Impact, Characterization, and Rescue of Pre-mRNA Splicing Mutations in Lysosomal Storage Disorders



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
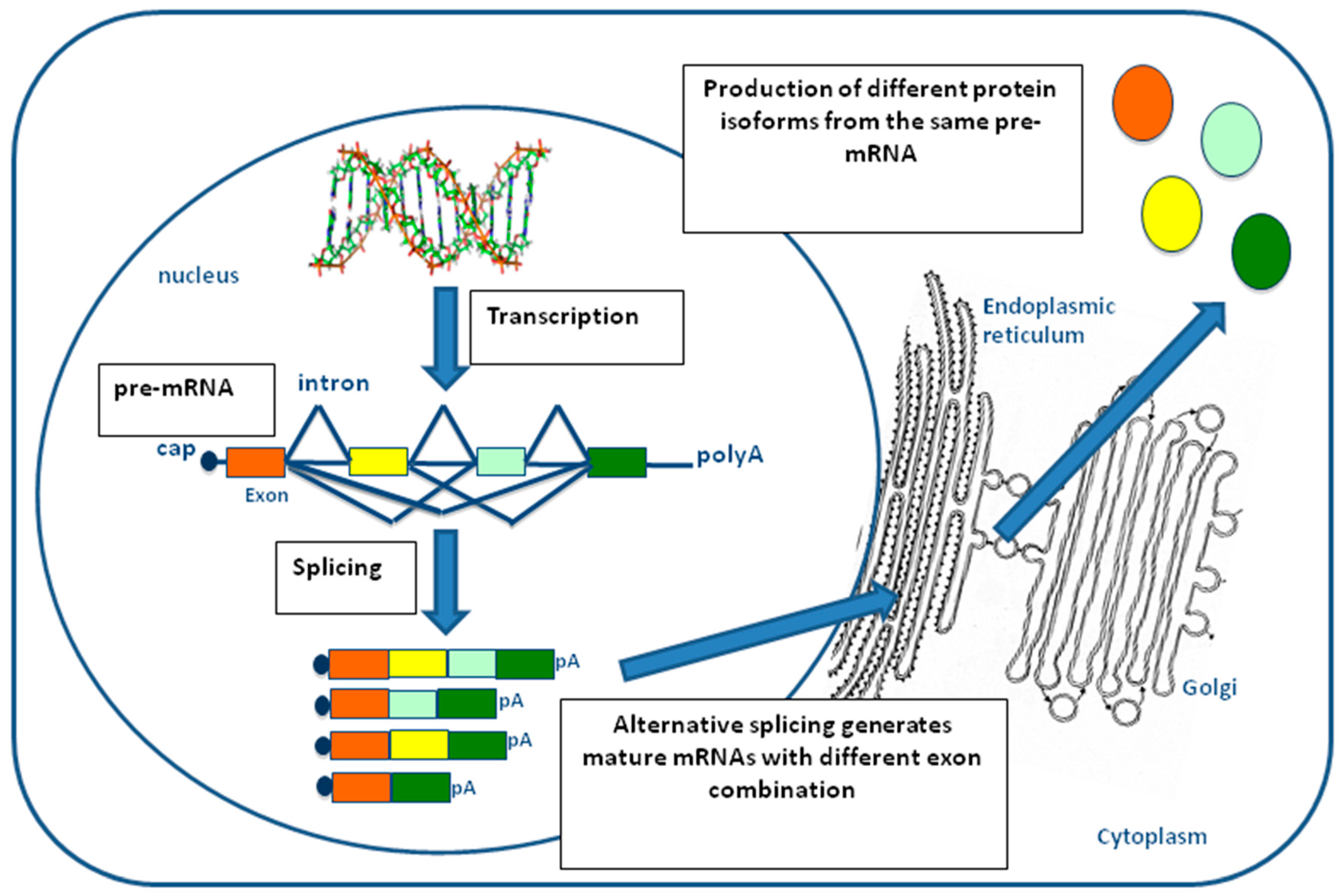
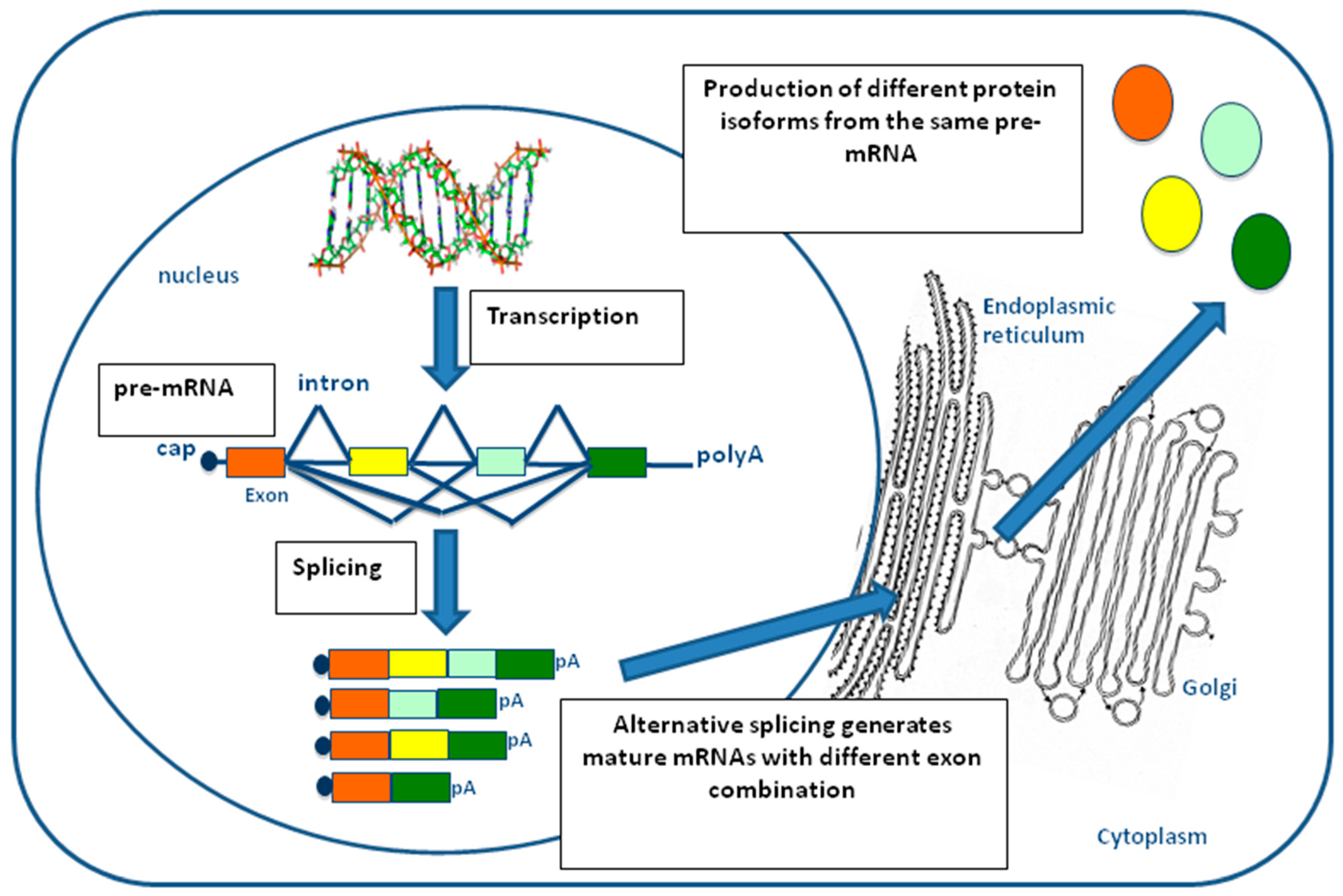
2. Pre-mRNA Splicing
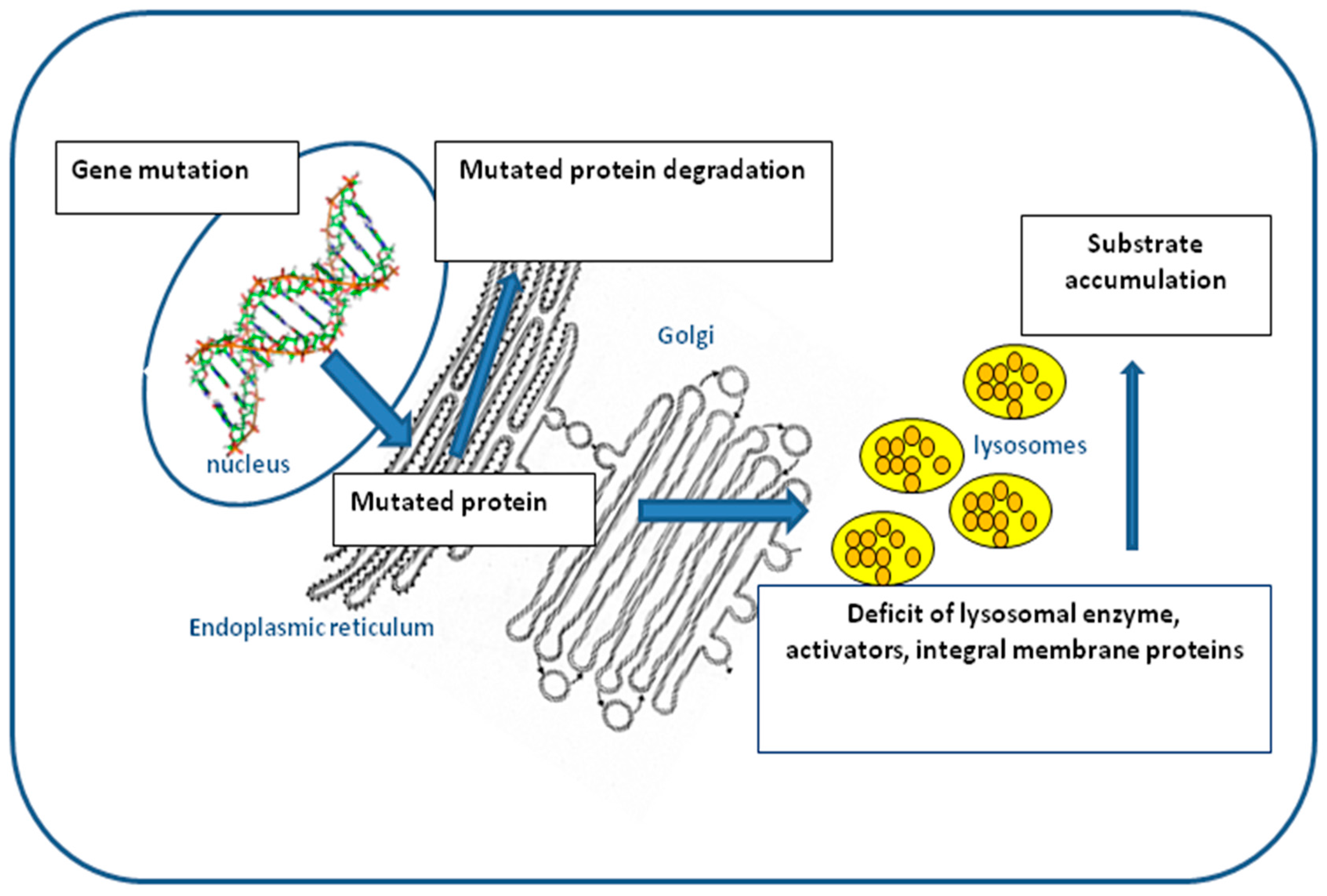
3. Splicing Mutations in LSDs
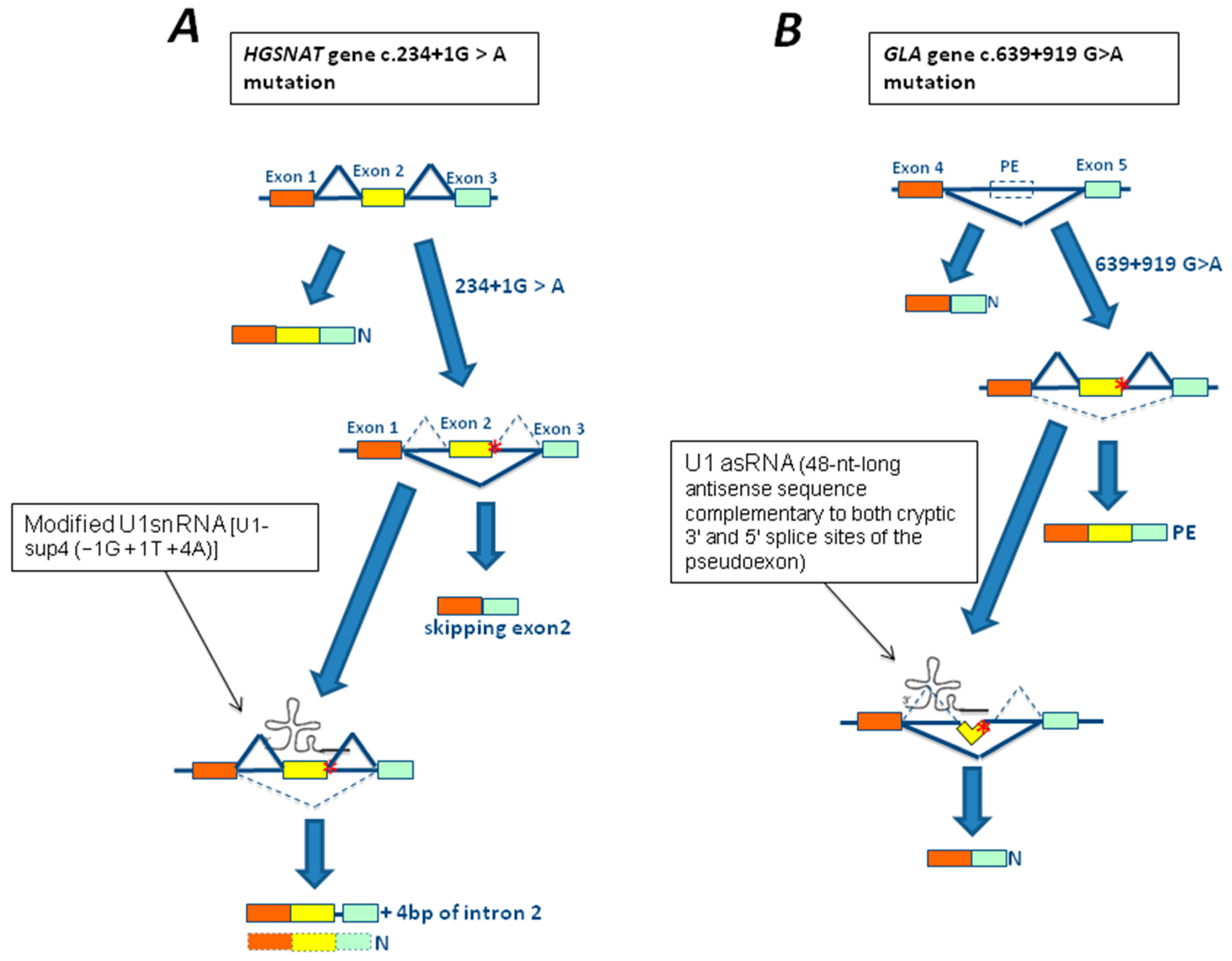
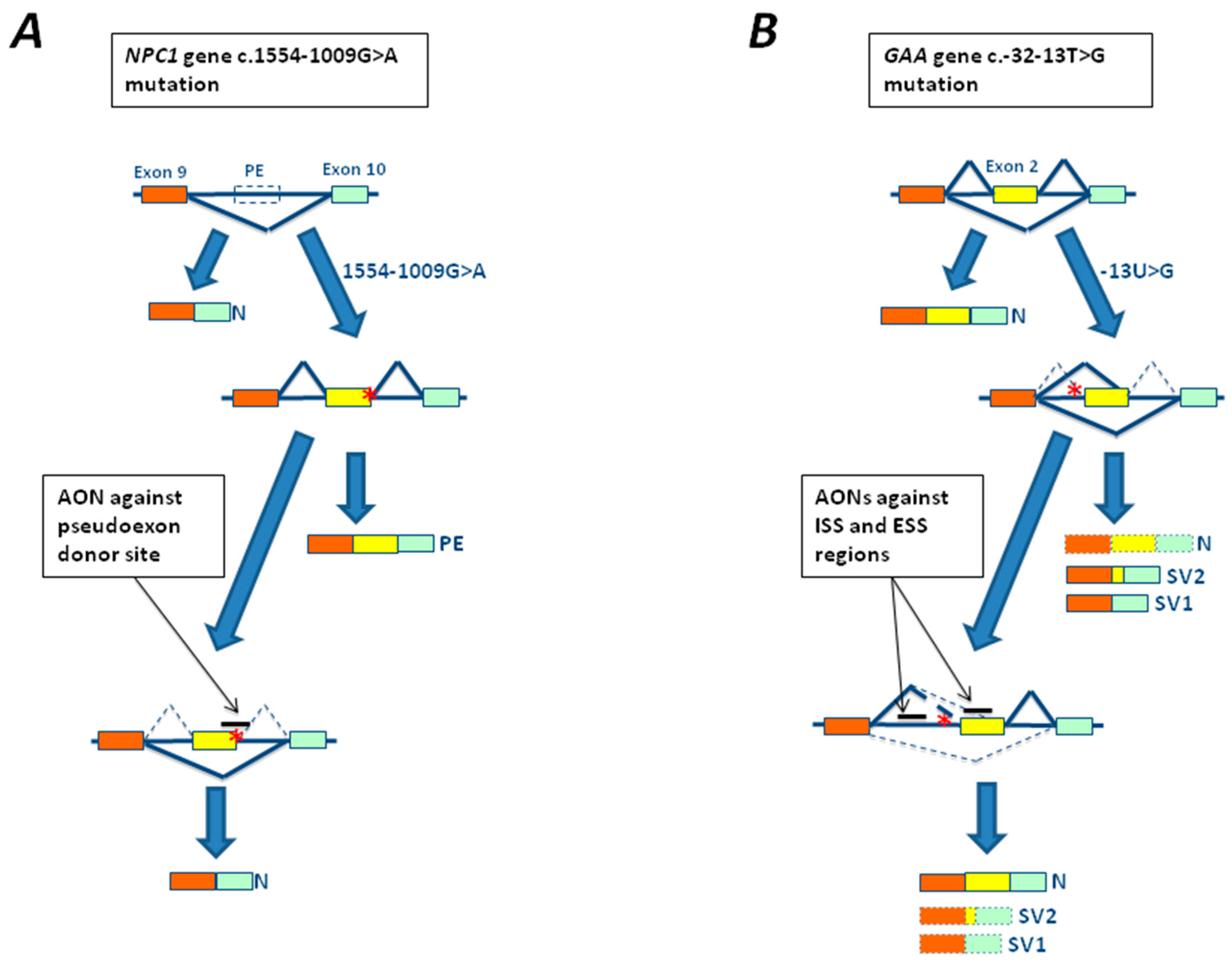
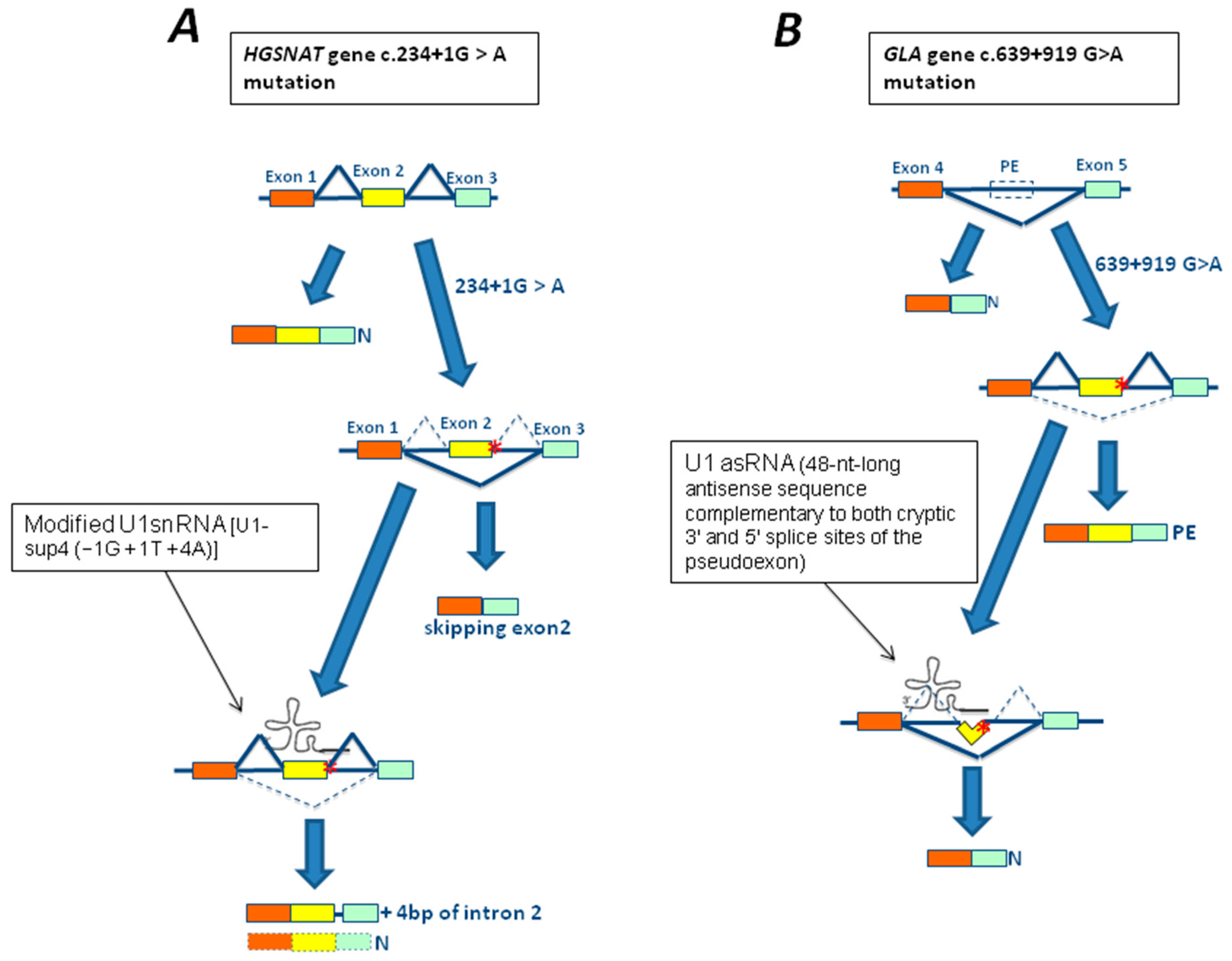
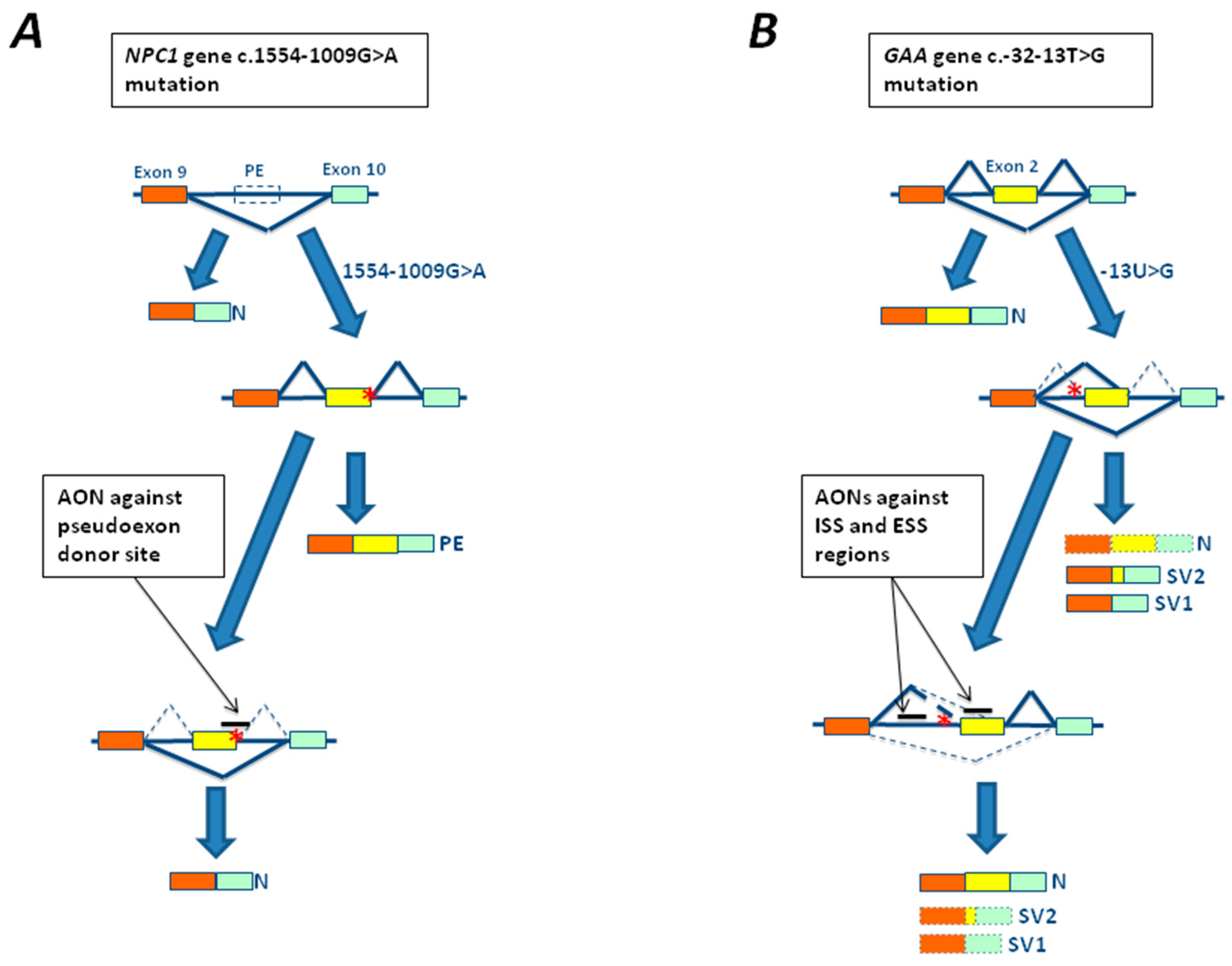
3.1. The c.-32-13T>G Mutation of GAA Gene
3.2. The c.894G>A Substitution of LIPA Gene
3.3. The 639+919G>A Mutation of GLA Gene
3.4. The c.2715+1G>A Mutation of the GNPTAB Gene
3.5. The c.459+5A>G Mutation of the HEXA Gene
4. Therapeutic Approaches for Splicing Defects in LSDs
4.1. In Vitro Studies Using Modified U1snRNA in LSDs
4.2. In Vitro Studies Using Antisense Oligonucleotides in LSDs
5. Open Issues and Conclusions
Acknowledgments
Conflicts of Interest
References
- Futerman, A.H.; van Meer, G. The Cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Meikle, P.J.; Hopwood, J.J. Fabry Disease: Perspectives from 5 years of FOS. In Epidemiology of Lysosomal Storage Diseases: An Overview; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Chapter 2. [Google Scholar]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; Wijburg, F.A. Treatment of lysosomal storage disorders: Successes and challenges. J. Inherit. Metab. Dis. 2014, 37, 587–598. [Google Scholar] [CrossRef] [PubMed]
- The Human Gene Mutation Database (HGMD). Available online: http://www.hgmd.cf.ac.uk (accessed on 15 December 2017).
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Papasaikas, P.; Valcárcel, J. The spliceosome: The ultimate RNA chaperone and sculptor. Trends Biochem. Sci. 2016, 41, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Galej, W.P.; Nguyen, T.H.; Newman, A.J.; Nagai, K. Structural studies of the spliceosome: Zooming into the heart of the machine. Curr. Opin. Struct. Biol. 2014, 25, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Fiszbein, A.; Kornblihtt, A.R. Alternative splicing switches: Important players in cell differentiation. Bioessays 2017, 39, 6. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Schor, I.E.; Alló, M.; Dujardin, G.; Petrillo, E.; Muñoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef]
- Baralle, D.; Lucassen, A.; Buratti, E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO Rep. 2009, 10, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Verot, L.; Chikh, K.; Freydière, E.; Honoré, R.; Vanier, M.T.; Millat, G. Niemann-Pick C disease: Functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin. Genet. 2007, 71, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Filocamo, M.; Buratti, E.; Stroppiano, M.; Vlahovicek, K.; Rosso, N.; Bignulin, E.; Regis, S.; Carnevale, F.; Bembi, B.; et al. Molecular and functional analysis of the HEXB gene in Italian patients affected with Sandhoff disease: Identification of six novel alleles. Neurogenetics 2009, 10, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Tappino, B.; Biancheri, R.; Mort, M.; Regis, S.; Corsolini, F.; Rossi, A.; Stroppiano, M.; Lualdi, S.; Fiumara, A.; Bembi, B.; et al. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum. Mutat. 2010, 31, E1894–E1914. [Google Scholar] [CrossRef] [PubMed]
- Fasano, T.; Pisciotta, L.; Bocchi, L.; Guardamagna, O.; Assandro, P.; Rabacchi, C.; Zanoni, P.; Filocamo, M.; Bertolini, S.; Calandra, S. Lysosomal lipase deficiency: Molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol. Genet. Metab. 2012, 105, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Cetin, H.; Wöhrer, A.; Rittelmeyer, I.; Gencik, M.; Zulehner, G.; Zimprich, F.; Ströbel, T.; Zimprich, A. The c.65-2A>G splice site mutation is associated with a mild phenotype in Danon disease due to the transcription of normal LAMP2 mRNA. Clin. Genet. 2016, 90, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, S.; Buratti, E.; Bembi, B.; Baralle, M.; Pittis, M.G. Characterization of two novel GBA mutations causing Gaucher disease that lead to aberrant RNA species by using functional splicing assays. Hum. Mutat. 2006, 27, 119. [Google Scholar] [CrossRef] [PubMed]
- Lualdi, S.; Pittis, M.G.; Regis, S.; Parini, R.; Allegri, A.E.; Furlan, F.; Bembi, B.; Filocamo, M. Multiple cryptic splice sites can be activated by IDS point mutations generating misspliced transcripts. J. Mol. Med. 2006, 84, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Buratti, E.; Dominissini, S.; Montalvo, A.L.; Pittis, M.G.; Bembi, B.; Dardis, A. Splicing mutations in glycogen-storage disease type II: Evaluation of the full spectrum of mutations and their relation to patients’ phenotypes. Eur. J. Hum. Genet. 2011, 19, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascau, L.; Coll, M.J.; Vilageliu, L.; Grinberg, D. Antisense oligonucleotide treatment for a pseudoexon-generating mutation in the NPC1 gene causing Niemann-Pick type C disease. Hum. Mutat. 2009, 30, E993–E1001. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Nakao, S.; Minamikawa-Tachino, R.; Desnick, R.J.; Fan, J.Q. Alternative splicing in the α-galactosidase A gene: Increased exon inclusion results in the Fabry cardiac phenotype. Am. J. Hum. Genet. 2002, 70, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, R.; Reuser, A.J.J. Glycogen storage disease type II: Acid α-glucosidase (acid maltase) deficiency. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume 3, pp. 3389–3420. [Google Scholar]
- Gungor, D.; Reuser, A.J. How to describe the clinical spectrum in Pompe disease? Am. J. Med. Genet. Part A 2013, 161A, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Van den Hout, H.M.; Hop, W.; van Diggelen, O.P.; Smeitink, J.A.; Smit, G.P.; Poll-The, B.T.; Bakker, H.D.; Loonen, M.C.; de Klerk, J.B.; Reuser, A.J.; et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003, 112, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Hwu, W.L.; Mandel, H.; Nicolino, M.; Yong, F.; Corzo, D.; Infantile-onset Pompe disease natural history study group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006, 148, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Hagemans, M.L.; Winkel, L.P.; Van Doorn, P.A.; Hop, W.J.; Loonen, M.C.; Reuser, A.J.; Van der Ploeg, A.T. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 2005, 128, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Herzog, A.; Hartung, R.; Reuser, A.J.; Hermanns, P.; Runz, H.; Karabul, N.; Gökce, S.; Pohlenz, J.; Kampmann, C.; Lampe, C.; et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: Molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J. Rare Dis. 2012, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuller, A.; Wenninger, S.; Strigl-Pill, N.; Schoser, B. Toward deconstructing the phenotype of late-onset Pompe disease. Ame. J. Med. Genet. Part C 2012, 160C, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Montalvo, A.L.; Bembi, B.; Donnarumma, M.; Filocamo, M.; Parenti, G.; Rossi, M.; Merlini, L.; Buratti, E.; De Filippi, P.; Dardis, A.; et al. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum. Mutat. 2006, 27, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Fanin, M.; Tasca, E.; Angelini, C. Molecular pathology and enzyme processing in various phenotypes of acid maltase deficiency. Neurology 2008, 70, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Gort, L.; Coll, M.J.; Chabas, A. Glycogen storage disease type II in Spanish patients: High frequency of c.1076-1G>C mutation. Mol. Genet. Metab. 2007, 92, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Lee, C.C.; Hsu, C.M.; Hwu, W.L.; Yang, C.C.; Tsai, C.H.; Tsai, F.J. Identification of eight novel mutations of the acid α-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J. Neurol. 2008, 255, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Glaser, D.; Schmidt, S.; Vorgerd, M.; Winterholler, M.; Eger, K.; Zierz, S.; Deschauer, M. Molecular diagnosis of German patients with late-onset glycogen storage disease type II. J. Inherit. Metab. Dis. 2008, 31 (Suppl. 2), S261–S265. [Google Scholar] [CrossRef] [PubMed]
- Huie, M.L.; Chen, A.S.; Tsujino, S.; Shanske, S.; DiMauro, S.; Engel, A.G.; Hirschhorn, R. Aberrant splicing in adult onset glycogen storage disease type II (GSDII): Molecular identification of an IVS1 (-13T-->G) mutation in a majority of patients and a novel IVS10 (+1GT-->CT) mutation. Hum. Mol. Genet. 1994, 3, 2231–2236. [Google Scholar] [PubMed]
- Boerkoel, C.F.; Exelbert, R.; Nicastri, C.; Nichols, R.C.; Miller, F.W.; Plotz, P.H.; Raben, N. Leaky splicing mutation in the acid maltase gene is associated with delayed onset of glycogenosis type II. Am. J. Hum. Genet. 1995, 56, 887–897. [Google Scholar] [PubMed]
- Raben, N.; Nichols, R.C.; Martiniuk, F.; Plotz, P.H. A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II). Hum. Mol. Genet. 1996, 5, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Dardis, A.; Zanin, I.; Zampieri, S.; Stuani, C.; Pianta, A.; Romanello, M.; Baralle, F.; Bembi, B.; Buratti, E. Functional characterization of the common c.-32-13T>G mutation of GAA gene: Identification of potential therapeutic agents. Nucleic Acids Res. 2014, 42, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Reed, S. Acid lipase cross-reacting material in Wolman disease and cholesterol ester storage disease. Am. J. Hum. Genet. 1981, 33, 203–208. [Google Scholar] [PubMed]
- Assmann, G.; Seedorf, U. Acid lipase deficiency: Wolman disease and cholesteryl ester storage disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3551–3572. [Google Scholar]
- Hoeg, J.M.; Demosky, S.J.; Pescovitz, O.H.; Brewer, H.B., Jr. Cholesteryl ester storage disease and Wolman disease: Phenotypic variants of lysosomal acid cholesteryl ester hydrolase deficiency. Am. J. Hum. Genet. 1984, 36, 1190–1203. [Google Scholar] [PubMed]
- Aslanidis, C.; Ries, S.; Fehringer, P.; Büchler, C.; Klima, H.; Schmitz, G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics 1996, 33, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Lohse, P.; Maas, S.; Lohse, P.; Elleder, M.; Kirk, J.M.; Besley, G.T.; Seidel, D. Compound heterozygosity for a Wolman mutation is frequent among patients with cholesteryl ester storage disease. J. Lipid Res. 2000, 41, 23–31. [Google Scholar] [PubMed]
- Pisciotta, L.; Fresa, R.; Bellocchio, A.; Pino, E.; Guido, V.; Cantafora, A.; Do Roccco, M.; Calandra, S.; Bertollini, S. Cholesteryl ester storage disease (CESD) due to novel mutations in the LIPA gene. Mol. Genet. Metab. 2009, 97, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chatrath, H.; Keilin, S.; Attar, B.M. Cholesterol ester storage disease (CESD) diagnosed in an asymptomatic adult. Dig. Dis. Sci. 2009, 54, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Aguisanda, F.; Thorne, N.; Zheng, W. Targeting Wolman disease and cholesteryl ester storage disease: disease pathogenesis and therapeutic development. Curr. Chem. Genom. Transl. Med. 2017, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Garcia, R.; Pariyarath, R.; Stuani, C.; Gridelli, B.; Paone, G.; Baralle, F.E. Expression of lysosomal acid lipase mutants detected in three patients with cholesteryl ester storage disease. Hum. Mol. Genet. 1996, 5, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Ameis, D.; Brokmann, G.; Knoblich, R.; Merkel, M.; Ostlund, R.E., Jr.; Yang, J.W.; Coates, P.M.; Cortner, J.A.; Feinman, S.V.; Greten, H. A 5′ splice-site mutation and a dinucleotide deletion in the lysosomal acid lipase gene in two patients with cholesteryl ester storage disease. J. Lipid Res. 1995, 36, 241–250. [Google Scholar] [PubMed]
- Pagani, F.; Pariyarath, R.; Garcia, R.; Stuani, C.; Burlina, A.B.; Ruotolo, G.; Rabusin, M.; Baralle, F.E. New lysosomal acid lipase gene mutants explain the phenotype of Wolman disease and cholesteryl ester storage disease. J. Lipid Res. 1998, 39, 1382–1388. [Google Scholar] [PubMed]
- Du, H.; Sheriff, S.; Bezerra, J.; Leonova, T.; Grabowski, G.A. Molecular and enzymatic analyses of lysosomal acid lipase in cholesteryl ester storage disease. Mol. Genet. Metab. 1998, 84, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.A.; Bryson, G.M.; Parks, J.S. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesteryl ester storage disease. Mol. Genet. Metab. 1999, 68, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Vom Dahl, S.; Hazer, K.; Rolfs, A.; Albrecht, B.; Niedereau, C.; Vogt, C.; van Weely, S.; Aerts, J.; Müller, G.; Häussinger, D. Hepatosplenomegalic lipidosis: What unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase—1 exon 8 splice junction mutation. J. Hepatol. 1999, 31, 741–746. [Google Scholar] [CrossRef]
- Desnick, R.; Ioannou, Y.; Eng, C. α-Galactosidase a deficiency: Fabry disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 2001, 38, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Filoni, C.; Caciotti, A.; Carraresi, L.; Donati, M.A.; Mignani, R.; Parini, R.; Filocamo, M.; Soliani, F.; Simi, L.; Guerrini, R.; et al. Unbalanced GLA mRNAs ratio quantified by real-time PCR in Fabry patients’ fibroblasts results in Fabry disease. Eur. J. Hum. Genet. 2008, 16, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, S.; Sly, W. I-cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 3469–3482. [Google Scholar]
- Liu, S.; Zhang, W.; Shi, H.; Yao, F.; Wei, M.; Qiu, Z. Mutation Analysis of 16 Mucolipidosis II and III α/β Chinese children revealed genotype-phenotype correlations. PLoS ONE 2016, 11, e0163204. [Google Scholar] [CrossRef] [PubMed]
- Paik, K.H.; Song, S.M.; Ki, C.S.; Yu, H.W.; Kim, J.S.; Min, K.H.; Chang, S.H.; Yoo, E.J.; Lee, I.J.; Kwan, E.K.; et al. Identification of mutations in the GNPTA (MGC4170) gene coding for GlcNAc-phosphotransferase α/β subunits in Korean patients with mucolipidosis type II or type IIIA. Hum. Mutat. 2005, 26, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Muramatsu, T.; Yorifuji, T.; Okuyama, T.; Nakabayashi, H.; Fukao, T.; Ohura, T.; Yoshino, M.; Tanaka, A.; Okamoto, N.; et al. Mucolipidosis II and III α/β: Mutation analysis of 40 Japanese patients showed genotype-phenotype correlation. J. Hum. Genet. 2009, 54, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Gravel, R.A.; Kaback, M.M.; Proia, R.L.; Sandhoff, K.; Suzuki, K.; Suzuki, K. The GM2 gangliosidoses. In The Metabolic and Molecular Bases of Inherited Diseases; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3827–3876. [Google Scholar]
- Gort, L.; de Olano, N.; Macías-Vidal, J.; Coll, M.A.; Spanish GM2 Working Group. GM2 gangliosidoses in Spain: Analysis of the HEXA and HEXB genes in 34 Tay-Sachs and 14 Sandhoff patients. Gene 2012, 506, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Montalvo, A.; Blanco, M.; Zanin, I.; Amartino, H.; Vlahovicek, K.; Szlago, M.; Schenone, A.; Pittis, G.; Bembi, B.; et al. Molecular analysis of HEXA gene in Argentinean patients affected with Tay-Sachs disease: Possible common origin of the prevalent c.459+5A>G mutation. Gene 2012, 499, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Akli, S.; Boue, J.; Sandhoff, K.; Kleijer, W.; Vamos, E.; Young, E.; Gatti, R.; Di Natale, P.; Motte, J.; Vanier, M.T.; et al. Collaborative study of the molecular epidemiology of Tay-Sachs disease in Europe. Eur. J. Hum. Genet. 1993, 1, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.; Yin, H.; McClorey, G. Modulating the expression of disease genes with RNA-based therapy. PLoS Genet. 2007, 3, e109. [Google Scholar] [CrossRef] [PubMed]
- Sumanasekera, C.; Watt, D.S.; Stamm, S. Substances that can change alternative splice-site selection. Biochem. Soc. Trans. 2008, 36, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; La Rosa, P.; Sette, C. Faulty RNA splicing: Consequences and therapeutic opportunities in brain and muscle disorders. Hum. Genet. 2017, 136, 1215–1235. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Walsh, C.E. Spliceosome-mediated RNA trans-splicing. Mol. Ther. 2005, 12, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, D. Novel roles of U1 snRNP in alternative splicing regulation. RNA Biol. 2010, 7, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Weiner, A.M. A compensatory base change in U1 snRNA suppresses a 5’ splice site mutation. Cell 1986, 46, 827–835. [Google Scholar] [CrossRef]
- Baralle, M.; Baralle, D.; De Conti, L.; Mattocks, C.; Whittaker, J.; Knezevich, A.; ffrench-Constant, C.; Baralle, F. Identification of a mutation that perturbs NF1 a gene splicing using genomic DNA samples and a minigene assay. J. Med. Genet. 2003, 40, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Susani, L.; Pangrazio, A.; Sobacchi, C.; Taranta, A.; Mortier, G.; Savarirayan, R.; Villa, A.; Orchard, P.; Vezzoni, P.; Albertini, A.; et al. TCIRG1-dependent recessive osteopetrosis: Mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum. Mutat. 2004, 24, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Pinotti, M.; Rizzotto, L.; Balestra, D.; Lewandowska, M.A.; Cavallari, N.; Marchetti, G.; Bernardi, F.; Pagani, F. U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency. Blood 2008, 2111, 2681–2684. [Google Scholar] [CrossRef] [PubMed]
- Tanner, G.; Glaus, E.; Barthelmes, D.; Ader, M.; Fleischhauer, J.; Pagani, F.; Berger, W.; Neidhardt, J. Therapeutic strategy to rescue mutation-induced exon skipping in rhodopsin by adaptation of U1 snRNA. Hum. Mutat. 2009, 30, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Balestra, D.; Faella, A.; Margaritis, P.; Cavallari, N.; Pagani, F.; Bernardi, F.; Arruda, V.R.; Pinotti, M. An engineered U1 small nuclear RNA rescues splicingdefective coagulation F7 gene expression in mice. J. Thromb. Haemost. 2014, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Rogalska, M.E.; Tajnik, M.; Licastro, D.; Bussani, E.; Camparini, L.; Mattioli, C.; Pagani, F. Therapeutic activity of modified U1 core spliceosomal particles. Nat. Commun. 2016, 7, 11168. [Google Scholar] [CrossRef] [PubMed]
- Denti, M.A.; Rosa, A.; Sthandier, O.; De Angelis, F.G.; Bozzoni, I. A new vector, based on the PolII promoter of the U1 snRNA gene, for the expression of siRNAs in mammalian cells. Mol. Ther. 2004, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.G.; Sthandier, O.; Berarducci, B.; Toso, S.; Galluzzi, G.; Ricci, E.; Cossu, G.; Bozzoni, I. Chimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in Δ48-50 DMD cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9456–9461. [Google Scholar] [CrossRef] [PubMed]
- Martone, J.; De Angelis, F.G.; Bozzoni, I. U1 snRNA as an effective vector for stable expression of antisense molecules and for the inhibition of the splicing reaction. Methods Mol. Biol. 2012, 867, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Seth, P.P.; Bennett, C.F. Antisense oligonucleotide-based therapies for diseases caused by pre-mRNA processing defects. Adv. Exp. Med. Biol. 2014, 825, 303–352. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Blanco, M.A. Making antisense of splicing. Curr. Opin. Mol. Ther. 2005, 7, 476–482. [Google Scholar] [PubMed]
- Aartsma-Rus, A.; van Ommen, G.J. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA 2007, 13, 1609–1624. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Eteplirsen: First global approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; de Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Brosseau, J.P.; Lucier, J.F.; Lamarche, A.A.; Shkreta, L.; Gendron, D.; Lapointe, E.; Thibault, P.; Paquet, E.; Perreault, J.P.; Abou Elela, S.; et al. Redirecting splicing with bifunctional oligonucleotides. Nucleic Acids Res. 2014, 42, e40. [Google Scholar] [CrossRef] [PubMed]
- Skordis, L.A.; Dunckley, M.G.; Yue, B.; Eperon, I.C.; Muntoni, F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl. Acad. Sci. USA 2003, 2100, 4114–4119. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; De Toledo, M.; Bonomi, S.; Valacca, C.; Gallo, S.; Apicella, M.; Eperon, I.; Tazi, J.; Biamonti, G. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: Therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol. 2010, 7, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Osman, E.Y.; Yen, P.F.; Lorson, C.L. Bifunctional RNAs targeting the intronic splicing silencer N1 increase SMN levels and reduce disease severity in an animal model of spinal muscular atrophy. Mol. Ther. 2012, 20, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Owen, N.; Zhou, H.; Malygin, A.A.; Sangha, J.; Smith, L.D.; Muntoni, F.; Eperon, I.C. Design principles for bifunctional targeted oligonucleotide enhancers of splicing. Nucleic Acids Res. 2011, 39, 7194–7208. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Canals, I.; Dridi, L.; Choi, Y.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Pérez, B.; Pshezhetsky, A.V.; Grinberg, D.; et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014, 9, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, L.; Covello, G.; Caciotti, A.; Guerrini, R.; Denti, M.A.; Morrone, A. Double-target antisense U1snRNAs correct mis-splicing due to c.639+861C>T and c.639+919G>A GLA Deep Intronic Mutations. Mol. Ther. Nucleic Acids 2016, 5, e380. [Google Scholar] [CrossRef] [PubMed]
- Goina, E.; Peruzzo, P.; Bembi, B.; Dardis, A.; Buratti, E. Glycogen reduction in myotubes of late-onset Pompe disease patients using antisense technology. Mol. Ther. 2017, 25, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, E.; Bergsma, A.J.; van Gestel, T.J.M.; In’t Groen, S.L.M.; Zaehres, H.; Araúzo-Bravo, M.J.; Schöler, H.R.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. GAA deficiency in Pompe disease is alleviated by exon inclusion in iPSC-derived skeletal muscle cells. Mol. Ther. Nucleic Acids 2017, 7, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, E.; Bergsma, A.J.; Pijnenburg, J.M.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Antisense oligonucleotides promote exon inclusion and correct the common c.-32-13T>G GAA splicing variant in Pompe disease. Mol. Ther. Nucleic Acids 2017, 7, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Grau, M.; Albaigès, J.; Casas, J.; Auladell, C.; Dierssen, M.; Vilageliu, L.; Grinberg, D. New murine Niemann-Pick type C models bearing a pseudoexon-generating mutation recapitulate the main neurobehavioural and molecular features of the disease. Sci. Rep. 2017, 7, 41931. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dardis, A.; Buratti, E. Impact, Characterization, and Rescue of Pre-mRNA Splicing Mutations in Lysosomal Storage Disorders. Genes 2018, 9, 73. https://doi.org/10.3390/genes9020073
Dardis A, Buratti E. Impact, Characterization, and Rescue of Pre-mRNA Splicing Mutations in Lysosomal Storage Disorders. Genes. 2018; 9(2):73. https://doi.org/10.3390/genes9020073
Chicago/Turabian StyleDardis, Andrea, and Emanuele Buratti. 2018. "Impact, Characterization, and Rescue of Pre-mRNA Splicing Mutations in Lysosomal Storage Disorders" Genes 9, no. 2: 73. https://doi.org/10.3390/genes9020073
APA StyleDardis, A., & Buratti, E. (2018). Impact, Characterization, and Rescue of Pre-mRNA Splicing Mutations in Lysosomal Storage Disorders. Genes, 9(2), 73. https://doi.org/10.3390/genes9020073