Development of Microplatforms to Mimic the In Vivo Architecture of CNS and PNS Physiology and Their Diseases

 , ,
, ,
Abstract
1. Introduction
1.1. Nervous System Cells: Their Roles and Microenvironment
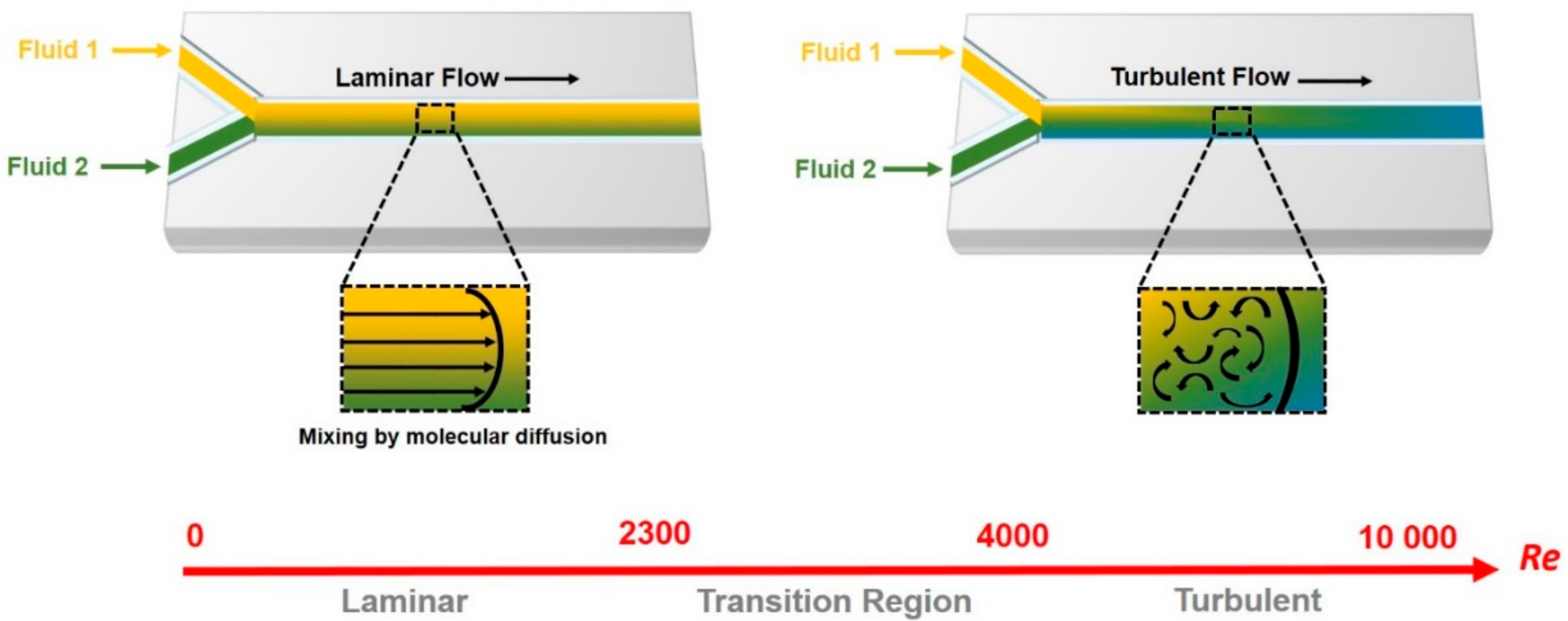
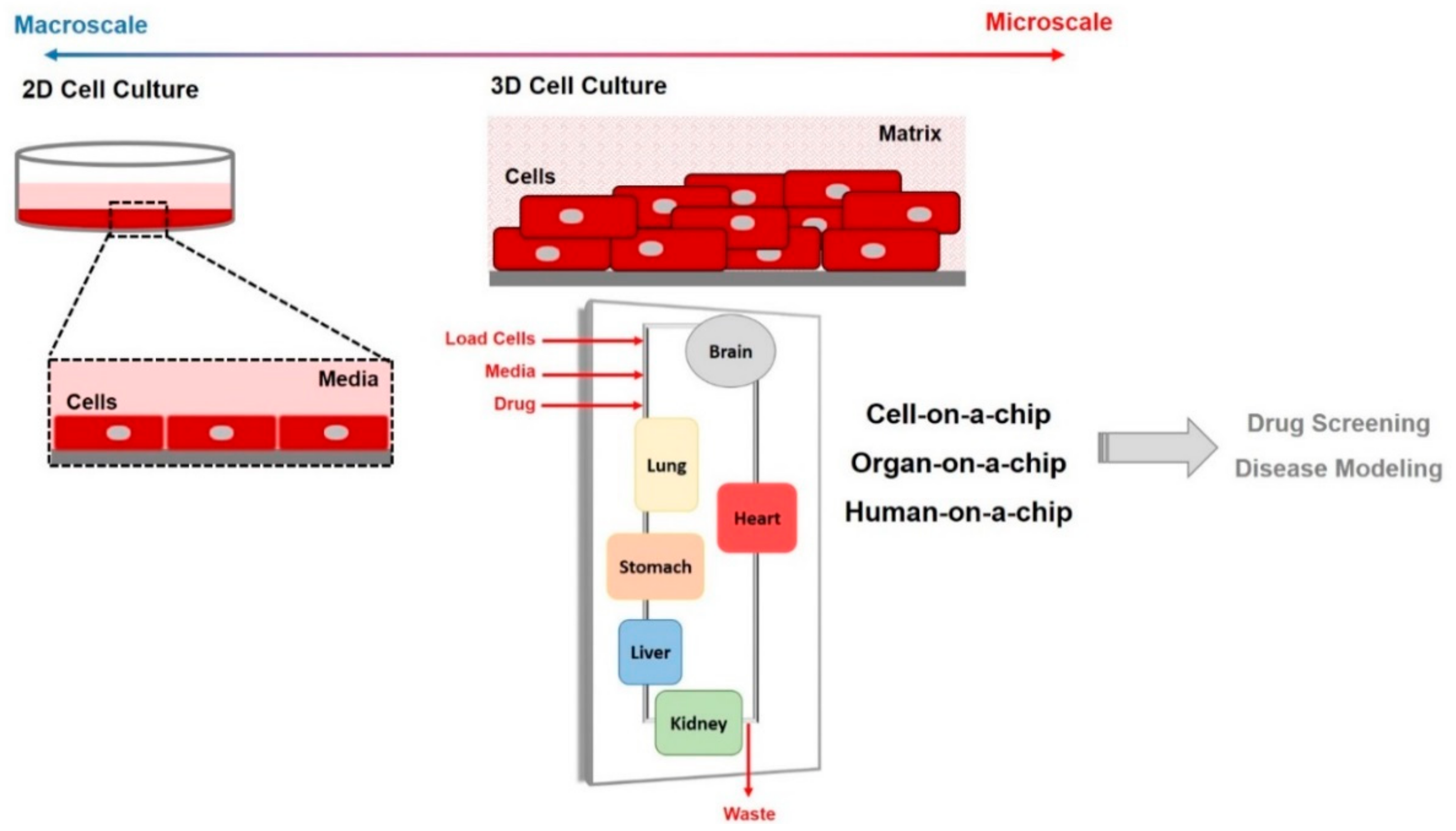
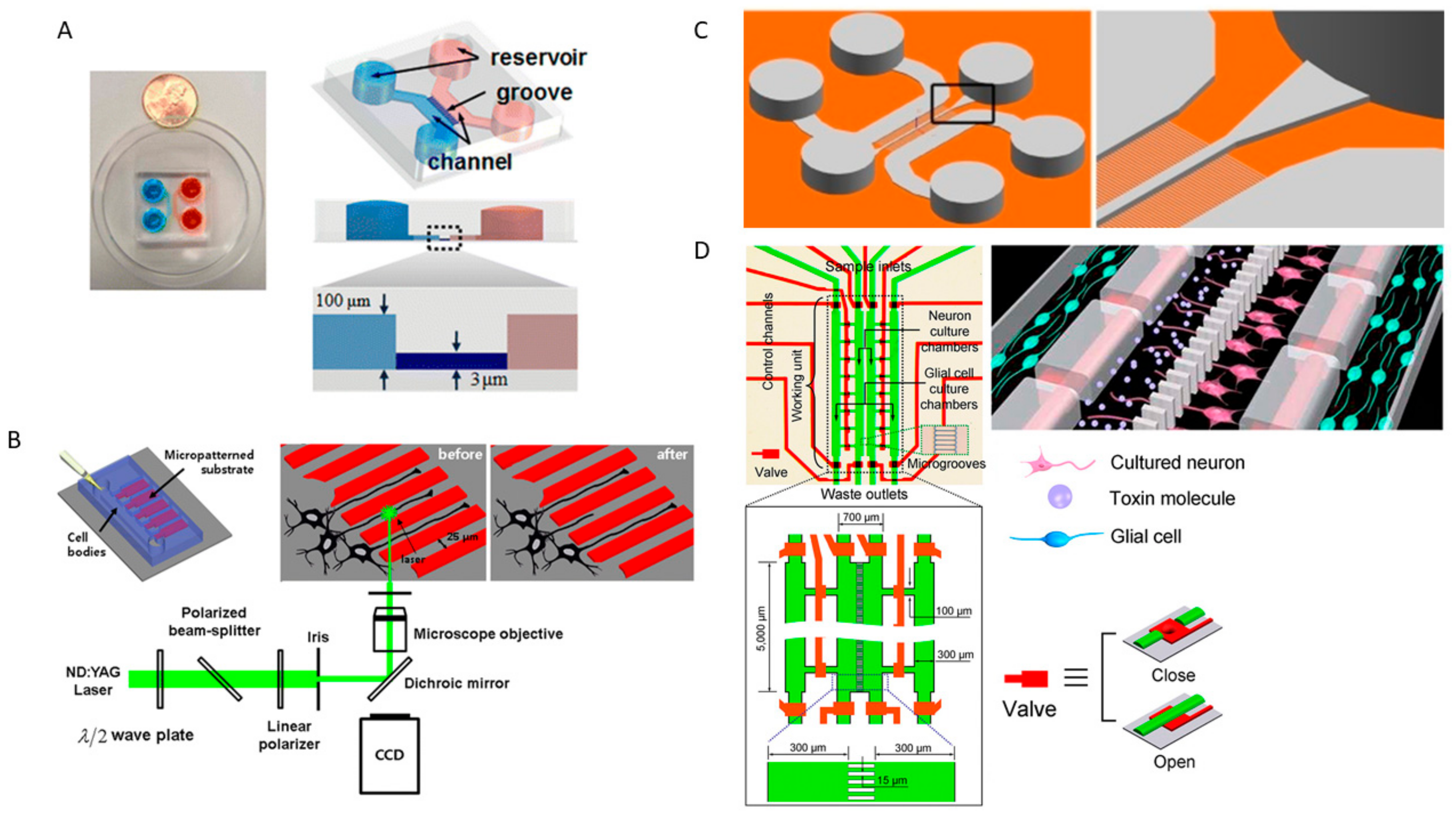
1.2. Microfluidic and Organ-on-Chip Technology
2. Nervous System-on-a Chip Models That Mimic the CNS and PNS Microenvironment and Its Physiology
2.1. Neurons and Skeletal Muscle Cells Co-Culture and Neuromuscular Junctions
2.2. Neuron Cultures, Neurogenesis and Synaptic Formation, and Neural Networks
2.3. Neuron and Glial Co-Culture and Neuron-Glial Interactions
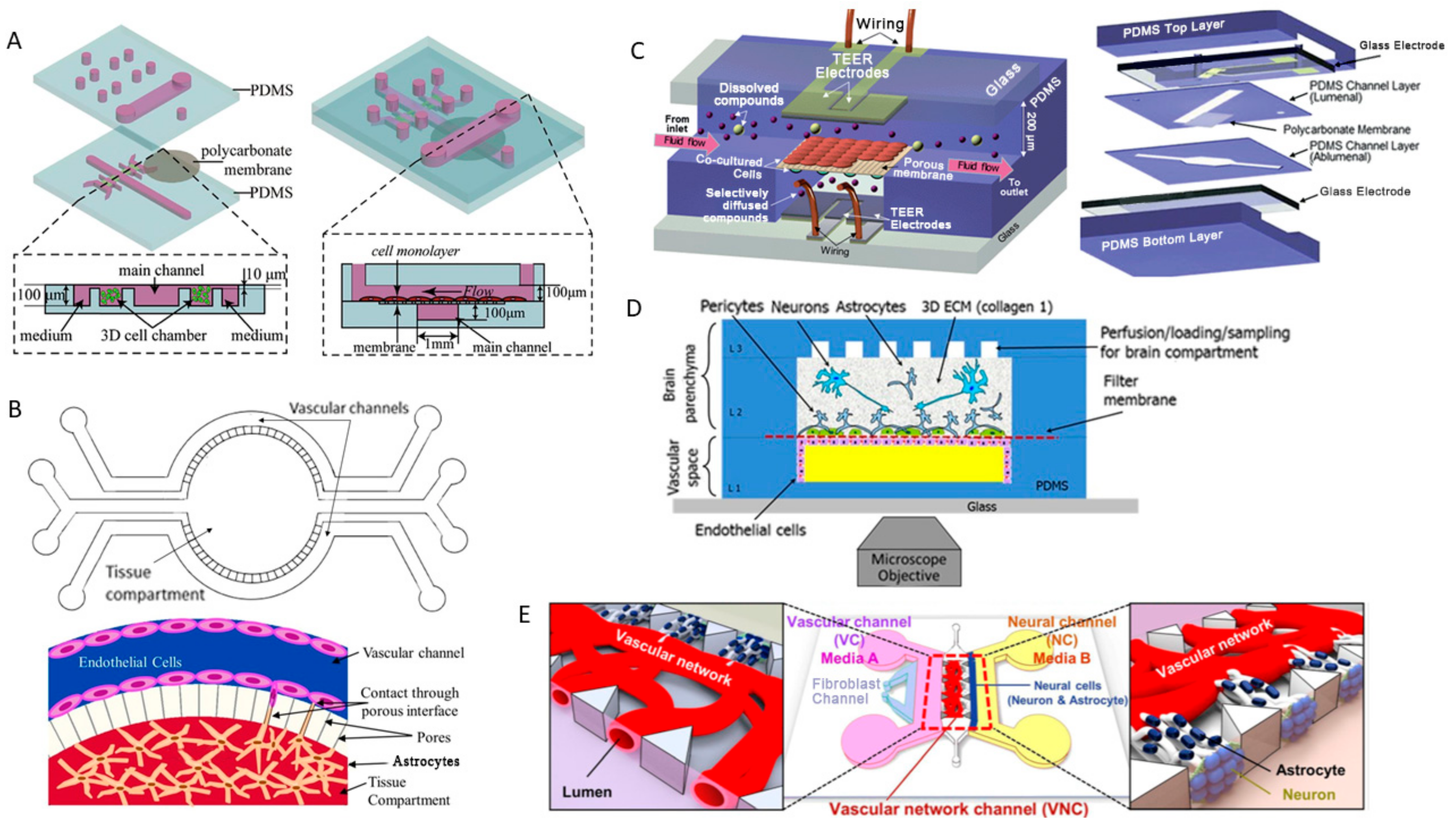
2.4. Blood-Brain Barrier-on-a-Chip and Drug Delivery
3. Nervous System Disease Models on Microplatforms to Replicate Cancer and Neurodegenerative Diseases
3.1. BBB Disruption and CNS Diseases
3.2. Brain Cancer and Metastasis
3.3. Alzheimer’s and Parkinson’s Diseases
3.4. Axon Regeneration and Neural Cell Biology
4. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Bear, M.F.; Connors, B.W.; Paradiso, M.A. Neuroscience: Exploring the Brain, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001. [Google Scholar]
- Auld, D.S.; Robitaille, R. Glial cells and neurotransmission: An inclusive view of synaptic function. Neuron 2003, 40, 389–400. [Google Scholar] [CrossRef]
- Araque, A.; Navarrete, M. Glial cells in neuronal network function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Jakel, S.; Dimou, L. Glial cells and their function in the adult brain: A journey through the history of their ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Bean, B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, G.W.; Gogos, J.A. Synaptic plasticity, neural circuits, and the emerging role of altered short-term information processing in schizophrenia. Front. Synaptic Neurosci. 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Ocker, G.K.; Litwin-Kumar, A.; Doiron, B. Self-organization of microcircuits in networks of spiking neurons with plastic synapses. PLoS Comput. Biol. 2015, 11, e1004458. [Google Scholar] [CrossRef] [PubMed]
- Grillner, S. The motor infrastructure: From ion channels to neuronal networks. Nat. Rev. Neurosci. 2003, 4, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Kiehn, O.; Kullander, K. Central pattern generators deciphered by molecular genetics. Neuron 2004, 41, 317–321. [Google Scholar] [CrossRef]
- Sidiropoulou, K.; Pissadaki, E.K.; Poirazi, P. Inside the brain of a neuron. EMBO Rep. 2006, 7, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Damiati, S.; Mhanna, R.; Kodzius, R.; Ehmoser, E.-K. Cell-free approaches in synthetic biology utilizing microfluidics. Genes 2018, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Damiati, S.; Kompella, U.B.; Damiati, S.A.; Kodzius, R. Microfluidic devices for drug delivery systems and drug screening. Genes 2018, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Valadez, A.V.; Zuo, P.; Nie, Z. Microfluidic 3D cell culture: Potential application for tissue-based bioassays. Bioanalysis 2012, 4, 1509–1525. [Google Scholar] [CrossRef] [PubMed]
- Vinuselvi, P.; Park, S.; Kim, M.; Park, J.M.; Kim, T.; Lee, S.K. Microfluidic technologies for synthetic biology. Int. J. Mol. Sci. 2011, 12, 3576–3593. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Schneider, P.; Schneider, G. Accessing new chemical entities through microfluidic systems. Angew. Chem. Int. Ed. 2014, 53, 5750–5758. [Google Scholar] [CrossRef] [PubMed]
- Wei, H. Microfluidic device with integrated porous membrane for cell sorting and separation. In Studying Cell Metabolism and Cell Interactions Using Microfluidic Devices Coupled with Mass Spectrometry; Springer: Berlin/Heidelberg, Germany, 2013; pp. 61–82. [Google Scholar]
- Van der Meer, A.D.; van den Berg, A. Organs-on-chips: Breaking the in vitro impasse. Integr. Biol. 2012, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Rouilly, V.; Niu, X.; Chappell, J.; Kitney, R.I.; Edel, J.B.; Freemont, P.S. Opportunities for microfluidic technologies in synthetic biology. J. R. Soc. Interface 2009. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.; Christodoulides, P.; Florides, G.; Kalli, K. Microfluidic flows and heat transfer and their influence on optical modes in microstructure fibers. Materials 2014, 7, 7566–7582. [Google Scholar] [CrossRef] [PubMed]
- Zhigaltsev, I.V.; Belliveau, N.; Hafez, I.; Leung, A.K.; Huft, J.; Hansen, C.; Cullis, P.R. Bottom-up design and synthesis of limit size lipid nanoparticle systems with aqueous and triglyceride cores using millisecond microfluidic mixing. Langmuir 2012, 28, 3633–3640. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, P.C. Microfluidic DNA microarray analysis: A review. Anal. Chim. Acta 2011, 687, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Yildiz-Ozturk, E.; Yesil-Celiktas, O. Diffusion phenomena of cells and biomolecules in microfluidic devices. Biomicrofluidics 2015, 9, 052606. [Google Scholar] [CrossRef] [PubMed]
- Elani, Y. Construction of membrane-bound artificial cells using microfluidics: A new frontier in bottom-up synthetic biology. Biochem. Soc. Trans. 2016, 44, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Wlodkowic, D.; Darzynkiewicz, Z. Microfluidics: Emerging prospects for anti-cancer drug screening. World J. Clin. Oncol. 2010, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Squires, T.M.; Quake, S.R. Microfluidics: Fluid physics at the nanoliter scale. Rev. Mod. Phys. 2005, 77, 977. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760. [Google Scholar] [CrossRef] [PubMed]
- Paoli, R.; Samitier, J. Mimicking the kidney: A key role in organ-on-chip development. Micromachines 2016, 7, 126. [Google Scholar] [CrossRef]
- Tehranirokh, M.; Kouzani, A.Z.; Francis, P.S.; Kanwar, J.R. Microfluidic devices for cell cultivation and proliferation. Biomicrofluidics 2013, 7, 051502. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.N. Cells and organs on chip—A revolutionary platform for biomedicine. In Lab-on-a-Chip Fabrication and Application; InTech: London, UK, 2016. [Google Scholar]
- Park, J.; Lee, B.K.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.-H. Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer’s disease. Lab Chip 2015, 15, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Goss, J.A.; Cho, A.; McCain, M.L.; Parker, K.K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip 2013, 13, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Ju, J.-W.; Kim, S.M.; Shin, Y.; Chung, S.; Pak, J.H. Clonorchis sinensis infestation promotes three-dimensional aggregation and invasion of cholangiocarcinoma cells. PLoS ONE 2014, 9, e110705. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ingber, D.E. Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. 2013, 5, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Torisawa, Y.-S.; Hamilton, G.A.; Kim, H.J.; Ingber, D.E. Microengineered physiological biomimicry: Organs-on-chips. Lab Chip 2012, 12, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, Q.; Gijs, M.A. In vitro micro-physiological models for translational immunology. Lab Chip 2015, 15, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control. Release 2012, 164, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Gurski, L.A.; Petrelli, N.J.; Jia, X.; Farach-Carson, M.C. 3D matrices for anti-cancer drug testing and development. Oncol. Issues 2010, 25, 20–25. [Google Scholar] [CrossRef]
- Kim, J.B. Three-dimensional tissue culture models in cancer biology. Semin. Cancer Biol. 2005, 15, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xiong, W.C.; Mei, L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 2010, 137, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Das, M.; Rumsey, J.; Gonzalez, M.; Stancescu, M.; Hickman, J. Neuromuscular junction formation between human stem-cell-derived motoneurons and rat skeletal muscle in a defined system. Tissue Eng. Part C Methods 2010, 16, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Gonzalez, M.; Stancescu, M.; Vandenburgh, H.H.; Hickman, J.J. Neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle in a defined system. Biomaterials 2011, 32, 9602–9611. [Google Scholar] [CrossRef] [PubMed]
- Mars, T.; Yu, K.J.; Tang, X.M.; Miranda, A.F.; Grubic, Z.; Cambi, F.; King, M.P. Differentiation of glial cells and motor neurons during the formation of neuromuscular junctions in cocultures of rat spinal cord explant and human muscle. J. Comp. Neurol. 2001, 438, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Kato-Negishi, M.; Onoe, H.; Takeuchi, S. Three-dimensional neuron–muscle constructs with neuromuscular junctions. Biomaterials 2013, 34, 9413–9419. [Google Scholar] [CrossRef] [PubMed]
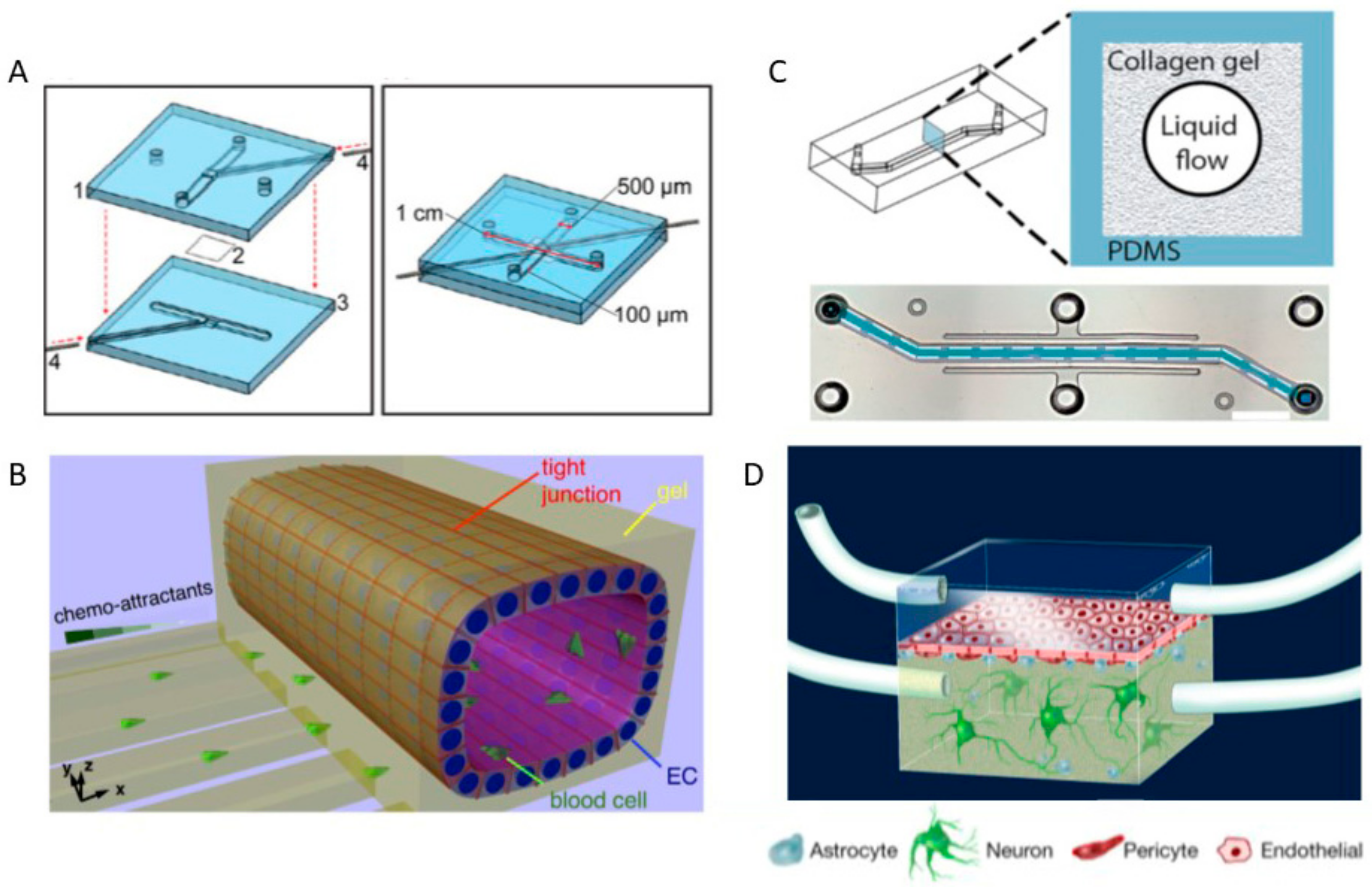
- Taylor, A.M.; Rhee, S.W.; Tu, C.H.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. Microfluidic multicompartment device for neuroscience research. Langmuir 2003, 19, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Liu, S.; McDonald, J.; Thakor, N.; Yang, I.H. Neuromuscular junction in a microfluidic device. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2013, 2013, 2833–2835. [Google Scholar] [PubMed]
- Zahavi, E.E.; Ionescu, A.; Gluska, S.; Gradus, T.; Ben-Yaakov, K.; Perlson, E. A compartmentalized microfluidic neuromuscular co-culture system reveals spatial aspects of GDNF functions. J. Cell Sci. 2015, 128, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Southam, K.A.; King, A.E.; Blizzard, C.A.; McCormack, G.H.; Dickson, T.C. Microfluidic primary culture model of the lower motor neuron–neuromuscular junction circuit. J. Neurosci. Methods 2013, 218, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Malin, S.A.; Davis, B.M.; Molliver, D.C. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat. Protoc. 2007, 2, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.V.; Bronner-Fraser, M. Vertebrate cranial placodes I. Embryonic induction. Dev. Biol. 2001, 232, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Covell, D.A., Jr.; Noden, D.M. Embryonic development of the chick primary trigeminal sensory-motor complex. J. Comp. Neurol. 1989, 286, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, G. Evolutionary origins of vertebrate placodes: Insights from developmental studies and from comparisons with other deuterostomes. J. Exp. Zool. B Mol. Dev. Evol. 2005, 304, 347–399. [Google Scholar] [CrossRef] [PubMed]
- Farinas, I.; Cano-Jaimez, M.; Bellmunt, E.; Soriano, M. Regulation of neurogenesis by neurotrophins in developing spinal sensory ganglia. Brain Res. Bull. 2002, 57, 809–816. [Google Scholar] [CrossRef]
- Lawson, S.N.; Biscoe, T.J. Development of mouse dorsal root ganglia: An autoradiographic and quantitative study. J. Neurocytol. 1979, 8, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.; Bayer, S.A. Development of the cranial nerve ganglia and related nuclei in the rat. Adv. Anat. Embryol. Cell Biol. 1982, 74, 1–90. [Google Scholar] [PubMed]
- Forbes, D.J.; Welt, C. Neurogenesis in the trigeminal ganglion of the albino rat: A quantitative autoradiographic study. J. Comp. Neurol. 1981, 199, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, R.W.; Enfiejian, H.L.; Chiaia, N.L.; Macdonald, G.J.; Miller, M.W.; McCann, P.; Goddard, C.M. Birthdates of trigeminal ganglion cells contributing axons to the infraorbital nerve and specific vibrissal follicles in the rat. J. Comp. Neurol. 1991, 307, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Buchman, V.L.; Davies, A.M. Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development 1993, 118, 989–1001. [Google Scholar] [PubMed]
- Davies, A.M.; Minichiello, L.; Klein, R. Developmental changes in NT3 signalling via TrkA and TrkB in embryonic neurons. EMBO J. 1995, 14, 4482–4489. [Google Scholar] [PubMed]
- Lindwall, C.; Kanje, M. The Janus role of c-Jun: Cell death versus survival and regeneration of neonatal sympathetic and sensory neurons. Exp. Neurol. 2005, 196, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Lumsden, A. Relation of target encounter and neuronal death to nerve growth factor responsiveness in the developing mouse trigeminal ganglion. J. Comp. Neurol. 1984, 223, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H.; Tansey, M.G.; Lampe, P.A.; Fahrner, T.J.; Enomoto, H.; Simburger, K.S.; Leitner, M.L.; Araki, T.; Johnson, E.M., Jr.; Milbrandt, J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3-RET receptor complex. Neuron 1998, 21, 1291–1302. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Yagil, Z.; Hagalili, Y.; Klein, H.; Lerman, O.; Behar, O. Semaphorin 3A and neurotrophins: A balance between apoptosis and survival signaling in embryonic DRG neurons. J. Neurochem. 2006, 96, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Maina, F.; Hilton, M.C.; Ponzetto, C.; Davies, A.M.; Klein, R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997, 11, 3341–3350. [Google Scholar] [CrossRef] [PubMed]
- Ryden, M.; Hempstead, B.; Ibanez, C.F. Differential modulation of neuron survival during development by nerve growth factor binding to the p75 neurotrophin receptor. J. Biol. Chem. 1997, 272, 16322–16328. [Google Scholar] [CrossRef] [PubMed]
- Campenot, R.B. NGF and the local control of nerve terminal growth. J. Neurobiol. 1994, 25, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Pierchala, B.A.; Ciarallo, C.L.; Ginty, D.D. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 1997, 277, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Liu, B.P.; Park, J.H.; Strittmatter, S.M. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 2004, 44, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.M. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci. 1988, 8, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Chierzi, S.; Codd, A.M.; Campbell, D.S.; Meyer, R.L.; Holt, C.E.; Fawcett, J.W. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J. Neurosci. 2005, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Q.; Kelly, T.K.; Chang, B.; Ryazantsev, S.; Rajasekaran, A.K.; Martin, K.C.; Twiss, J.L. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J. Neurosci. 2001, 21, 9291–9303. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.Y.; Hengst, U.; Cox, L.J.; Macosko, E.Z.; Jeromin, A.; Urquhart, E.R.; Jaffrey, S.R. Local translation of RhoA regulates growth cone collapse. Nature 2005, 436, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.N.; Winter, J.; James, I.F.; Rang, H.P.; Yeats, J.; Bevan, S. Capsaicin-induced ion fluxes in dorsal root ganglion cells in culture. J. Neurosci. 1988, 8, 3208–3220. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.E.; Bautista, D.M.; Chuang, H.H.; McKemy, D.D.; Zygmunt, P.M.; Hogestatt, E.D.; Meng, I.D.; Julius, D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 2004, 427, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Peier, A.M.; Reeve, A.J.; Andersson, D.A.; Moqrich, A.; Earley, T.J.; Hergarden, A.C.; Story, G.M.; Colley, S.; Hogenesch, J.B.; McIntyre, P.; et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 2002, 296, 2046–2049. [Google Scholar] [CrossRef] [PubMed]
- Cesare, P.; McNaughton, P. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc. Natl. Acad. Sci. USA 1996, 93, 15435–15439. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Flonta, M.L. Physiology. Cold current in thermoreceptive neurons. Nature 2001, 413, 480. [Google Scholar] [CrossRef] [PubMed]
- McCarter, G.C.; Reichling, D.B.; Levine, J.D. Mechanical transduction by rat dorsal root ganglion neurons in vitro. Neurosci. Lett. 1999, 273, 179–182. [Google Scholar] [CrossRef]
- Chen, M.; Li, Y.; Yang, M.; Chen, X.; Chen, Y.; Yang, F.; Lu, S.; Yao, S.; Zhou, T.; Liu, J.; et al. A new method for quantifying mitochondrial axonal transport. Protein Cell 2016, 7, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Dinh, N.D.; Chiang, Y.Y.; Hardelauf, H.; Baumann, J.; Jackson, E.; Waide, S.; Sisnaiske, J.; Frimat, J.P.; van Thriel, C.; Janasek, D.; et al. Microfluidic construction of minimalistic neuronal co-cultures. Lab Chip 2013, 13, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.S.; Kim, H.J.; Sim, S.J.; Jeon, N.L. Shear stress effect on transfection of neurons cultured in microfluidic devices. J. Nanosci. Nanotechnol. 2009, 9, 7330–7335. [Google Scholar] [CrossRef] [PubMed]
- Park, T.H.; Shuler, M.L. Integration of cell culture and microfabrication technology. Biotechnol. Prog. 2003, 19, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Degenaar, P.; Pioufle, B.L.; Griscom, L.; Tixier, A.; Akagi, Y.; Morita, Y.; Murakami, Y.; Yokoyama, K.; Fujita, H.; Tamiya, E. A method for micrometer resolution patterning of primary culture neurons for SPM analysis. J. Biochem. 2001, 130, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bomba-Warczak, E.; Vevea, J.D.; Brittain, J.M.; Figueroa-Bernier, A.; Tepp, W.H.; Johnson, E.A.; Yeh, F.L.; Chapman, E.R. Interneuronal transfer and distal action of tetanus toxin and botulinum neurotoxins a and d in central neurons. Cell Rep. 2016, 16, 1974–1987. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, X.; Lv, X.; Geng, L.; Li, Y.; Qin, K.; Deng, Y. Microvalve controlled multi-functional microfluidic chip for divisional cell co-culture. Anal. Biochem. 2017, 539, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Fehlauer, H.; Nekimken, A.L.; Kim, A.A.; Pruitt, B.L.; Goodman, M.B.; Krieg, M. Using a microfluidics device for mechanical stimulation and high resolution imaging of C. elegans. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Gross, C.G. Neurogenesis in adult mammals: Some progress and problems. J. Neurosci. 2002, 22, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Aimone, J.B.; Li, Y.; Lee, S.W.; Clemenson, G.D.; Deng, W.; Gage, F.H. Regulation and function of adult neurogenesis: From genes to cognition. Physiol. Rev. 2014, 94, 991–1026. [Google Scholar] [CrossRef] [PubMed]
- Morshead, C.M.; Reynolds, B.A.; Craig, C.G.; McBurney, M.W.; Staines, W.A.; Morassutti, D.; Weiss, S.; van der Kooy, D. Neural stem cells in the adult mammalian forebrain: A relatively quiescent subpopulation of subependymal cells. Neuron 1994, 13, 1071–1082. [Google Scholar] [CrossRef]
- Doetsch, F.; Caille, I.; Lim, D.A.; García-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef]
- Johansson, C.B.; Momma, S.; Clarke, D.L.; Risling, M.; Lendahl, U.; Frisén, J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell 1999, 96, 25–34. [Google Scholar] [CrossRef]
- Nottebohm, F. Why are some neurons replaced in adult brain? J. Neurosci. 2002, 22, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Zupanc, G.K. A comparative approach towards the understanding of adult neurogenesis. Brain Behav. Evol. 2001, 58, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Cameron, H.A.; Dayer, A.G. New interneurons in the adult neocortex: Small, sparse, but significant? Biol. Psychiatry 2008, 63, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Gould, E. How widespread is adult neurogenesis in mammals? Nat. Rev. Neurosci. 2007, 8, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Reeves, A.J.; Graziano, M.S.; Gross, C.G. Neurogenesis in the neocortex of adult primates. Science 1999, 286, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Kokoeva, M.V.; Yin, H.; Flier, J.S. Neurogenesis in the hypothalamus of adult mice: Potential role in energy balance. Science 2005, 310, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Battista, D.; Ferrari, C.C.; Gage, F.H.; Pitossi, F.J. Neurogenic niche modulation by activated microglia: Transforming growth factor β increases neurogenesis in the adult dentate gyrus. Eur. J. Neurosci. 2006, 23, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Ziv, Y.; Schwartz, A.; Landa, G.; Talpalar, A.E.; Pluchino, S.; Martino, G.; Schwartz, M. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 2006, 31, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, P.A.; Palmer, T.D. Immune influence on adult neural stem cell regulation and function. Neuron 2009, 64, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Barkho, B.Z.; Song, H.; Aimone, J.B.; Smrt, R.D.; Kuwabara, T.; Nakashima, K.; Gage, F.H.; Zhao, X. Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev. 2006, 15, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; McCloskey, M.A.; Blong, C.C.; Bendickson, L.; Nilsen-Hamilton, M.; Sakaguchi, D.S. Astrocyte-derived interleukin-6 promotes specific neuronal differentiation of neural progenitor cells from adult hippocampus. J. Neurosci. Res. 2010, 88, 2798–2809. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Stevens, C.F.; Gage, F.H. Astroglia induce neurogenesis from adult neural stem cells. Nature 2002, 417, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Rudge, J.S.; Pasnikowski, E.M.; Holst, P.; Lindsay, R.M. Changes in neurotrophic factor expression and receptor activation following exposure of hippocampal neuron/astrocyte cocultures to kainic acid. J. Neurosci. 1995, 15, 6856–6867. [Google Scholar] [CrossRef] [PubMed]
- Rudge, J.S.; Alderson, R.F.; Pasnikowski, E.; McClain, J.; Ip, N.Y.; Lindsay, R.M. Expression of ciliary neurotrophic factor and the neurotrophins-nerve growth factor, brain-derived neurotrophic factor and neurotrophin 3-in cultured rat hippocampal astrocytes. Eur. J. Neurosci. 1992, 4, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.D.; Takahashi, J.; Gage, F.H. The adult rat hippocampus contains primordial neural stem cells. Mol. Cell. Neurosci. 1997, 8, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Dranovsky, A.; Picchini, A.M.; Moadel, T.; Sisti, A.C.; Yamada, A.; Kimura, S.; Leonardo, E.D.; Hen, R. Experience dictates stem cell fate in the adult hippocampus. Neuron 2011, 70, 908–923. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhong, C.; Bonaguidi, M.A.; Sun, G.J.; Hsu, D.; Gu, Y.; Meletis, K.; Huang, Z.J.; Ge, S.; Enikolopov, G.; et al. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 2012, 489, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Veena, J.; Rao, B.S.; Srikumar, B.N. Regulation of adult neurogenesis in the hippocampus by stress, acetylcholine and dopamine. J. Nat. Sci. Biol. Med. 2011, 2, 26–37. [Google Scholar] [PubMed]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Hollenberg, S.M.; Snider, L.; Turner, D.L.; Lipnick, N.; Weintraub, H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science 1995, 268, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Gould, E. Serotonin and hippocampal neurogenesis. Neuropsychopharmacology 1999, 21, 46s–51s. [Google Scholar] [CrossRef]
- Olah, M.; Ping, G.; De Haas, A.H.; Brouwer, N.; Meerlo, P.; Van Der Zee, E.A.; Biber, K.; Boddeke, H.W. Enhanced hippocampal neurogenesis in the absence of microglia T cell interaction and microglia activation in the murine running wheel model. Glia 2009, 57, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.M.; Churchwell, J.C.; Kesner, R.P.; Gilbert, P.E. Selective lesions of the dentate gyrus produce disruptions in place learning for adjacent spatial locations. Neurobiol. Learn. Mem. 2012, 97, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, I.H.; Maxwell, F.; Glode, L.M. Regulated expression of a diphtheria toxin A-chain gene transfected into human cells: Possible strategy for inducing cancer cell suicide. Cancer Res. 1986, 46, 4660–4664. [Google Scholar] [PubMed]
- Goebbels, S.; Bode, U.; Pieper, A.; Funfschilling, U.; Schwab, M.H.; Nave, K.A. Cre/loxP-mediated inactivation of the bHLH transcription factor gene NeuroD/BETA2. Genesis 2005, 42, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Uwamori, H.; Higuchi, T.; Arai, K.; Sudo, R. Integration of neurogenesis and angiogenesis models for constructing a neurovascular tissue. Sci. Rep. 2017, 7, 17349. [Google Scholar] [CrossRef] [PubMed]
- Adriani, G.; Ma, D.; Pavesi, A.; Kamm, R.D.; Goh, E.L. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood-brain barrier. Lab Chip 2017, 17, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Liu, Y.; Qi, H.; Ma, Q.; Xu, L.; Chen, W.; Chen, G.; Xu, X. A novel brain neurovascular unit model with neurons, astrocytes and microvascular endothelial cells of rat. Int. J. Biol. Sci. 2013, 9, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, A.; Tanaka, K.; Niwa, M. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Bicker, J.; Alves, G.; Fortuna, A.; Falcao, A. Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: A review. Eur. J. Pharm. Biopharm. 2014, 87, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Seo, J.H.; Wong, K.H.; Terasaki, Y.; Park, J.; Bong, K.; Arai, K.; Lo, E.H.; Irimia, D. Three-dimensional blood-brain barrier model for in vitro studies of neurovascular pathology. Sci. Rep. 2015, 5, 15222. [Google Scholar] [CrossRef] [PubMed]
- Juttner, R.; Rathjen, F.G. Molecular analysis of axonal target specificity and synapse formation. Cell. Mol. Life Sci. 2005, 62, 2811–2827. [Google Scholar] [CrossRef] [PubMed]
- Salie, R.; Niederkofler, V.; Arber, S. Patterning molecules; multitasking in the nervous system. Neuron 2005, 45, 189–192. [Google Scholar] [PubMed]
- Waites, C.L.; Craig, A.M.; Garner, C.C. Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci. 2005, 28, 251–274. [Google Scholar] [CrossRef] [PubMed]
- Colon-Ramos, D.A. Synapse formation in developing neural circuits. Curr. Top. Dev. Biol. 2009, 87, 53–79. [Google Scholar] [PubMed]
- O’Leary, D.D.; Chou, S.J.; Sahara, S. Area patterning of the mammalian cortex. Neuron 2007, 56, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Polleux, F.; Ince-Dunn, G.; Ghosh, A. Transcriptional regulation of vertebrate axon guidance and synapse formation. Nat. Rev. Neurosci. 2007, 8, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Plachez, C.; Richards, L.J. Mechanisms of axon guidance in the developing nervous system. Curr. Top. Dev. Biol. 2005, 69, 267–346. [Google Scholar] [PubMed]
- Tessier-Lavigne, M.; Goodman, C.S. The molecular biology of axon guidance. Science 1996, 274, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J.; Doyle, J.L.; Serafini, T.; Kennedy, T.E.; Tessier-Lavigne, M.; Goodman, C.S.; Dickson, B.J. Genetic analysis of Netrin genes in Drosophila: Netrins guide CNS commissural axons and peripheral motor axons. Neuron 1996, 17, 203–215. [Google Scholar] [CrossRef]
- Adler, C.E.; Fetter, R.D.; Bargmann, C.I. UNC-6/Netrin induces neuronal asymmetry and defines the site of axon formation. Nat. Neurosci. 2006, 9, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.E.; Tessier-Lavigne, M. Guidance and induction of branch formation in developing axons by target-derived diffusible factors. Curr. Opin. Neurobiol. 1995, 5, 83–90. [Google Scholar] [CrossRef]
- Akins, M.R.; Biederer, T. Cell-cell interactions in synaptogenesis. Curr. Opin. Neurobiol. 2006, 16, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.L.; Colman, D.R.; Huntley, G.W. Molecules, maps and synapse specificity. Nat. Rev. Neurosci. 2001, 2, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Dalva, M.B.; Takasu, M.A.; Lin, M.Z.; Shamah, S.M.; Hu, L.; Gale, N.W.; Greenberg, M.E. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 2000, 103, 945–956. [Google Scholar] [CrossRef]
- Scheiffele, P. Cell-cell signaling during synapse formation in the CNS. Annu. Rev. Neurosci. 2003, 26, 485–508. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Sanes, J.R.; Weiner, J.A. Synaptic adhesion molecules. Curr. Opin. Cell Biol. 2003, 15, 621–632. [Google Scholar] [CrossRef]
- Fields, R.D.; Stevens-Graham, B. New insights into neuron-glia communication. Science 2002, 298, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Koito, H.; Li, J.; Han, A. A multi-compartment CNS neuron-glia Co-culture microfluidic platform. J. Vis. Exp. 2009. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Koito, H.; Li, J.; Han, A. Microfluidic compartmentalized co-culture platform for CNS axon myelination research. Biomed. Microdevices 2009, 11, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Koito, H.; Li, J.; Han, A. Multi-compartment neuron-glia co-culture platform for localized CNS axon-glia interaction study. Lab Chip 2012, 12, 3296–3304. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, H.; Yang, Y. Imaging analysis of neuron to glia interaction in microfluidic culture platform (MCP)-based neuronal axon and glia co-culture system. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Majumdar, D.; Jovanovic, B.; Shaifer, C.; Lin, P.C.; Zijlstra, A.; Webb, D.J.; Li, D. A versatile valve-enabled microfluidic cell co-culture platform and demonstration of its applications to neurobiology and cancer biology. Biomed. Microdevices 2011, 13, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Tonna, N.; Lovchik, R.D.; Mastrangelo, R.; Morini, R.; Ruiz, A.; Delamarche, E.; Matteoli, M. Overflow microfluidic networks: Application to the biochemical analysis of brain cell interactions in complex neuroinflammatory scenarios. Anal. Chem. 2012, 84, 9833–9840. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, L.M.; Sakiyama-Elbert, S.E. GDNF preconditioning can overcome Schwann cell phenotypic memory. Exp. Neurol. 2015, 265, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Simon, M.J.; Cancel, L.M.; Shi, Z.-D.; Ji, X.; Tarbell, J.M.; Morrison, B.; Fu, B.M. Permeability of Endothelial and Astrocyte Cocultures: In Vitro Blood–Brain Barrier Models for Drug Delivery Studies. Ann. Biomed. Eng. 2010, 38, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood–brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.; Saha, S. Nanotechnology for the detection and therapy of stroke. Adv. Healthc. Mater. 2014, 3, 1703–1720. [Google Scholar] [CrossRef] [PubMed]
- Bonakdar, M.; Graybill, P.; Davalos, R. A microfluidic model of the blood–brain barrier to study permeabilization by pulsed electric fields. RSC Adv. 2017, 7, 42811–42818. [Google Scholar] [CrossRef] [PubMed]
- Siddharthan, V.; Kim, Y.V.; Liu, S.; Kim, K.S. Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. 2007, 1147, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Gao, D.; Chen, Y.; Jin, F.; Hu, G.; Jiang, Y.; Liu, H. Development of a blood-brain barrier model in a membrane-based microchip for characterization of drug permeability and cytotoxicity for drug screening. Anal. Chim. Acta 2016, 934, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Yeon, J.H.; Na, D.; Choi, K.; Ryu, S.-W.; Choi, C.; Park, J.-K. Reliable permeability assay system in a microfluidic device mimicking cerebral vasculatures. Biomed. Microdevices 2012, 14, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Deosarkar, S.P.; Prabhakarpandian, B.; Wang, B.; Sheffield, J.B.; Krynska, B.; Kiani, M.F. A novel dynamic neonatal blood-brain barrier on a chip. PLoS ONE 2015, 10, e0142725. [Google Scholar] [CrossRef] [PubMed]
- Booth, R.; Kim, H. Characterization of a microfluidic in vitro model of the blood-brain barrier (μBBB). Lab Chip 2012, 12, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Pensabene, V.; Markov, D.A.; Allwardt, V.; Neely, M.D.; Shi, M.; Britt, C.M.; Hoilett, O.S.; Yang, Q.; Brewer, B.M. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015, 9, 054124. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Lee, S.-R.; Ko, J.; Son, K.; Tahk, D.; Ahn, J.; Im, C.; Jeon, N.L. A low permeability microfluidic blood-brain barrier platform with direct contact between perfusable vascular network and astrocytes. Sci. Rep. 2017, 7, 8083. [Google Scholar] [CrossRef] [PubMed]
- Koo, Y.; Hawkins, B.T.; Yun, Y. Three-dimensional (3D) tetra-culture brain on chip platform for organophosphate toxicity screening. Sci. Rep. 2018, 8, 2841. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Griep, L.M.; Wolbers, F.; de Wagenaar, B.; ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; van der Meer, A.D.; van den Berg, A. BBB ON CHIP: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Herland, A.; van der Meer, A.D.; FitzGerald, E.A.; Park, T.E.; Sleeboom, J.J.; Ingber, D.E. Distinct contributions of astrocytes and pericytes to neuroinflammation identified in a 3D human blood-brain barrier on a chip. PLoS ONE 2016, 11, e0150360. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Codreanu, S.G.; Shi, M.; Sherrod, S.D.; Markov, D.A.; Neely, M.D.; Britt, C.M.; Hoilett, O.S.; Reiserer, R.S.; Samson, P.C. Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J. Neuroinflamm. 2016, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kievit, F.M.; Florczyk, S.J.; Leung, M.C.; Veiseh, O.; Park, J.O.; Disis, M.L.; Zhang, M. Chitosan–alginate 3D scaffolds as a mimic of the glioma tumor microenvironment. Biomaterials 2010, 31, 5903–5910. [Google Scholar] [CrossRef] [PubMed]
- Cucullo, L.; Hossain, M.; Puvenna, V.; Marchi, N.; Janigro, D. The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neurosci. 2011, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Czupalla, C.J.; Liebner, S.; Devraj, K. In vitro models of the blood-brain barrier. Methods Mol. Biol. 2014, 1135, 415–437. [Google Scholar] [PubMed]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Forster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Prabhakarpandian, B.; Shen, M.C.; Nichols, J.B.; Mills, I.R.; Sidoryk-Wegrzynowicz, M.; Aschner, M.; Pant, K. SyM-BBB: A microfluidic Blood Brain Barrier model. Lab Chip 2013, 13, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Janigro, D.; Hossain, M.; Oby, E.; Rapp, E.; Cucullo, L. Side by side comparison between dynamic versus static models of blood-brain barrier in vitro: A permeability study. Brain Res. 2006, 1109, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Korjamo, T.; Heikkinen, A.T.; Monkkonen, J. Analysis of unstirred water layer in in vitro permeability experiments. J. Pharm. Sci. 2009, 98, 4469–4479. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T. Drug permeation through biomembranes: Cyclodextrins and the unstirred water layer. Pharmazie 2012, 67, 363–370. [Google Scholar] [PubMed]
- Denkins, Y.; Reiland, J.; Roy, M.; Sinnappah-Kang, N.D.; Galjour, J.; Murry, B.P.; Blust, J.; Aucoin, R.; Marchetti, D. Brain metastases in melanoma: Roles of neurotrophins. Neuro Oncol. 2004, 6, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Schmid, B.C.; Rezniczek, G.A.; Fabjani, G.; Yoneda, T.; Leodolter, S.; Zeillinger, R. The neuronal guidance cue Slit2 induces targeted migration and may play a role in brain metastasis of breast cancer cells. Breast Cancer Res. Treat. 2007, 106, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Barker, J.; Zhou, C.; Li, W.; Zhang, J.; Lin, B.; Foltz, G.; Kublbeck, J.; Honkakoski, P. Towards personalized medicine with a three-dimensional micro-scale perfusion-based two-chamber tissue model system. Biomaterials 2012, 33, 4353–4361. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Lee, K.H.; Lee, J.; Choi, H.; Lee, D.; Park, Y.; Lee, S.H. Integration of microfluidic chip with biomimetic hydrogel for 3D controlling and monitoring of cell alignment and migration. J. Biomed. Mater. Res. A 2014, 102, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Tourovskaia, A.; Fauver, M.; Kramer, G.; Simonson, S.; Neumann, T. Tissue-engineered microenvironment systems for modeling human vasculature. Exp. Biol. Med. 2014, 239, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Blasig, I.E.; Giese, H.; Schroeter, M.L.; Sporbert, A.; Utepbergenov, D.I.; Buchwalow, I.B.; Neubert, K.; Schonfelder, G.; Freyer, D.; Schimke, I.; et al. *NO and oxyradical metabolism in new cell lines of rat brain capillary endothelial cells forming the blood-brain barrier. Microvasc. Res. 2001, 62, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Lv, Y.; Zeng, M.; Fu, B.M. Non-invasive measurement of solute permeability in cerebral microvessels of the rat. Microvasc. Res. 2009, 77, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Logan, A. CNS growth factors. Br. J. Hosp. Med. 1990, 43, 428–437. [Google Scholar] [PubMed]
- Lei, Y.; Li, J.; Wang, N.; Yang, X.; Hamada, Y.; Li, Q.; Zheng, W.; Jiang, X. An on-chip model for investigating the interaction between neurons and cancer cells. Integr. Biol. 2016, 8, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, Z.; Yu, Y.; Sizdahkhani, S.; Ho, W.S.; Yin, F.; Wang, L.; Zhu, G.; Zhang, M.; Jiang, L.; et al. A dynamic in vivo-like organotypic blood-brain barrier model to probe metastatic brain tumors. Sci. Rep. 2016, 6, 36670. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A.; Skinner, R.A.; Abbott, N.J. Establishment of a simplified in vitro porcine blood-brain barrier model with high transendothelial electrical resistance. Brain Res. 2013, 1521, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, E.; Guo, Z.; Yu, R.; Hao, H.; Xu, Y.; Sun, Z.; Li, X.; Lyu, J.; Wang, Q. Design and construction of a multi-organ microfluidic chip mimicking the in vivo microenvironment of lung cancer metastasis. ACS Appl. Mater. Interfaces 2016, 8, 25840–25847. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, Z.; Guo, Y.; Peng, X.; Qin, J. Probing the response of lung tumor cells to inflammatory microvascular endothelial cells on fluidic microdevice. Electrophoresis 2017, 38, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Terrell-Hall, T.B.; Ammer, A.G.; Griffith, J.I.; Lockman, P.R. Permeability across a novel microfluidic blood-tumor barrier model. Fluids Barriers CNS 2017, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Terrell-Hall, T.B.; Nounou, M.I.; El-Amrawy, F.; Griffith, J.I.G.; Lockman, P.R. Trastuzumab distribution in an in-vivo and in-vitro model of brain metastases of breast cancer. Oncotarget 2017, 8, 83734–83744. [Google Scholar] [CrossRef] [PubMed]
- Shumakovich, M.A.; Mencio, C.P.; Siglin, J.S.; Moriarty, R.A.; Geller, H.M.; Stroka, K.M. Astrocytes from the brain microenvironment alter migration and morphology of metastatic breast cancer cells. FASEB J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Potier, M.C.; Delatour, B. Alzheimer disease models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 5–38. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O. Altered neurogenesis in mouse models of Alzheimer disease. Neurogenesis 2017, 4, e1327002. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 703. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.M. Parkinson’s disease: A review. Front. Biosci. 2014, 6, 65–74. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Gazewood, J.D.; Richards, D.R.; Clebak, K. Parkinson disease: An update. Am. Fam. Physician 2013, 87, 267–273. [Google Scholar] [PubMed]
- Stewart, D.; Morgan, E.; Burn, D.; Grosset, D.; Chaudhuri, K.R.; MacMahon, D.; Needleman, F.; Macphee, G.; Heywood, P. Dopamine agonist switching in Parkinson’s disease. Hosp. Med. 2004, 65, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Osaki, T.; Shin, Y.; Sivathanu, V.; Campisi, M.; Kamm, R.D. In vitro microfluidic models for neurodegenerative disorders. Adv. Healthc. Mater. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Joshi, P.; Mastrangelo, R.; Francolini, M.; Verderio, C.; Matteoli, M. Testing Aβ toxicity on primary CNS cultures using drug-screening microfluidic chips. Lab Chip 2014, 14, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Lovchik, R.D.; Bianco, F.; Tonna, N.; Ruiz, A.; Matteoli, M.; Delamarche, E. Overflow microfluidic networks for open and closed cell cultures on chip. Anal. Chem. 2010, 82, 3936–3942. [Google Scholar] [CrossRef] [PubMed]
- Kunze, A.; Meissner, R.; Brando, S.; Renaud, P. Co-pathological connected primary neurons in a microfluidic device for Alzheimer studies. Biotechnol. Bioeng. 2011, 108, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Shimohama, S.; Tamura, Y.; Kawamura, T.; Akaike, A.; Kimura, J. Methylphenylpyridium ion (MPP+) enhances glutamate-induced cytotoxicity against dopaminergic neurons in cultured rat mesencephalon. J. Neurosci. Res. 1996, 43, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Kosaka, T.; Kakimura, J.I.; Matsuoka, Y.; Kohno, Y.; Nomura, Y.; Taniguchi, T. Protective effects of the antiparkinsonian drugs talipexole and pramipexole against 1-methyl-4-phenylpyridinium-induced apoptotic death in human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1998, 54, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Auerbach, J.M.; Rodriguez-Gomez, J.A.; Velasco, I.; Gavin, D.; Lumelsky, N.; Lee, S.H.; Nguyen, J.; Sanchez-Pernaute, R.; Bankiewicz, K.; et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature 2002, 418, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.G.; Flanagan, L.A.; Rhee, S.W.; Schwartz, P.H.; Lee, A.P.; Monuki, E.S.; Jeon, N.L. Human neural stem cell growth and differentiation in a gradient-generating microfluidic device. Lab Chip 2005, 5, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Millet, L.J.; Stewart, M.E.; Nuzzo, R.G.; Gillette, M.U. Guiding neuron development with planar surface gradients of substrate cues deposited using microfluidic devices. Lab Chip 2010, 10, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Blurton-Jones, M.; Rhee, S.W.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.L.; Ambravaneswaran, V.; Agrawal, N.; Bilodeau, M.; Toner, M.; Tompkins, R.G.; Fagan, S.; Irimia, D. Burn injury reduces neutrophil directional migration speed in microfluidic devices. PLoS ONE 2010, 5, e11921. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, S.K.; Woo, D.H.; Lee, E.J.; Kim, J.H.; Lee, S.H. Differentiation of neural progenitor cells in a microfluidic chip-generated cytokine gradient. Stem Cells 2009, 27, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Chae, S.; Kim, J.H.; Barald, K.F.; Park, J.Y.; Lee, S.-H. Neurotoxic amyloid beta oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow. Sci. Rep. 2013, 3, 1921. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Choi, H.; Jung, E.S.; Lee, W.; Oh, S.; Jeon, N.L.; Mook-Jung, I. HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PLoS ONE 2012, 7, e42983. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Brugg, B.; Peyrin, J.-M.; Bartha, R.; Beauchet, O. Combination of memantine and vitamin D prevents axon degeneration induced by amyloid-beta and glutamate. Neurobiol. Aging 2014, 35, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, Y.C.; Meng, J.; Zhu, H.; Zhang, X. A microfluidics-based mobility shift assay to identify new inhibitors of β-secretase for Alzheimer’s disease. Anal. Bioanal. Chem. 2017, 409, 6635–6642. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.Y. Exogenous α-synuclein fibrils induce lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.Y.; Gordon, T. The cellular and molecular basis of peripheral nerve regeneration. Mol. Neurobiol. 1997, 14, 67–116. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.-M.; Yang, I.H.; Kim, D.-H.; Byun, J.; Xu, W.-L.; Nicovich, P.R.; Cheong, R.; Levchenko, A.; Thakor, N.; Zhou, F.-Q. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc. Natl. Acad. Sci. USA 2011, 108, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Ide, C. Peripheral nerve regeneration. Neurosci. Res. 1996, 25, 101–121. [Google Scholar] [CrossRef]
- Terenghi, G. Peripheral nerve regeneration and neurotrophic factors. J. Anat. 1999, 194, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, J.W.; Park, J.W.; Byun, J.H.; Vahidi, B.; Rhee, S.W.; Jeon, N.L. Integrated microfluidics platforms for investigating injury and regeneration of CNS axons. Ann. Biomed. Eng. 2012, 40, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.N.; Vahidi, B.; Kim, H.J.; Mismar, W.; Steward, O.; Jeon, N.L.; Venugopalan, V. Examination of axonal injury and regeneration in micropatterned neuronal culture using pulsed laser microbeam dissection. Lab Chip 2010, 10, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, D.; Peyrin, J.-M.; Soubeyre, V.; Magnifico, S.; Saias, L.; Viovy, J.-L.; Brugg, B. Wallerian-like degeneration of central neurons after synchronized and geometrically registered mass axotomy in a three-compartmental microfluidic chip. Neurotox. Res. 2011, 19, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ren, L.; Liu, W.; Wang, J.-C.; Wang, Y.; Tu, Q.; Xu, J.; Liu, R.; Zhang, Y.; Yuan, M.-S. Spatiotemporally controlled and multifactor involved assay of neuronal compartment regeneration after chemical injury in an integrated microfluidics. Anal. Chem. 2012, 84, 6444–6453. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D Cell Culture | Cellular Characteristics | 3D Cell Culture |
|---|---|---|
| Flat and stretched cells on monolayer | Morphology | Form natural shape in aggregate or spheroid structures |
| Faster rate than in vivo | Proliferation | Depends on the cell type and 3D model system |
| Exhibits differential gene/protein expression levels | Gene/Protein Expression | Similar to in vivo tissue models |
| Only on edges | Cell-to-Cell contact | Dominant |
| Most cells are at the same stage (usually proliferating stage) | Stage of Cell Cycle | Different stages: proliferating, hypoxia, and necrotic cells |
| Grow and adhere on a flat substrate | Growth Conditions | Grow on matrix or in suspension media |
| No | Diffusion gradient of O2, nutrients, drugs, waste | Yes |
| No | Show resistivity to anticancer drugs | Yes |
| No | Mimicking in vivo environment | Yes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saliba, J.; Daou, A.; Damiati, S.; Saliba, J.; El-Sabban, M.; Mhanna, R. Development of Microplatforms to Mimic the In Vivo Architecture of CNS and PNS Physiology and Their Diseases. Genes 2018, 9, 285. https://doi.org/10.3390/genes9060285
Saliba J, Daou A, Damiati S, Saliba J, El-Sabban M, Mhanna R. Development of Microplatforms to Mimic the In Vivo Architecture of CNS and PNS Physiology and Their Diseases. Genes. 2018; 9(6):285. https://doi.org/10.3390/genes9060285
Chicago/Turabian StyleSaliba, John, Arij Daou, Samar Damiati, Jessica Saliba, Marwan El-Sabban, and Rami Mhanna. 2018. "Development of Microplatforms to Mimic the In Vivo Architecture of CNS and PNS Physiology and Their Diseases" Genes 9, no. 6: 285. https://doi.org/10.3390/genes9060285
APA StyleSaliba, J., Daou, A., Damiati, S., Saliba, J., El-Sabban, M., & Mhanna, R. (2018). Development of Microplatforms to Mimic the In Vivo Architecture of CNS and PNS Physiology and Their Diseases. Genes, 9(6), 285. https://doi.org/10.3390/genes9060285