Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA and RNA Isolation
2.3. Multiplex Ligand Probe Amplification Analysis
2.4. Complementary DNA and Genomic DNA Sequencing
2.5. Next Generation Sequencing
2.6. Bioinformatic Analyses
2.7. Statistical Analysis
3. Results
3.1. Patients’ Screening
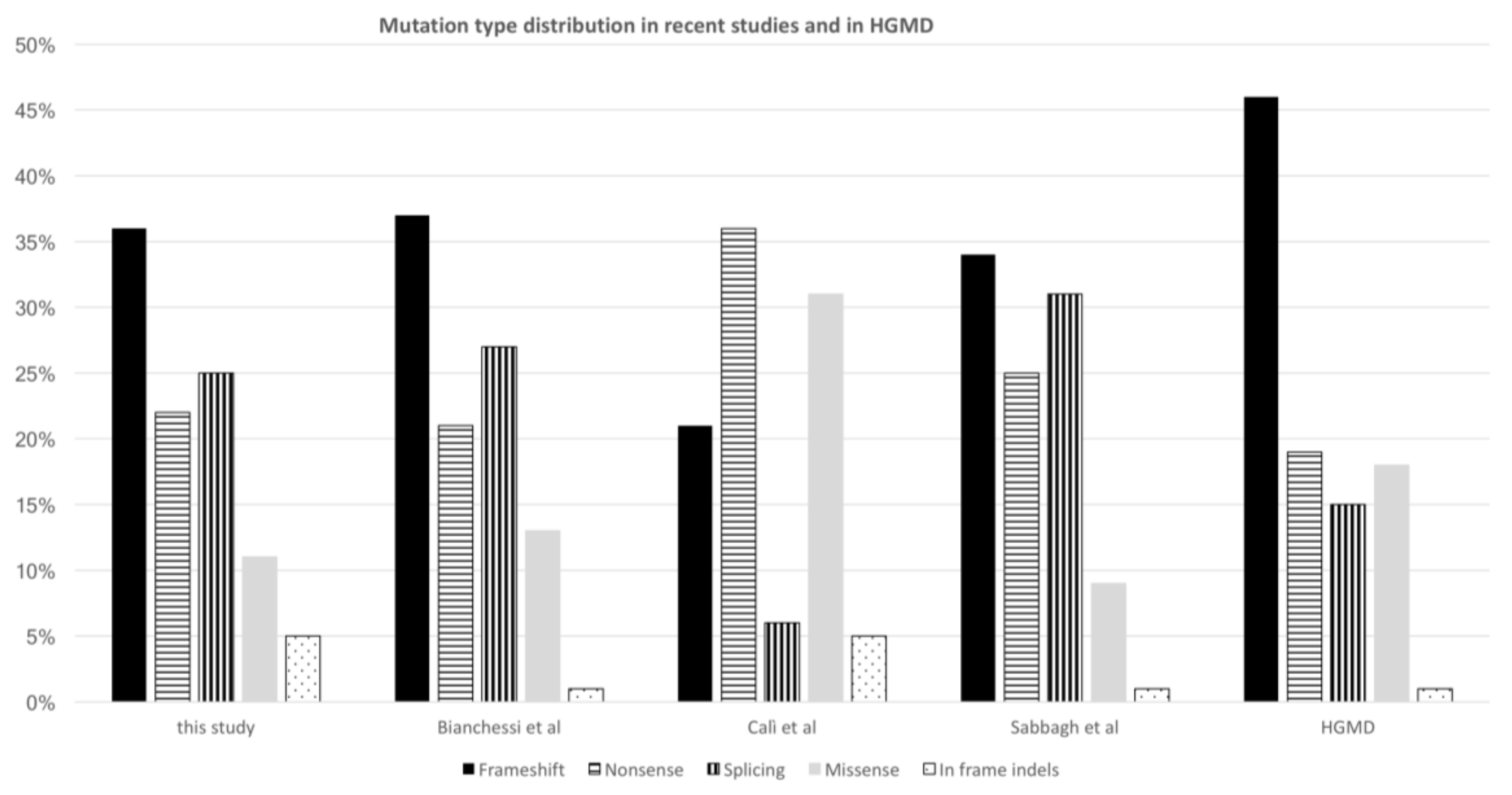
3.2. Molecular Analyses and Classification of Identified Variants
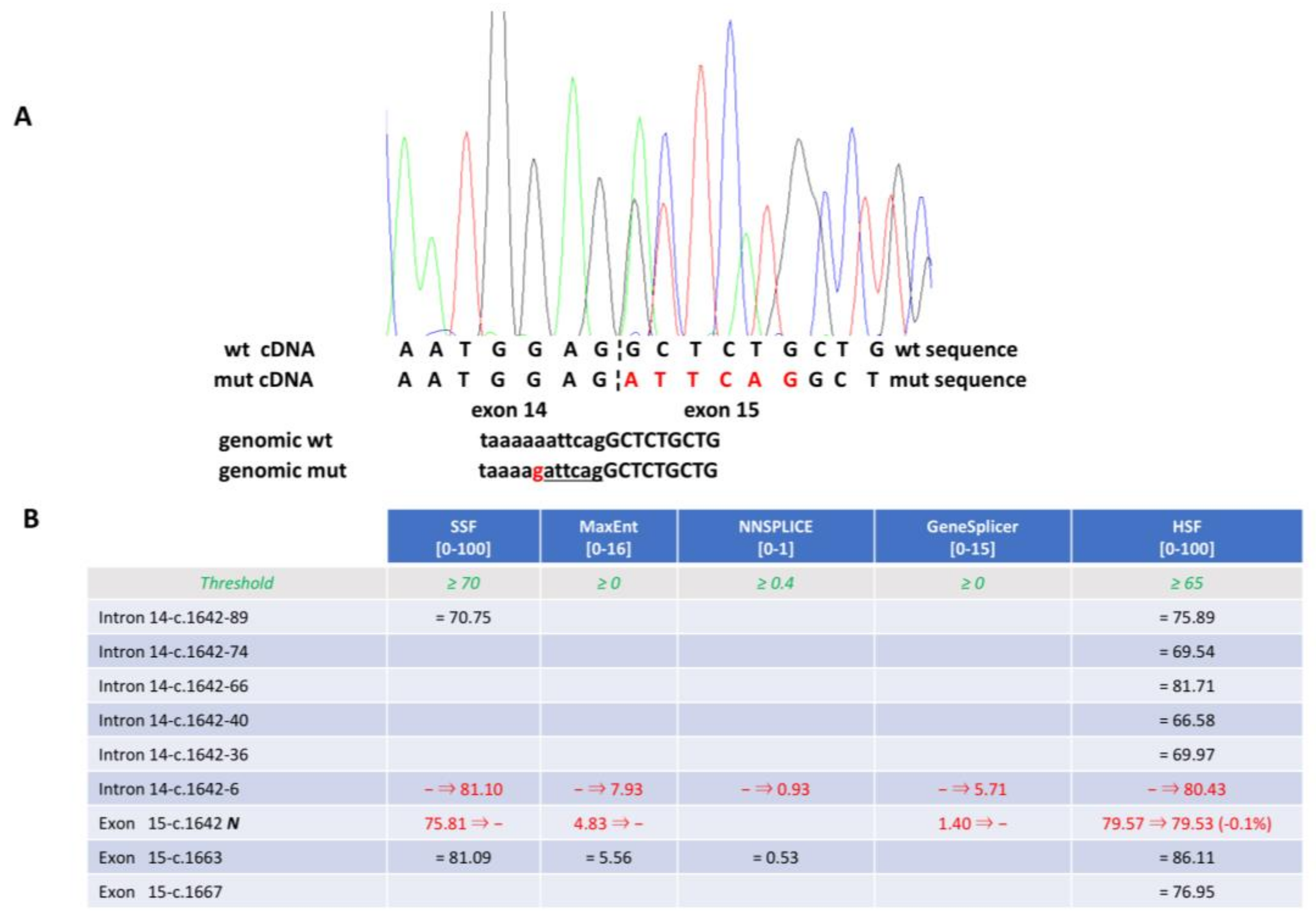
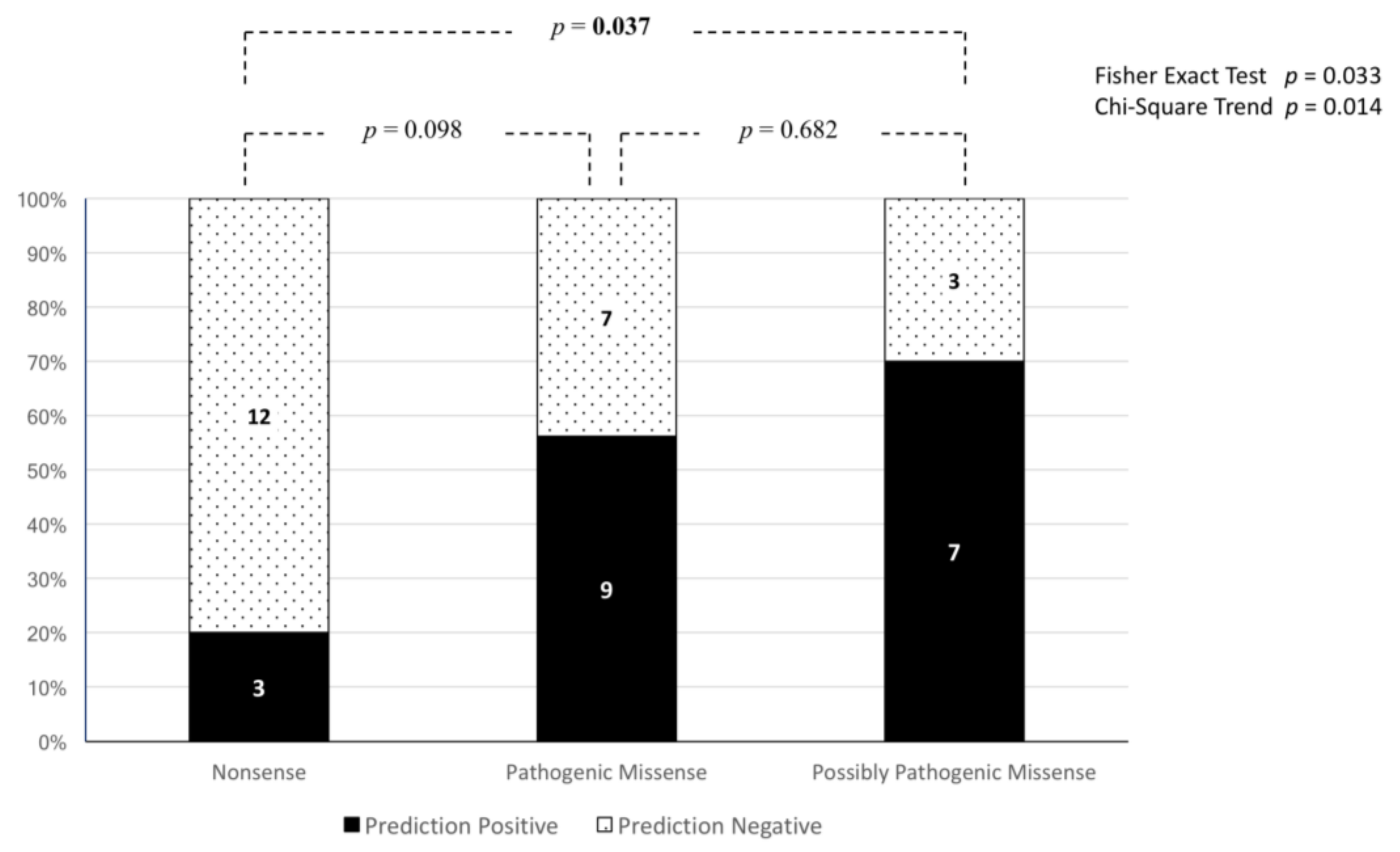
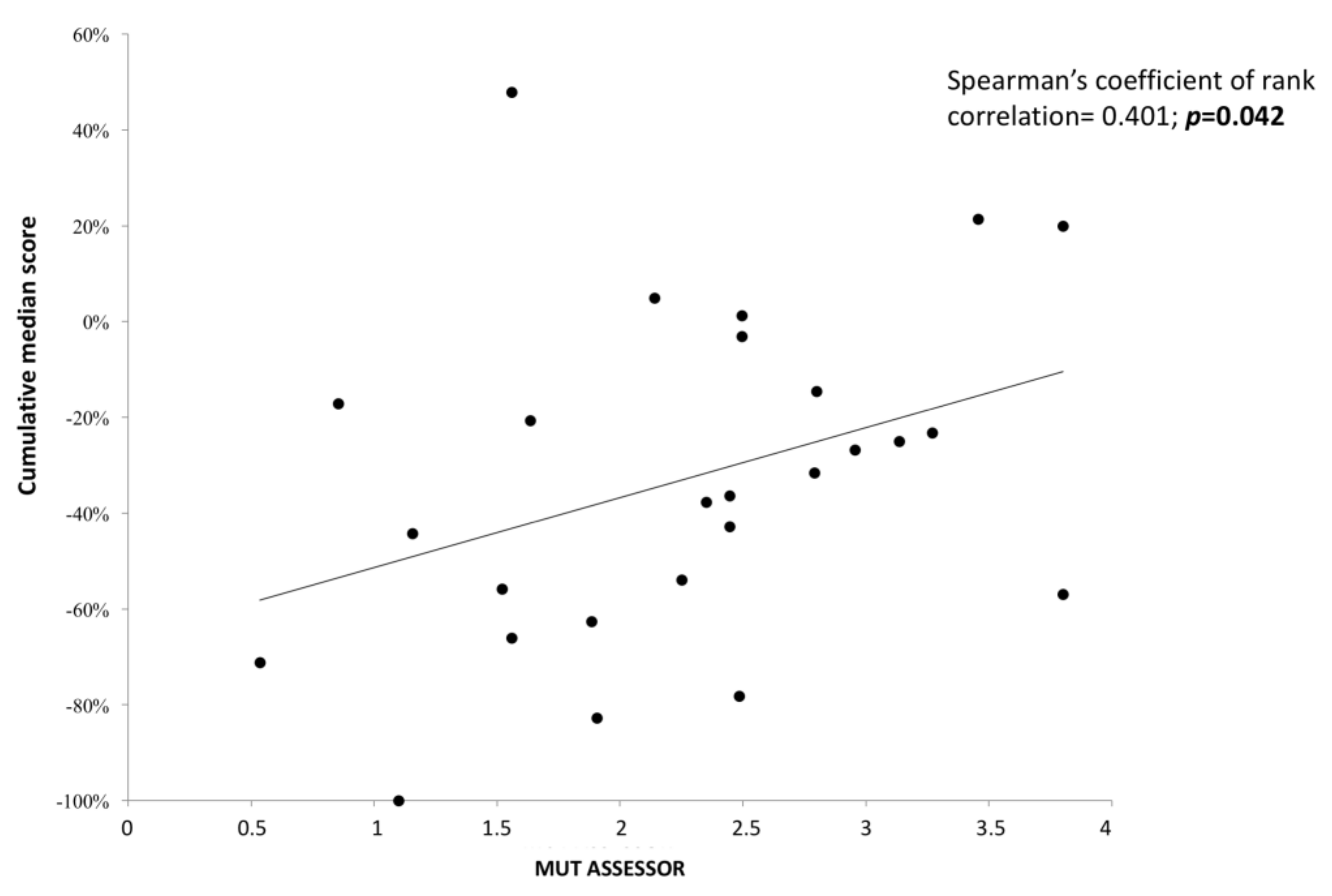
3.3. Reassessment of Predicted Consequences in Reported Variants
3.4. Genotype–Phenotype Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Evans, D.G.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, E.; Rantanen, M.; Kallionpää, R.A.; Pöyhönen, M.; Leppävirta, J.; Ylä-Outinen, H.; Riccardi, V.M.; Pukkala, E.; Pitkäniemi, J.; Peltonen, S.; et al. Distinctive cancer associations in patients with neurofibromatosis type 1. J. Clin. Oncol. 2016, 34, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health Consensus Development Conference Statement: Neurofibromatosis. Arch. Neurol. 1988, 45, 575–578.
- Okumura, A.; Ozaki, M.; Niida, Y. Development of a practical NF1 genetic testing method through the pilot analysis of five Japanese families with neurofibromatosis type 1. Brain Dev. 2015, 37, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Valero, M.C.; Martín, Y.; Hernández-Imaz, E.; Marina Hernández, A.; Meleán, G.; Valero, A.M.; Javier Rodríguez-Álvarez, F.; Tellería, D.; Hernández-Chico, C. A highly sensitive genetic protocol to detect NF1 mutations. J. Mol. Diagn. 2011, 13, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Fahsold, R.; Hoffmeyer, S.; Mischung, C.; Gille, C.; Ehlers, C.; Kücükceylan, N.; Abdel-Nour, M.; Gewies, A.; Peters, H.; Kaufmann, D.; et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am. J. Hum. Genet. 2000, 66, 790–818. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Cooper, D.N.; Upadhyaya, M.N. Evaluation of denaturing high performance liquid chromatography (DHPLC) for the mutational analysis of the neurofibromatosis type 1 (NF1) gene. Hum. Genet. 2001, 109, 487–497. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Buccino, A.; Gianni, D.; Mangino, M.; Giustini, S.; Richetta, A.; Divona, L.; Calvieri, S.; Mingarelli, R.; Dallapiccola, B. NF1 gene analysis based on DHPLC. Hum. Mutat. 2003, 21, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Van Minkelen, R.; van Bever, Y.; Kromosoeto, J.N.; Withagen-Hermans, C.J.; Nieuwlaat, A.; Halley, D.J.; van den Ouweland, A.M. A clinical and genetic overview of 18 years neurofibromatosis type 1 molecular diagnostics in the Netherlands. Clin. Genet. 2014, 85, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Pros, E.; Gómez, C.; Martín, T.; Fábregas, P.; Serra, E.; Lázaro, C. Nature and mRNA effect of 282 different NF1 point mutations: Focus on splicing alterations. Hum. Mutat. 2008, 29, E173–E193. [Google Scholar] [CrossRef] [PubMed]
- Calì, F.; Chiavetta, V.; Ruggeri, G.; Piccione, M.; Selicorni, A.; Palazzo, D.; Bonsignore, M.; Cereda, A.; Elia, M.; Failla, P.; et al. Mutation spectrum of NF1 gene in Italian patients with neurofibromatosis type 1 using Ion Torrent PGM™ platform. Eur. J. Med. Genet. 2017, 60, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Bianchessi, D.; Morosini, S.; Saletti, V.; Ibba, M.C.; Natacci, F.; Esposito, S.; Cesaretti, C.; Riva, D.; Finocchiaro, G.; Eoli, M. 126 novel mutations in Italian patients with neurofibromatosis type 1. Mol. Genet. Genomic Med. 2015, 7, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; McLellan, M.D.; Hussain, I.; Wallis, J.W.; Fulton, L.L.; Fulton, R.S.; Magrini, V.; Demeter, R.; Wylie, T.; Kandoth, C.; et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013, 23, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Bowers, N.; Burkitt-Wright, E.; Miles, E.; Garg, S.; Scott-Kitching, V.; Penman-Splitt, M.; Dobbie, A.; Howard, E.; Ealing, J.; et al. Comprehensive RNA Analysis of the NF1 Gene in Classically Affected NF1 Individuals Meeting NIH Criteria has High Sensitivity and Mutation Negative Testing is Reassuring in Isolated Cases With Pigmentary Features Only. EBioMedicine 2016, 7, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Caminsky, N.; Mucaki, E.J.; Rogan, P.K. Interpretation of mRNA splicing mutations in genetic disease: Review of the literature and guidelines for information-theoretical analysis. F1000Research 2014, 3, 282. [Google Scholar] [CrossRef] [PubMed]
- Brent, M.R.; Guigò, R. Recent advances in gene structure predictions. Curr. Opin. Struct. Biol. 2004, 3, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Altman, R.B.; Bromberg, Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics 2013, 14 (Suppl. 3). [Google Scholar] [CrossRef] [PubMed]
- Vihinen, M. Majority vote and other problems when using computational tools. Hum. Mutat. 2014, 35, 912–914. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Wimmer, K.; Roca, X.; Beiglböck, H.; Callens, T.; Etzler, J.; Rao, A.R.; Krainer, A.R.; Fonatsch, C.; Messiaen, L. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5′ splice-site disruption. Hum. Mutat. 2007, 28, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M. The Molecular Biology of Neurofibromatosis Type 1; Morgan & Claypool: Williston, VT, USA, 2014. [Google Scholar]
- Torres Nupan, M.M.; Velez Van Meerbeke, A.; López Cabra, C.A.; Herrera Gomez, P.M. Cognitive and Behavioral Disorders in Children with Neurofibromatosis Type 1. Front. Pediatr. 2017, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Penas, J.J. Learning disorders in neurofibromatosis type 1. Rev. Neurol. 2017, 64, S59–S63. [Google Scholar] [PubMed]
- Alkindy, A.; Chuzhanova, N.; Kini, U.; Cooper, D.N.; Upadhyaya, M. Genotype-phenotype associations in neurofibromatosis type 1 (NF1): An increased risk of tumor complications in patients with NF1 splice-site mutations? Hum. Genomics 2012, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Huson, S.M.; Harper, P.S.; Compston, D.A. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain 1988, 111, 1355–1381. [Google Scholar] [CrossRef] [PubMed]
- Ehara, Y.; Yamamoto, O.; Kosaki, K.; Yoshida, Y. Natural course and characteristics of cutaneous neurofibromas in neurofibromatosis 1. J. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shofty, B.; Constantini, S.; Ben-Shachar, S. Advances in Molecular Diagnosis of Neurofibromatosis Type 1. Semin. Pediatr. Neurol. 2015, 22, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef] [PubMed]
- Parisi, P.; Persechino, S.; Paolino, M.C.; Nicita, F.; Torrente, I.; Bozzao, A.; Villa, M.P. Four-year follow-up study in a NF1 boy with a focal pontine hamartoma. Ital. J. Pediatr. 2013, 39, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Cheon, C.K. Neurofibromatosis type 1: A single center’s experience in Korea. Korean J. Pediatr. 2014, 57, 410–415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Bella, K.; Poskitt, K.; Szudek, J.; Friedman, J.M. Use of “unidentified bright objects” on MRI for diagnosis of neurofibromatosis 1 in children. Neurology 2000, 54, 1646–1650. [Google Scholar] [CrossRef]
- Hervey-Jumper, S.L.; Singla, N.; Gebarski, S.S.; Robertson, P.; Maher, C.O. Diffuse pontine lesions in children with neurofibromatosis type 1: Making a case for unidentified bright objects. Pediatr. Neurosurg. 2013, 49, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Tadini, G.; Milani, D.; Menni, F.; Pezzani, L.; Sabatini, C.; Esposito, S. Is it time to change the neurofibromatosis 1 diagnostic criteria? Eur. J. Int. Med. 2014, 25, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Castle, B.; Baser, M.E.; Huson, S.M.; Cooper, D.N.; Upadhyaya, M. Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J. Med. Genet. 2003, 40, e109. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients | Total (n = 58) | <12 Years (n = 26) | >12 Years (n = 32) | p-Value |
|---|---|---|---|---|
| CALs | 53 (91) | 25 (96) | 28 (87) | NS |
| Freckling | 32 (55) | 15 (58) | 17 (52) | NS |
| Lisch nodules | 14 (24) | 5 (21) | 9 (26) | NS |
| Neurofibromas | 34 (58) | 11 (42) | 23 (71) | 0.03 |
| First degree relative | 8 (13) | 0 (0) | 8 (23) | 0.013 |
| Optic glioma | 3 (5) | 1 (4) | 2 (6) | NS |
| Bone lesions | 16 (27) | 10 (37) | 6 (19) | NS |
| Sphenoid dysplasia | 3 (5) | 1 (4) | 2 (6) | NS |
| Intellectual disability | 7(13) | 6 (21) | 2 (6) | NS |
| Amartomas | 5 (9) | 3 (12) | 2 (6) | NS |
| Deafness incl. neurinomas | 5 (9) | 0 (0) | 5 (16) | NS |
| MUTATION | DIST To NEAREST SS a | SIFT b | POLYPHEN2 c | MUTATION ASSESSOR d | REVEL e | CADD PHRED | SSF f | MES f | NNSPLICE f | GENESPLICER f | HSF f | NOVEL | CLASS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.565A>C (p.Lys189Gln) | 20 (D) | D (0.00) | PoD (0.54) | M (2.095) | 0.612 | 25 | No change | No change | No change | No change | No change | Y | MISSENSE |
| c.1642-7A>G | 7 (A) | NE | NE | NE | NE | 4.1 | SD abrogation | SD abrogation | No change | SD abrogation | No change | Y | SPLICING |
| c.1658A>C (p.His553Pro) * | 17 (A) | T (0.06) | PD (0.99) | M (2.395) | 0.778 | 27.4 | No change | No change | NA | −14% | No change | CM076335 | MISSENSE |
| c.2990G>A (p.Arg997Lys) | 0 (D) | T (0.35) | PoD (0.902) | L (1.535) | 0.269 | 24.7 | −13% | −24% | −2% | −79% | −26% | LOVD | SPLICING |
| c.3250C>T (p.Pro1084Ser) | 52 (A) | T (0.08) | PD (1.00) | M (3.235) | 0.833 | 24.5 | +3% | +13% | +11% | +137% | +1% | CM143367 | MISSENSE |
| c.3827G>A (p.Arg1276Gln) | 44 (D) | D (0.00) | PD (1.00) | H (3.820) | 0.887 | 34 | +1% | SA creation | NA | NA | +1% | CM000802 | MISSENSE |
| c.4267A>G (p.Lys1423Glu) | 3 (D) | D (0.00) | PD (1.00) | H (3.80) | 0.905 | 34 | −3% | −16% | SD abrogation | No change | −1% | CM920506 | SPLICING |
| c.4768C>T (p.Arg1590Trp) | 4 (D) | D (0.01) | PD (1.00) | M (2.72) | 0.533 | 35 | No change | No change | +0.2% | NA | No change | CM971051 | MISSENSE |
| c.5546+5G>A | 5 (D) | NE | NE | NE | NE | 10.3 | SD abrogation | SD abrogation | SD abrogation | −14% | −73% | CS076638 | SPLICING |
| c.6755A>G (p.Lys2252Arg) | 2 (D) | T (0.75) | PD (0.99) | L (0.84) | 0.676 | 9.4 | SD abrogation | −50% | SD abrogation | −6% | −71% | CM143467 | SPLICING |
| c.6756+3A>G | 3 (D) | NE | NE | NE | NE | 26.5 | −6% | −37% | −50% | −11% | −41% | CS031795 | SPLICING |
| DNA Change | DIST to SS | mRNA Effect | Consequences at Protein Level | SIFT | POLYPHEN | MUT ASSESSOR | SSF | MES | NNPLICE | GENESPLICER | HSF | Predicted to Alter Splicing |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.58C>T | −3 SD | r.57_60del4 | Gln58* | NE | NE | NE | −5.1% | −23.1% | −28.0% | −35.2% | −2.3% | YES |
| c.730G>A | −1 SD | r.731_888del | Glu244Lys | T (0.45) | B (0.081) | N (−0.60) | −14.0% | −46.6% | SD abrogation | NA | −11.3% | YES |
| c.1062G>A | −1 SD | r.1063_1185del | Lys354Lys | NE | NE | NE | SD abrogation | −79.1% | SD abrogation | NA | −12.5% | YES |
| c.1845G>T | −1 SD | r.1846_2001del | Lys615Asn | T (0.48) | B (0.397) | L (1.845) | −13.4% | −40.1% | −2.6% | SD abrogation | −11.2% | YES |
| c.2251G>A | −1 SD | r.2252_2325del | Gly751Arg | D (0.01) | PD (1.00) | M (2.325) | −13.8% | −49.3% | −29.5% | SD abrogation | −12.0% | YES |
| c.2325G>T | −1 SD | r.2326_2409del | Glu775Asp | T (0.24) | PoD (0.863) | L (1.50) | SD abrogation | −59.3% | SD abrogation | −76.9% | −12.5% | YES |
| c.3113G>C | −1 SD | r.3114_3197del | Arg1038Thr | D (0.02) | PD (0.998) | M (3.27) | −14.5% | −12.0% | −6.4% | SD abrogation | −11.4% | YES |
| c.4269G>C | −1 SD | r.4270_4367del | Lys1423Asn | D (0.00) | PD (1.00) | H (3.80) | SD abrogation | SD abrogation | SD abrogation | NA | −12.8% | YES |
| c.5546G>A | −1 SD | r.5547_5749del | Arg1849Gln | D (0.04) | PD (0.998) | M (3.345) | SD abrogation | −44.1% | SD abrogation | SD abrogation | −12.3% | YES |
| DNA Change | DIST to SS a | PROTEIN CHANGE | SIFT b | Polyphen2 c | Mutation Assessor d | HGMD CLASS e | SSF f | MES f | NNSPLICE f | GENESPLICER f | HSF f | Predicted to Alter Splicing |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.62T>G | +2 SA | Leu21Arg | D (0.00) | PoD (0.902) | M (2.495) | DM | no change | 3.50% | −0.40% | 3.90% | −0.60% | NO |
| c.62T>C | +2 SA | Leu21Pro | D (0.00) | PD (0.99) | M (2.495) | DM | no change | −9.40% | −0.30% | −5% | −0.90% | NO |
| c.479G>T | −1 SD | Arg160Met | D (0.01) | PD (0.99) | M (2.445) | DM? | −14% | SD abrogation | −51.90% | NA | −11.40% | YES |
| c.479G>C | −1 SD | Arg160Thr | D (0.04) | PD (0.97) | M (2.445) | DM? | −14.80% | −94.50% | −24.50% | NA | −11.60% | YES |
| c.482T>A | +3 SA | Stop | NE | NE | NE | NONSENSE | no change | −2.10% | no change | 11.50% | no change | NO |
| c.482T>G | +3 SA | Stop | NE | NE | NE | NONSENSE | no change | −1.50% | no change | NA | no change | NO |
| c.586G>T | −1 SD | Stop | NE | NE | NE | NONSENSE | −13.90% | −19.10% | −1.50% | NA | −11.50% | YES |
| c.887A>G | −2 SD | Lys296Arg | T (0.24) | PD (0.97) | L (1.635) | DM? | −9.50% | −12.90% | −0.30% | −75.40% | −5% | NO |
| c.1062G>C | −1 SD | Lys394Asn | T (0.07) | PD (0.98) | L (1.520) | DM? | SD abrogation | −55% | −55.10% | NA | −13% | YES |
| c.1060A>T | −3 SD | Stop | NE | NE | NE | NONSENSE | −4.70% | −30.00% | −16.40% | NA | −1.20% | NO |
| c.1639G>T | −3 SD | Stop | NE | NE | NE | NONSENSE | −1.20% | −9.50% | −0.80% | −75.70% | −0.20% | NO |
| c.1720A>G | −2 SD | Ser574Gly | T (0.12) | PoD (0.954) | L (1.560) | DM | SD abrogation | −58.10% | SD abrogation | NA | −6.30% | YES |
| c.1720A>C | −2 SD | Ser574Arg | T (0.14) | PD (0.99) | M (2.250) | DM | SD abrogation | −9.60% | SD abrogation | NA | −6.20% | NO |
| c.1721G>A | −1 SD | Ser574Asn | T (0.12) | PD (0.981) | L (1.905) | DM | SD abrogation | SD abrogation | SD abrogation | SD abrogation | −13.70% | YES |
| c.1722C>G | +1 SA | Ser574Arg | T (0.14) | PD (0.994) | L (1.560) | DM | 6.70% | 27.80% | SA creation | SA creation | 4.90% | NO |
| c.1846C>G | +1 SA | Gln616Glu | T (1.00) | B (0.257) | L (1.10) | DM | NA | SA abrogation | SA abrogation | SA abrogation | −11.0% | YES |
| c.1846C>T | +1 SA | Stop | NE | NE | NE | NONSENSE | −0.60% | −11.00% | −0.10% | SA abrogation | −0.20% | NO |
| c.2252G>T | +1 SA | Gly751Val | D (0.03) | PD (1.00) | M (2.35) | DM? | −7.50% | −41.80% | SA abrogation | −33.50% | −5.40% | YES |
| c.2407C>T | −3 SD | Stop | NE | NE | NE | NONSENSE | −4.10% | −11% | −0.10% | −79.60% | −2% | NO |
| c.2848C>T | −3 SD | Stop | NE | NE | NE | NONSENSE | −5% | −28.20% | −30.20% | SD abrogation | −2.30% | YES |
| c.2851G>T | +1 SA | Val951Phe | D (0.01) | PD (0.994) | M (2.800) | DM | −6.90% | −25% | −3.20% | −32.80% | −4.80% | NO |
| c.2990G>C | −1 SD | Arg997Thr | T (0.21) | PD (0.981) | M (2.955) | DM? | −14.10% | −33.50% | −0.90% | −74.20% | −11.30% | YES |
| c.2992T>G | +2 SA | Tyr998Asp | T (0.55) | PD (0.997) | M (3.135) | DM? | SD abrogation | SD abrogation | SD abrogation | NA | SD abrogation | YES |
| c.3114G>T | +1 SA | Arg1038Ser | D (0.01) | PD (0.981) | M (3.270) | DM | −8.10% | −40.50% | −7.30% | −55% | −5.30% | NO |
| c.3197G>A | −1 SD | Arg1066Lys | D (0.00) | PoD (0.902) | L (1.885) | DM | −14.80% | −85.90% | SD abrogation | SD abrogation | −12.50% | YES |
| c.3494T>C | −3 SD | Ile1165Thr | D (0.01) | PD (0.96) | M (2.14) | DM? | 4.80% | 32.70% | 22.10% | −37.20% | 2.10% | NO |
| c.3497G>A | +1 SA | Gly1166Lys | T (0.89) | PD (1.00) | L (0.855) | DM | −4.90% | −19.50% | −21% | −36.20% | −3.90% | YES |
| c.3707G>A | −2 SD | Stop | NE | NE | NE | NONSENSE | 10.30% | −5.60% | 0.20% | 17.60% | 5.20% | NO |
| c.3974G>A | −1 SD | Arg1325Lys | T (0.50) | PoD (0.902) | N (0.535) | DM | SD abrogation | −72.30% | SD abrogation | NA | −12.30% | YES |
| c.3974G>C | −1 SD | Arg1325Thr | D (0.02) | PD (0.981) | M (2.485) | DM | SD abrogation | SD abrogation | SD abrogation | NA | −12.80% | YES |
| c.3977T>G | +3 SA | Stop | NE | NE | NE | NONSENSE | no change | 1.20% | no change | 10.80% | no change | NO |
| c.4108C>T | −3 SD | Stop | NE | NE | NE | NONSENSE | −4.90% | −19.60% | −2.30% | NA | −2.20% | NO |
| c.4269G>T | −1 SD | Lys1423Asn | D (0.00) | PD (0.989) | H (3.8) | DM | −15.20% | SD abrogation | SD abrogation | NA | −12.70% | YES |
| c.4267A>C | −3 SD | Lys1423Gln | D (0.00) | PD (0.989) | M (3.455) | DM? | 0.40% | 3.40% | 2% | SD creation | 1.10% | NO |
| c.4267A>G | −3 SD | Lys1423Glu | D (0.00) | PD (0.974) | H (3.8) | DM | −3.30% | −15.80% | SD creation | NA | −0.90% | NO |
| c.4267A>T | −3 SD | Stop | NE | NE | NE | NONSENSE | −4.60% | −40.30% | SD creation | NA | −1.20% | NO |
| c.4367G>C | −1 SD | Arg1456Thr | D (0.00) | PD (0.981) | L (1.155) | DM? | −15.20% | −49.30% | SD abrogation | NA | −12.60% | YES |
| c.5203A>T | −3 SD | Stop | NE | NE | NE | NONSENSE | −4.40% | −17% | −5.30% | SD abrogation | −1.20% | NO |
| c.6364G>A | −1 SD | Glu2122Lys | D (0.03) | PD (0.974) | M (2.79) | DM | −13.70% | −22.70% | −7.60% | SD abrogation | −11.30% | YES |
| c.6640A>T | −2 SD | Stop | NE | NE | NE | NONSENSE | −9.50% | −14.70% | −0.60% | −51.10% | −5.30% | NO |
| c.7552G>T | −1 SD | Stop | NE | NE | NE | NONSENSE | SD abrogation | SD abrogation | SD abrogation | NA | −12.30% | YES |
| Number of Patients | Frameshifting (n = 62) | In-Frame (n = 22) | p-Value |
|---|---|---|---|
| CALs | 55 (88.7) | 21 (95.4) | NS |
| Freckling | 30 (46.7) | 14 (63.6) | NS |
| Lisch nodules | 14 (22.6) | 2 (9.1) | NS |
| Neurofibromas | 46 (74.2) | 12 (54.5) | NS |
| Optic glioma | 3 (4.2) | 1 (4.8) | NS |
| Bone lesions | 16 (25.8) | 6 (28.6) | NS |
| Intellectual disability | 8 (12.9) | 2 (9.5) | NS |
| Hamartomas | 6 (9.6) | 2 (9.5) | NS |
| Deafness incl. neurinomas | 4 (6.4) | 0 (0) | NS |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, A.; Lastella, P.; Loconte, D.C.; Bukvic, N.; Varvara, D.; Patruno, M.; Bagnulo, R.; Lovaglio, R.; Bartolomeo, N.; Serio, G.; et al. Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1. Genes 2018, 9, 216. https://doi.org/10.3390/genes9040216
Stella A, Lastella P, Loconte DC, Bukvic N, Varvara D, Patruno M, Bagnulo R, Lovaglio R, Bartolomeo N, Serio G, et al. Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1. Genes. 2018; 9(4):216. https://doi.org/10.3390/genes9040216
Chicago/Turabian StyleStella, Alessandro, Patrizia Lastella, Daria Carmela Loconte, Nenad Bukvic, Dora Varvara, Margherita Patruno, Rosanna Bagnulo, Rosaura Lovaglio, Nicola Bartolomeo, Gabriella Serio, and et al. 2018. "Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1" Genes 9, no. 4: 216. https://doi.org/10.3390/genes9040216
APA StyleStella, A., Lastella, P., Loconte, D. C., Bukvic, N., Varvara, D., Patruno, M., Bagnulo, R., Lovaglio, R., Bartolomeo, N., Serio, G., & Resta, N. (2018). Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1. Genes, 9(4), 216. https://doi.org/10.3390/genes9040216