliferation and heart regeneration. Dev. Cell 2015, 34, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Haubner, B.J.; Adamowicz-Brice, M.; Khadayate, S.; Tiefenthaler, V.; Metzler, B.; Aitman, T.; Penninger, J.M. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 2012, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P.; et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ. Res. 2012, 110, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Poss, K.D. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 2012, 484, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abderrahman, Y.; Chen, R.; Garcia, J.A.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015, 523, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Yang, C.C.; Chen, I.H.; Liu, Y.M.; Chang, S.J.; Chuang, Y.J. Treatment of glucocorticoids inhibited early immune responses and impaired cardiac repair in adult zebrafish. PLoS ONE 2013, 8, e66613. [Google Scholar] [CrossRef] [PubMed]
- De Preux Charles, A.S.; Bise, T.; Baier, F.; Marro, J.; Jazwinska, A. Distinct effects of inflammation on preconditioning and regeneration of the adult zebrafish heart. Open Biol. 2016, 6, 160102. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Godwin, J.W.; Pinto, A.R.; Rosenthal, N.A. Chasing the recipe for a pro-regenerative immune system. Semin. Cell Dev. Biol. 2017, 61, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Morioka, M.; Kimura, S.; Tasaki, M.; Inohaya, K.; Kudo, A. Differential reparative phenotypes between zebrafish and medaka after cardiac injury. Dev. Dyn. 2014, 243, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Navis, A.; Cox, B.D.; Dickson, A.L.; Gemberling, M.; Karra, R.; Bagnat, M.; Poss, K.D. Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 2016, 143, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Wagner, D.E.; McKenna, A.; Pandey, S.; Klein, A.M.; Shendure, J.; Gagnon, J.A.; Schier, A.F. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 2018, 36, 442. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Findlay, G.M.; Gagnon, J.A.; Horwitz, M.S.; Schier, A.F.; Shendure, J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 2016, 353, aaf7907. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Heart Malformation | Identified Disease Genes | Reference |
|---|---|---|
| 362 parent-offspring trios with severe CHD (excluded isolated ventricular septal defects, atrial septal defects, patent ductus arteriosus and pulmonic stenosis) and 264 control trios | Genes involved in production, removal and reading of methylation of H3K4 (H3K4me): MML2, KDM6A, CHD7, WDR5, RNF20, UBE2B, SMAD2 | [44] |
| 2871 CHD cases including parent-offspring trios and 1789 controls | GDF1, MYH6, FLT4 CHD7, KMT2D, PTPN11, FLT4, NOTCH1, RBFOX2, SMAD6, GATA6, ELN, CCDC154, SLOCO1B3, GPBAR1, PTEN, RPL5, NSD1, SAMD11, C210RF2, NODAL, SMAD2, H1F00, FRYL, KDM5B, POGZ, SOS1, TBX18 | [41] |
| 715 CHD parent-offspring trios and 416 healthy individuals | Mosaic KMT2D mutations | [46] |
| TOF, combined with Cleft Lip and Palate | G586A and G196S variant of MTHFR | [47] |
| 8 TOF families | Novel causative mutations of PEX5, NACA, ATXN2, CELA1, PCDHB4, CTBP1 | [48] |
| 4 families with ASD, 2 families with PDA, 2 families with TOF and 1 family with dysplastic pulmonary valve | GATA4 G115W (ASD), TLL1 I263V (ASD kindred), MYH11 (ductus arteriosus). | [49] |
| 4-generation ASD family | Novel GATA4 A899C, and K300T variants | [50] |
| 79 subjects with BAV | GATA4/5, APC, JAG1, NOTCH1/2/3, PAX8, SOX9, TBX5, WNT4 | [37] |
| Left-sided lesions | 17 genes not previously associated with human cardiovascular malformation including ACVR1, JARID2, NR2F2, PLRG1, SMURF1. Gene established syndromic association with CHD such as KMT2D, NF1, TBX20, ZEB2. | [51] |
| 181 individuals from 41 families with LVOTO | NOTCH1, ARHGAP31, MAML1, SMARCA4, JARID2, JAG1. | [40] |
| 32 BAV patients | NOTCH1 G4297A and putative pathogenic ADMTS5 C935A. | [38] |
| Twins with heterotaxy | MMP21 | [52] |
| 1,365 trios with CHD, 68 probands from 32 multisibling families, and 458 singleton probands, 12,031 controls | CHD4, CDK13, PRKD1 | [53] |
| 59 CHD trios and 59 control trios, 100 affected singletons, 533 unaffected singletons | Novel NR1D2 R175W mutation associated with AVSD, previously associated with CDH: ZFPM2, NSD1, NOTCH1, VCAN, and MYH6. | [54] |
| 3 family members with LVNC | Novel genetic variant of MYH7 gene | [55] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlak, M.; Niescierowicz, K.; Winata, C.L. Decoding the Heart through Next Generation Sequencing Approaches. Genes 2018, 9, 289. https://doi.org/10.3390/genes9060289
Pawlak M, Niescierowicz K, Winata CL. Decoding the Heart through Next Generation Sequencing Approaches. Genes. 2018; 9(6):289. https://doi.org/10.3390/genes9060289
Chicago/Turabian StylePawlak, Michal, Katarzyna Niescierowicz, and Cecilia Lanny Winata. 2018. "Decoding the Heart through Next Generation Sequencing Approaches" Genes 9, no. 6: 289. https://doi.org/10.3390/genes9060289
APA StylePawlak, M., Niescierowicz, K., & Winata, C. L. (2018). Decoding the Heart through Next Generation Sequencing Approaches. Genes, 9(6), 289. https://doi.org/10.3390/genes9060289