Exploring the Variable Phenotypes of RPGR Carrier Females in Assessing Their Potential for Retinal Gene Therapy

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
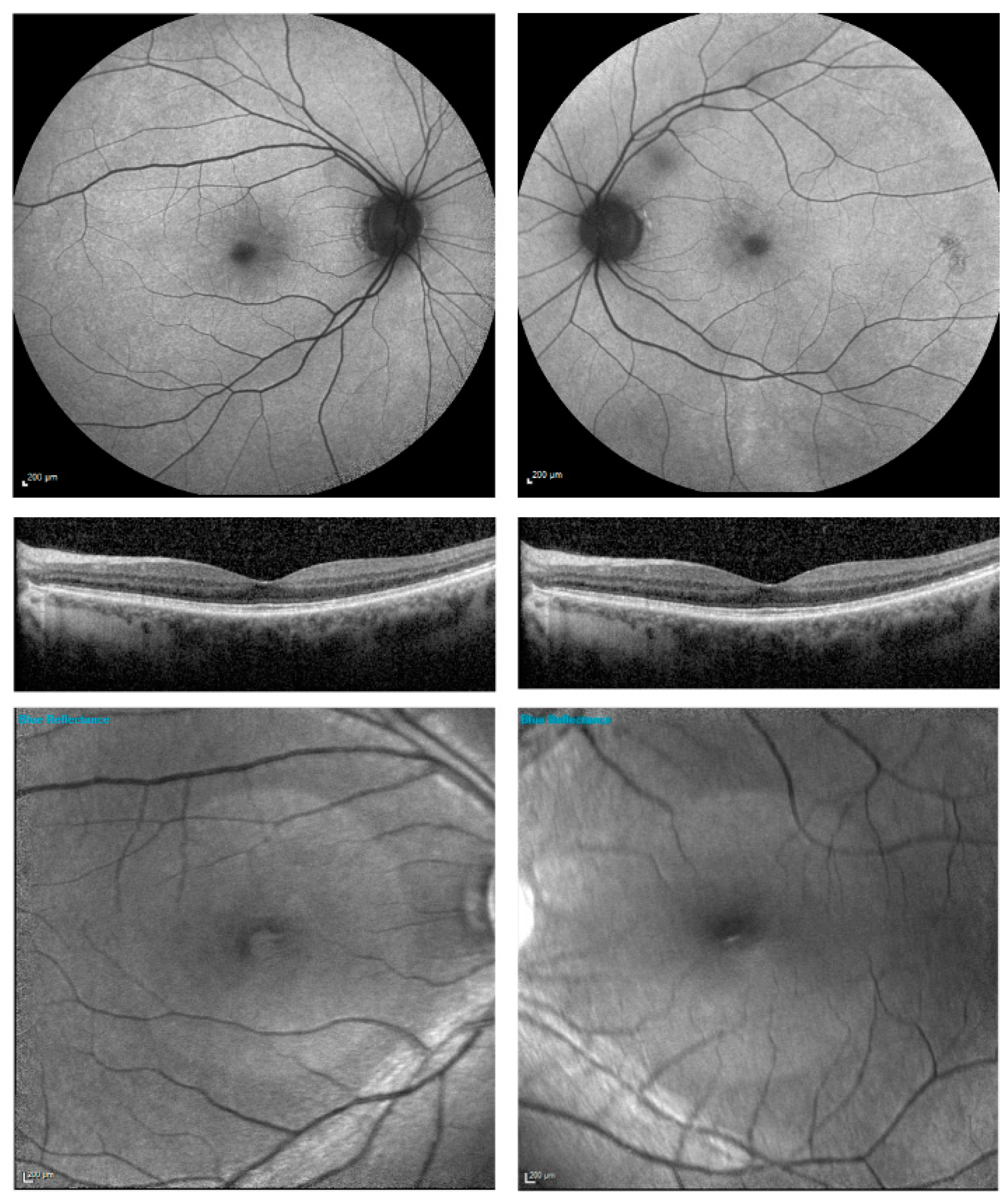
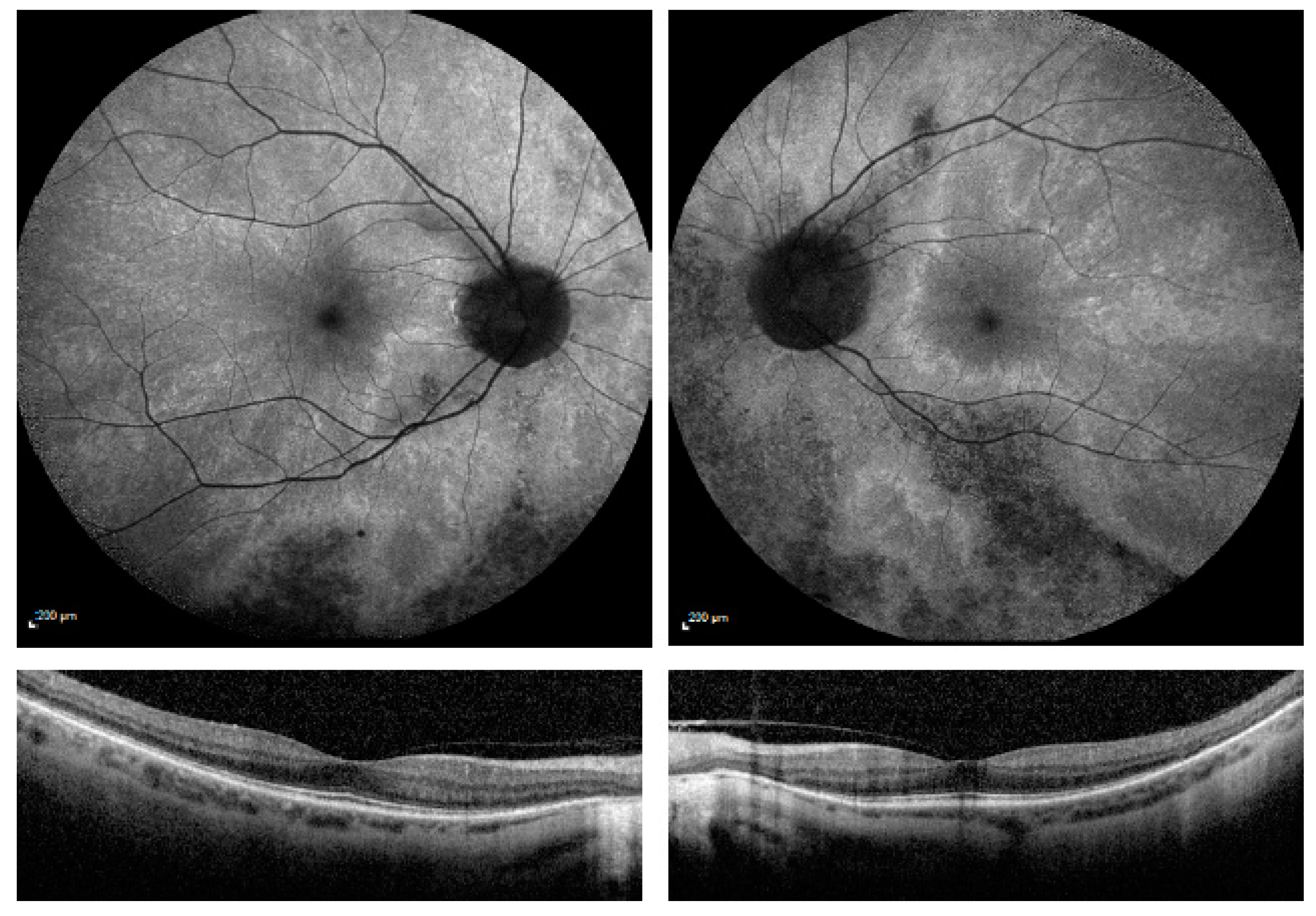
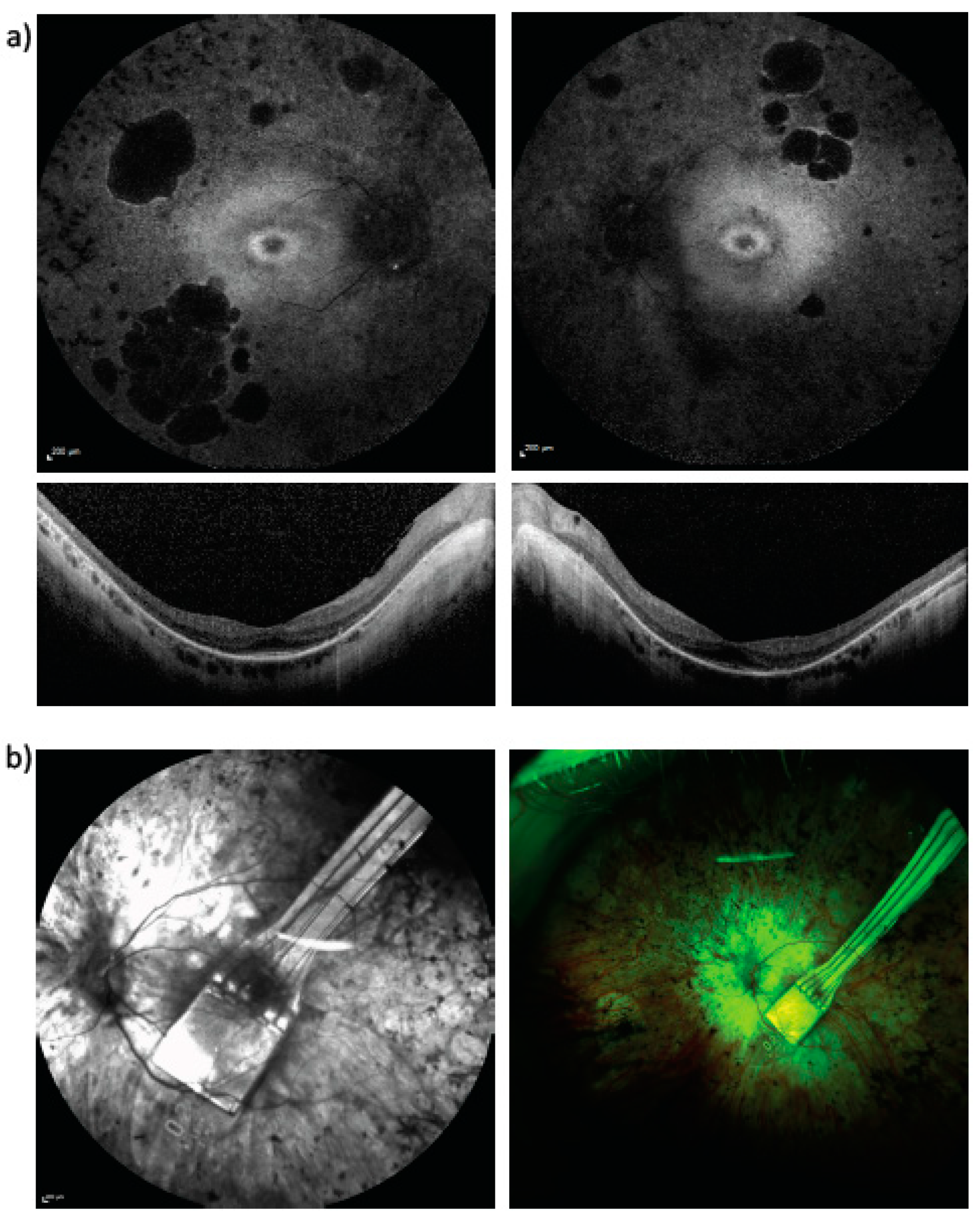
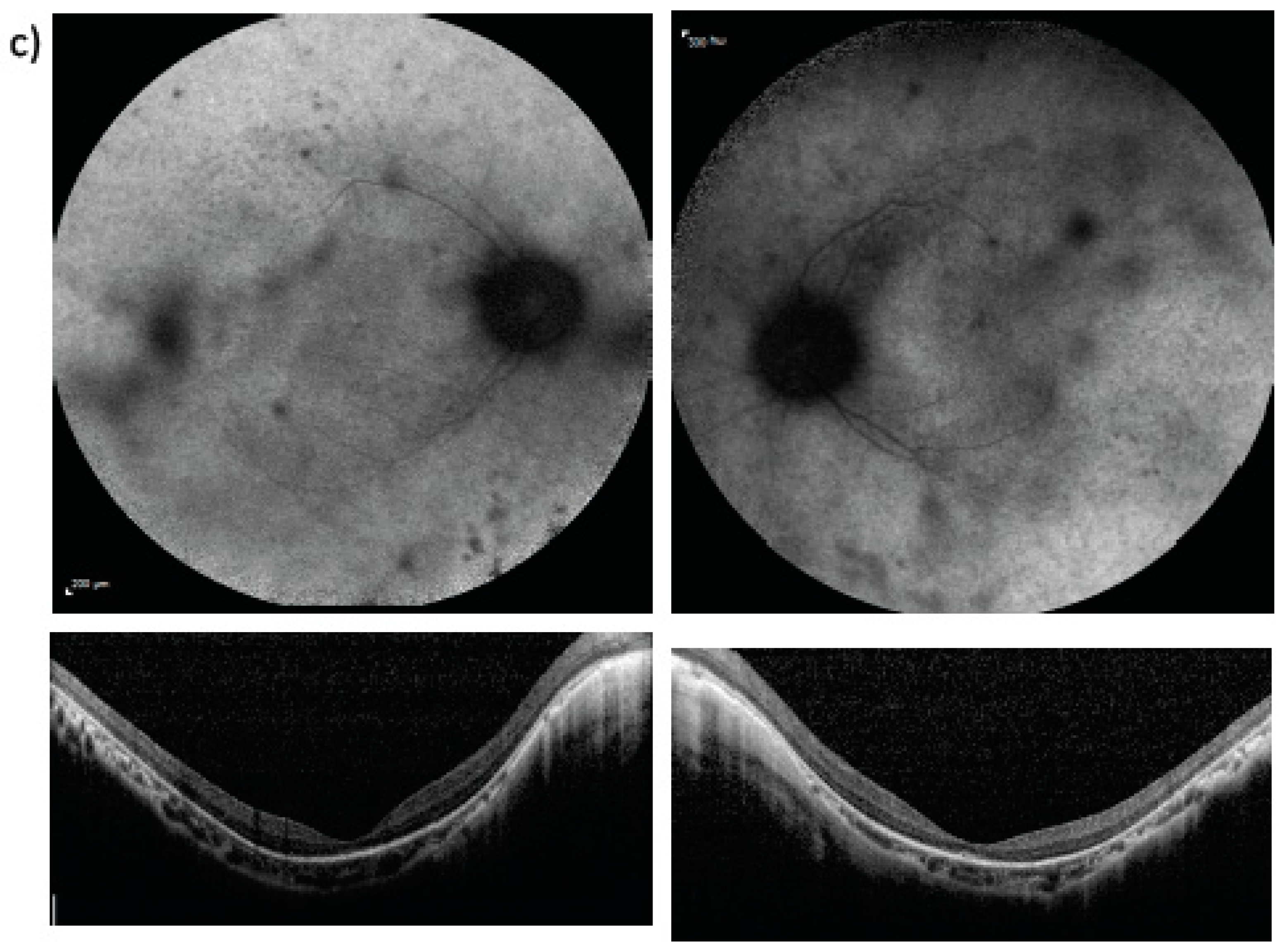
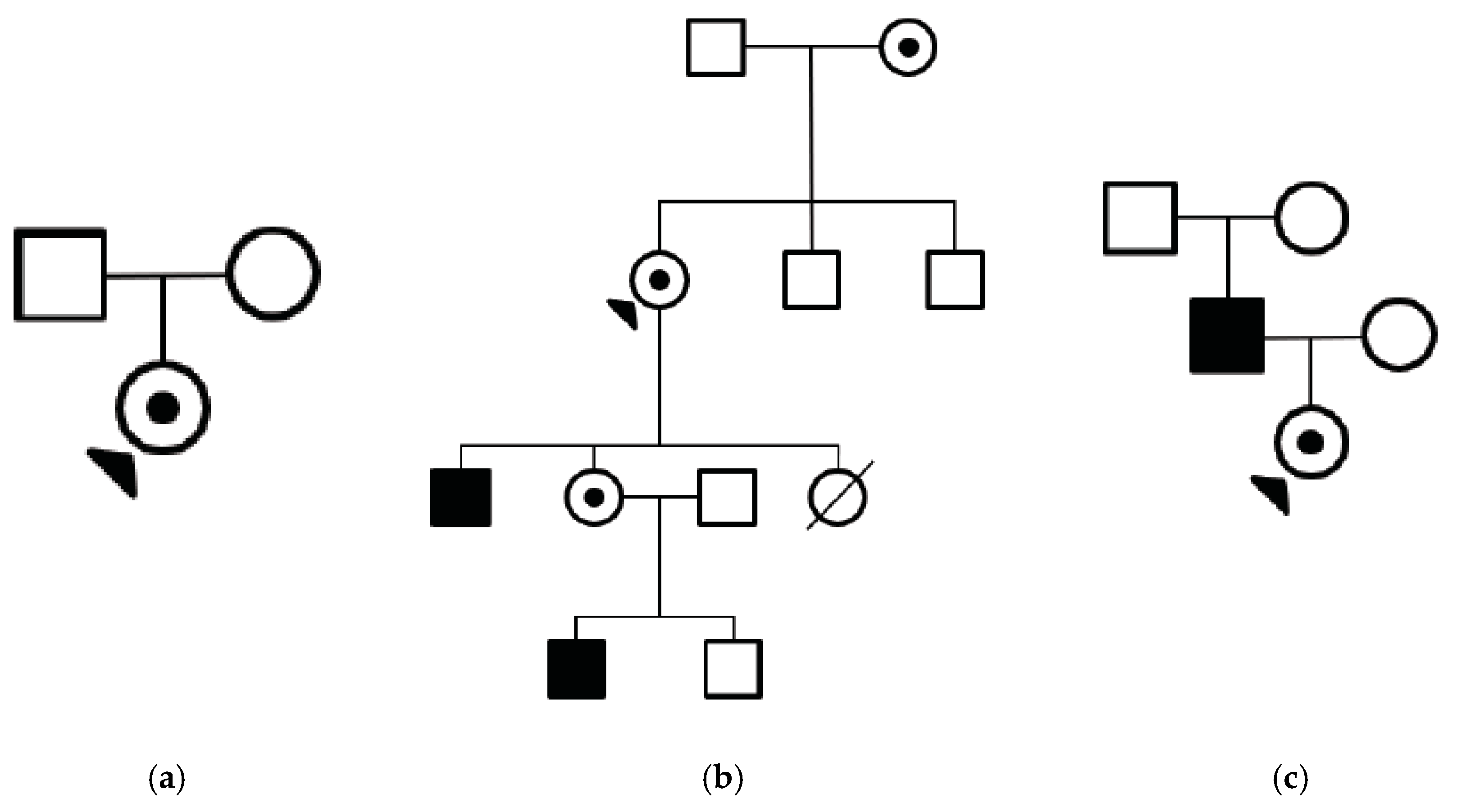
3. Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Tee, J.J.; Smith, A.J.; Hardcastle, A.J.; Michaelides, M. RPGR-associated retinopathy: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2016, 100, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Flaxel, C.; Jay, M.; Thiselton, D.; Nayudu, M.; Hardcastle, A.; Wright, A.; Bird, A. Difference between RP2 and RP3 phenotypes in X linked retinitis pigmentosa. Br. J. Ophthalmol. 1999, 83, 1144–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.C. X-linked retinitis pigmentosa. Br. J. Ophthalmol. 1975, 59, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.T.; Daiger, S.P. The role of X-chromosome inactivation in retinal development and disease. Adv. Exp. Med. Biol. 2016, 854, 325–331. [Google Scholar] [CrossRef]
- Churchill, J.D.; Bowne, S.J.; Sullivan, L.S.; Lewis, R.A.; Wheaton, D.K.; Birch, D.G.; Branham, K.E.; Heckenlively, J.R.; Daiger, S.P. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Investig. Ophthalmolo. Vis. Sci. 2013, 54, 1411–1416. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Yagasaki, K.; Feuer, W.J.; Roman, A.J. Interocular asymmetry of visual function in heterozygotes of X-linked retinitis pigmentosa. Exp. Eye Res. 1989, 48, 679–691. [Google Scholar] [CrossRef]
- Comander, J.; Weigel-DiFranco, C.; Sandberg, M.A.; Berson, E.L. Visual function in carriers of X-linked Retinitis Pigmentosa. Ophthalmology 2015, 122, 1899–1906. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; Van Cauwenbergh, C.; Wijnholds, J.; Ten Brink, J.B.; Florijn, R.J.; Schalij-Delfos, N.E.; Dagnelie, G.; van Genderen, M.M.; De Baere, E.; et al. The spectrum of structural and functional abnormalities in female carriers of pathogenic variants in the RPGR gene. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4123–4133. [Google Scholar] [CrossRef]
- Kalitzeos, A.; Samra, R.; Kasilian, M.; Tee, J.J.L.; Strampe, M.; Langlo, C.; Webster, A.R.; Dubra, A.; Carroll, J.; Michaelides, M. Cellular imaging of the tapetal-like reflex in carriers of RPGR-associated retinopathy. Retina 2017. [Google Scholar] [CrossRef]
- Stingl, K.; Bartz-Schmidt, K.U.; Besch, D.; Chee, C.K.; Cottriall, C.L.; Gekeler, F.; Groppe, M.; Jackson, T.L.; MacLaren, R.E.; Koitschev, A.; et al. Subretinal visual implant alpha IMS—Clinical trial interim report. Vis. Res. 2015, 111, 149–160. [Google Scholar] [CrossRef]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its role in photoreceptor physiology, human disease, and future therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Pullirsch, D.; Leeb, M.; Wutz, A. Xist and the order of silencing. EMBO Rep. 2007, 8, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Heard, E. Recent advances in X-chromosome inactivation. Curr. Opin. Cell Biol. 2004, 16, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Banin, E.; Mizrahi-Meissonnier, L.; Neis, R.; Silverstein, S.; Magyar, I.; Abeliovich, D.; Roepman, R.; Berger, W.; Rosenberg, T.; Sharon, D. A non-ancestral RPGR missense mutation in families with either recessive or semi-dominant X-linked retinitis pigmentosa. Am. J. Med. Genet. Part A 2007, 143a, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
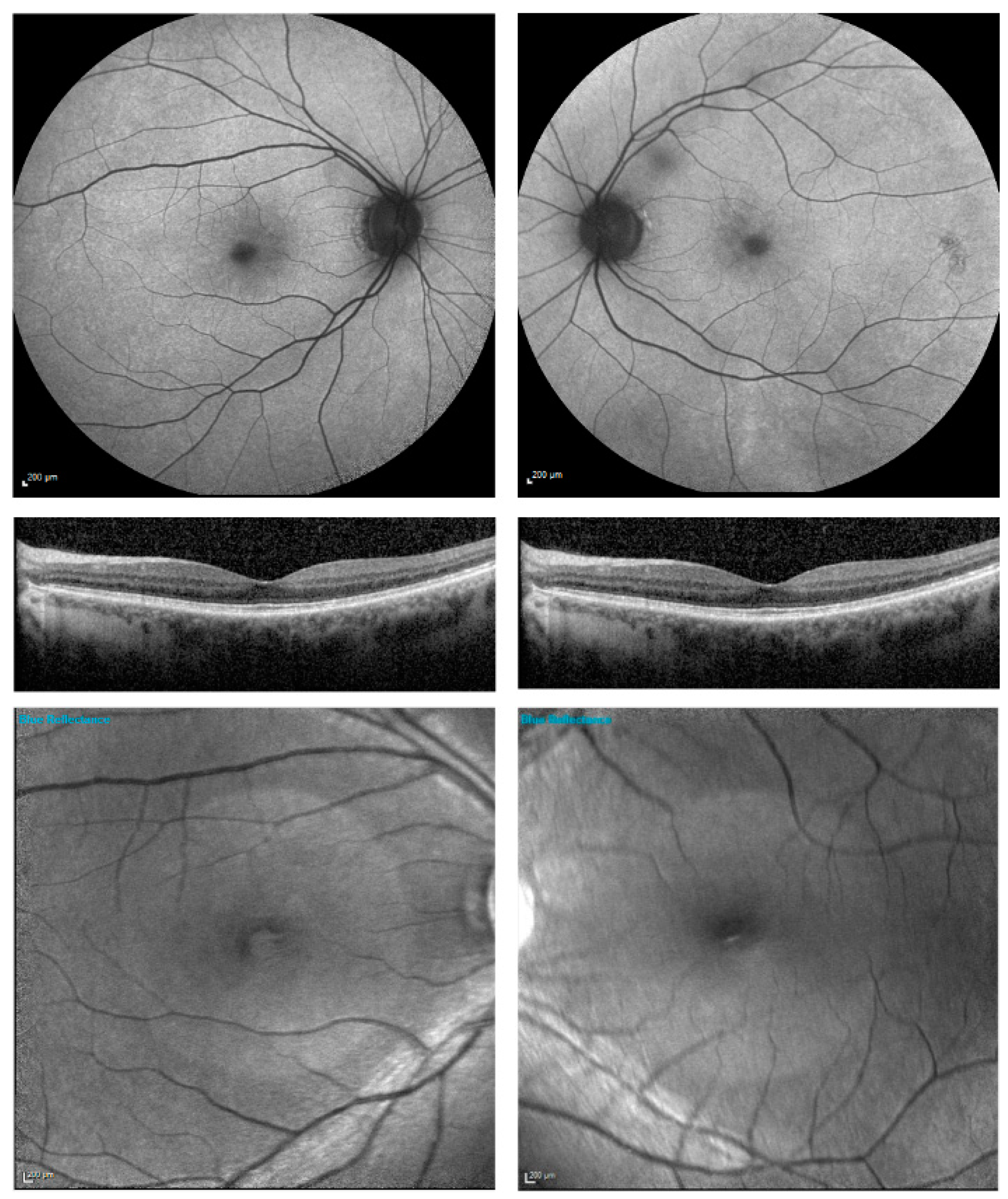
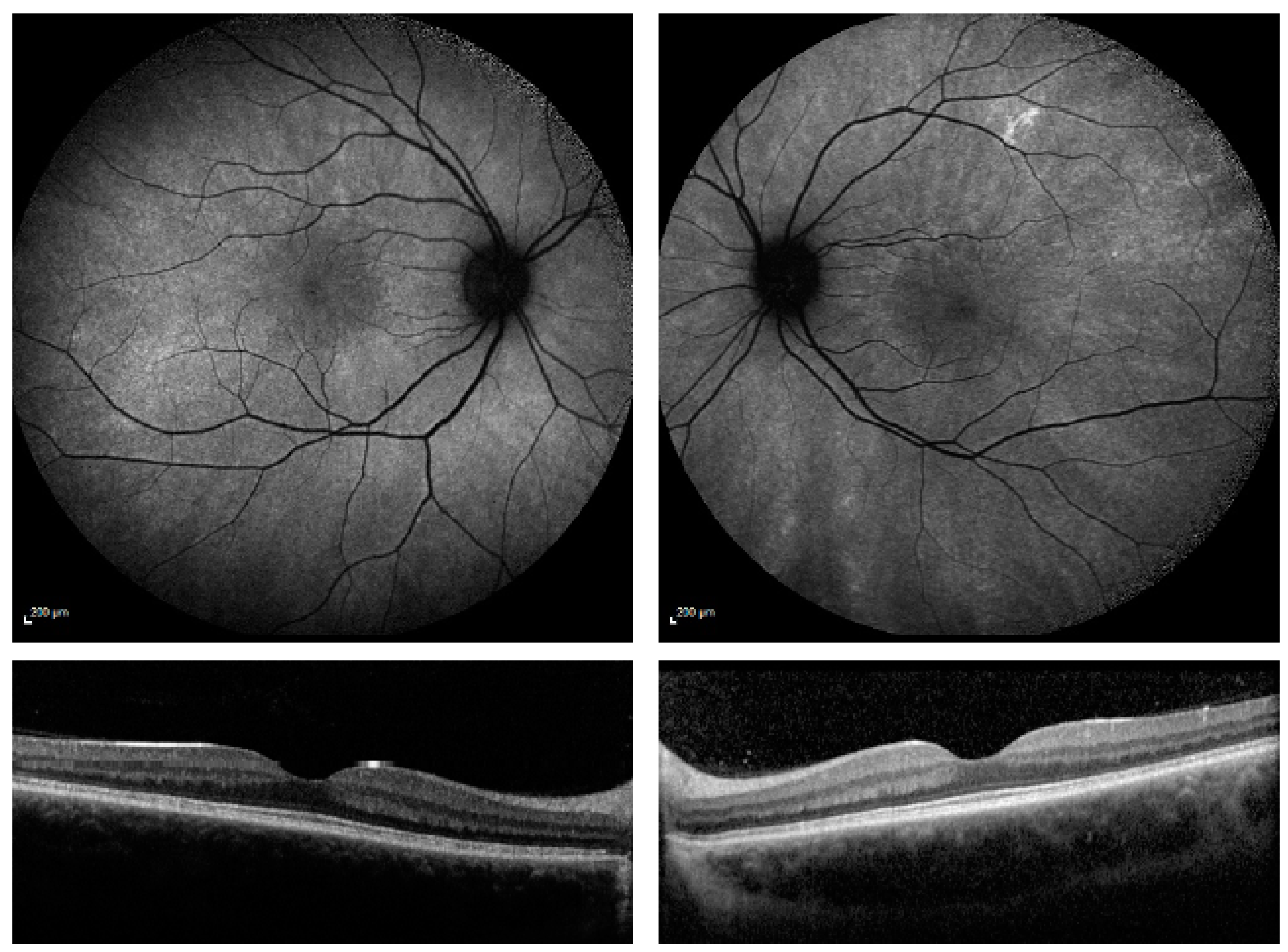
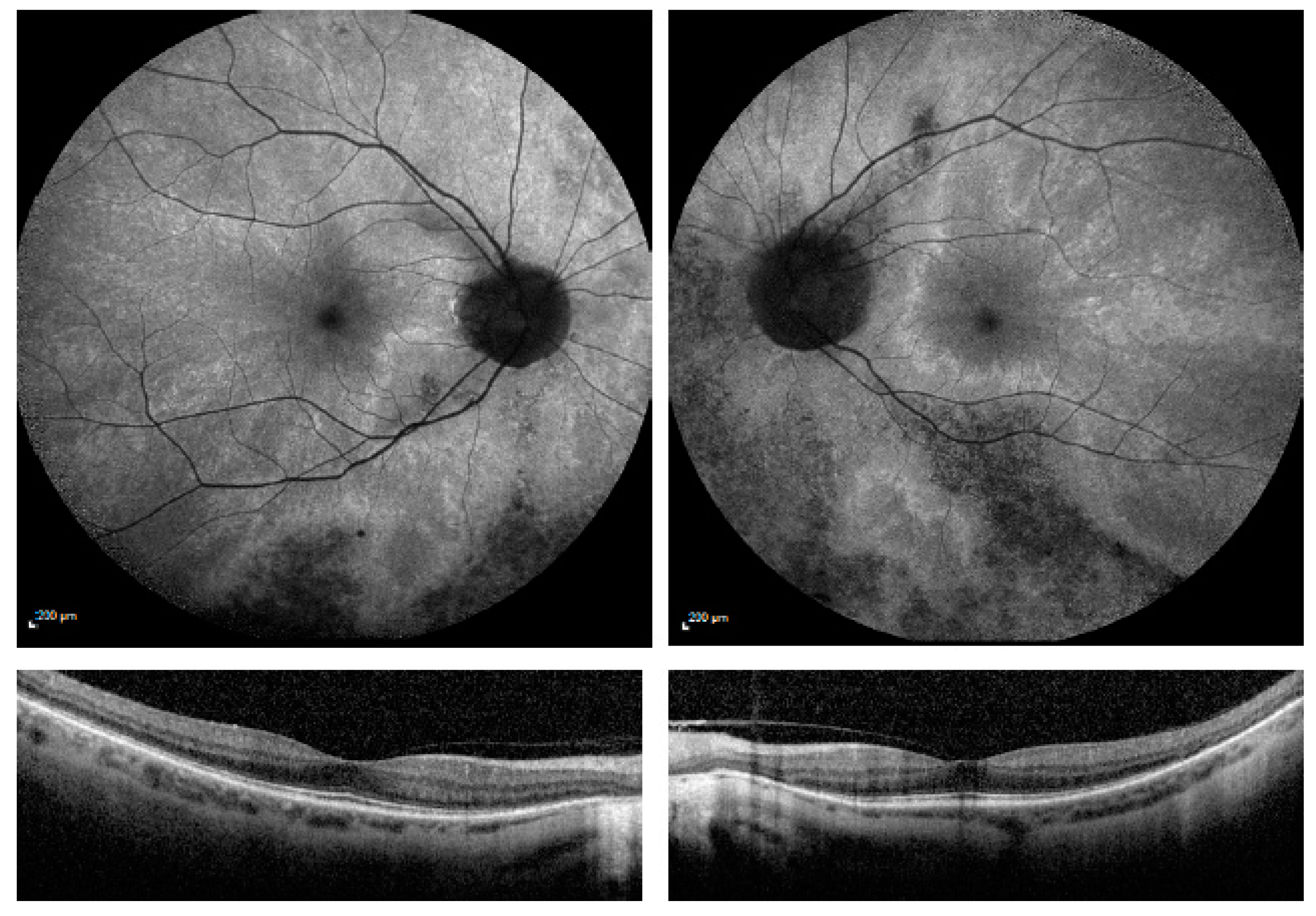
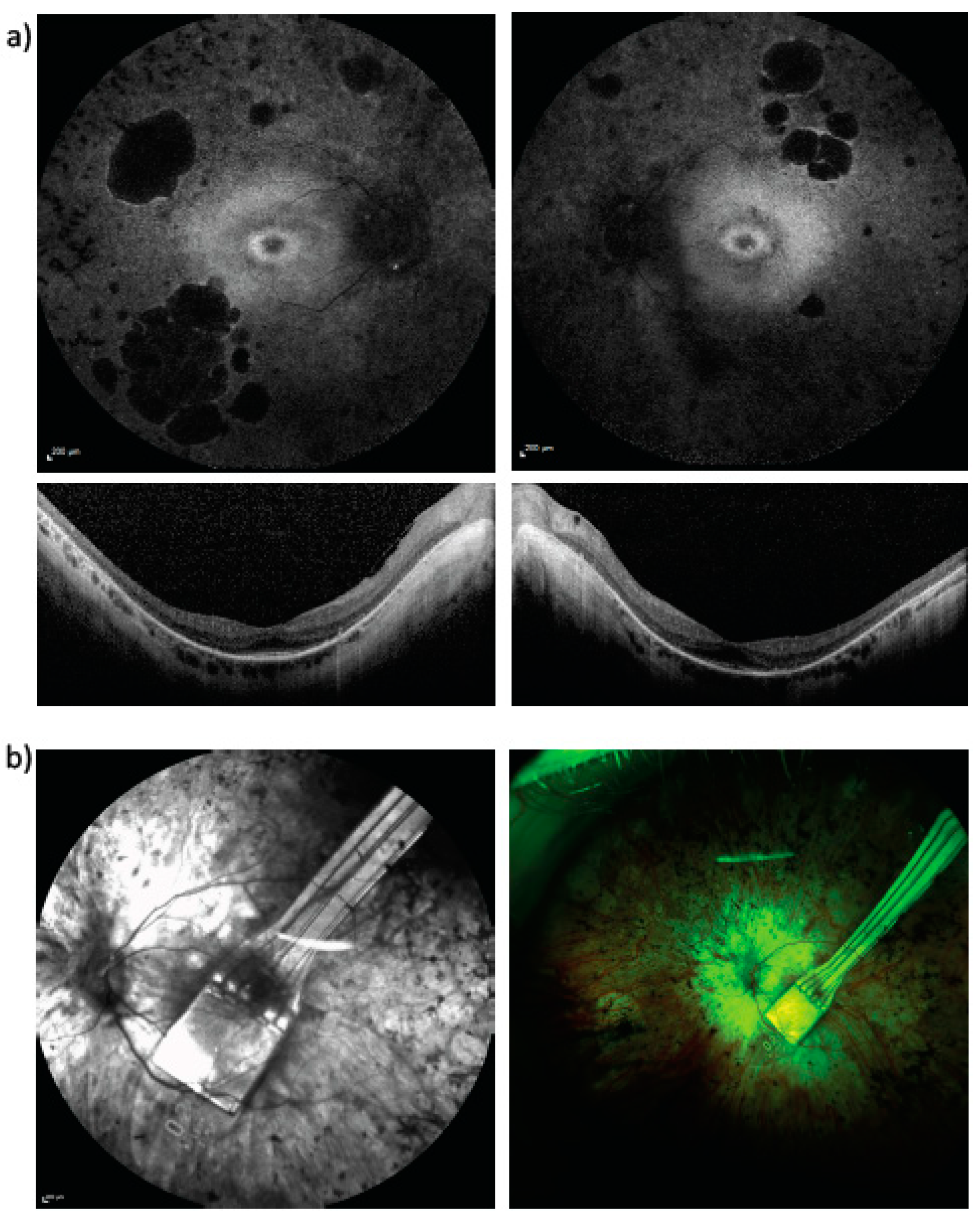
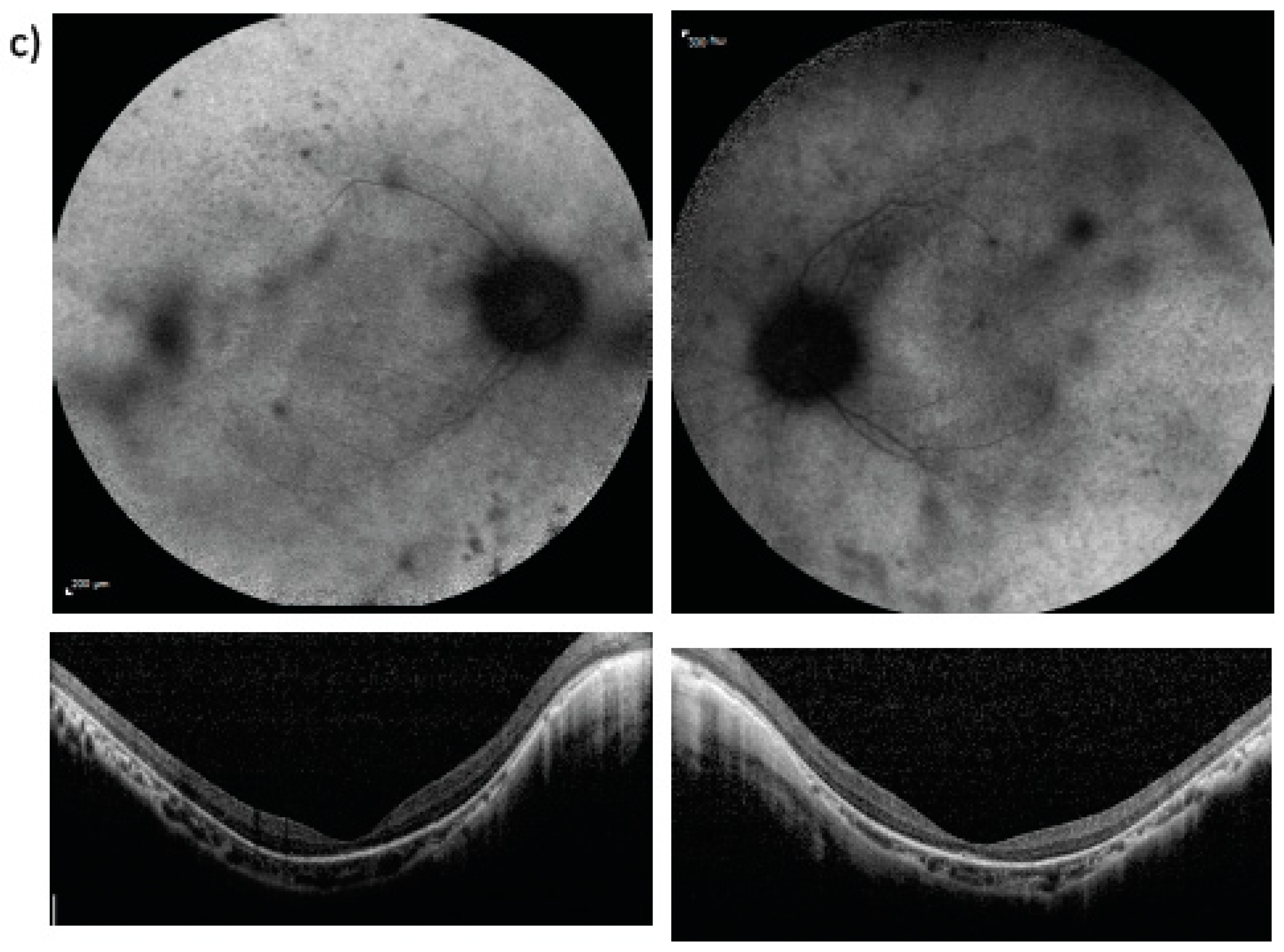
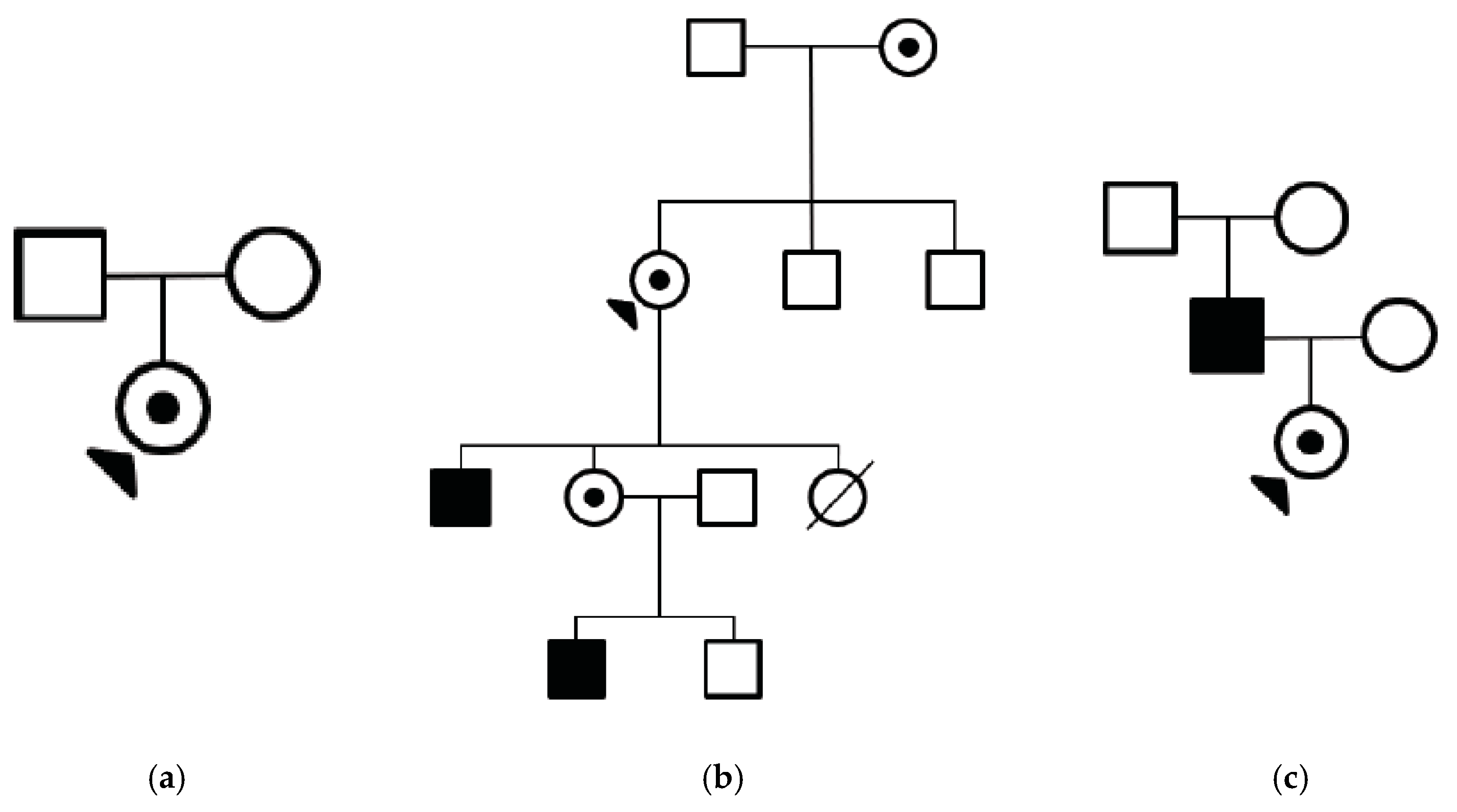

{kind=link}
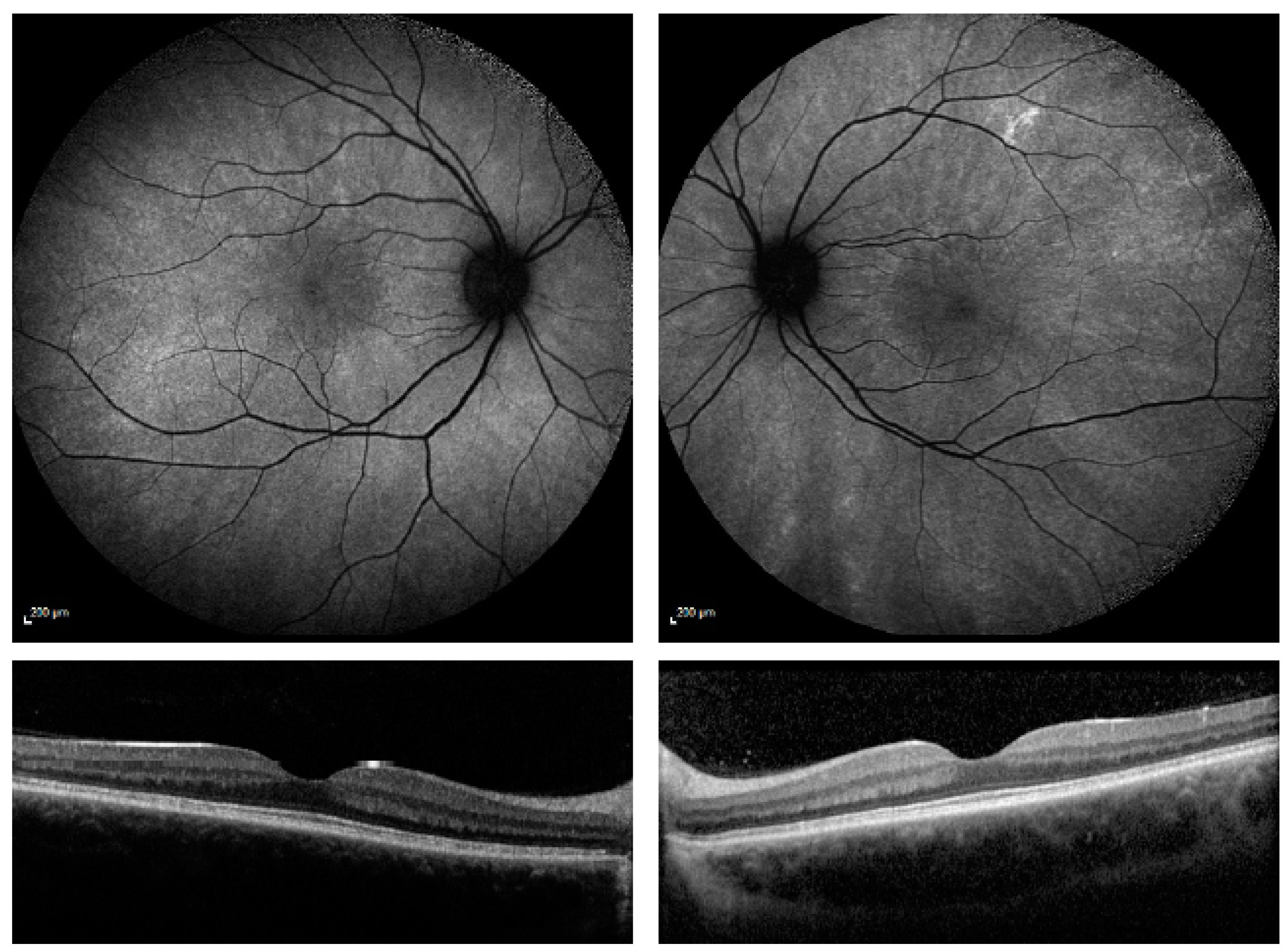{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Current Age | RPGR Mutation Confirmed in Male Relative Suffering with RPGR Retinitis Pigmentosa | RPGR Exon Location | Autofluorescence Imaging Category |
|---|---|---|---|---|
| 1 | 51 | c.581G > A (p.Trp194Ter) | 6 | Normal |
| 2 | 52 | c.779-5T > G | 8 | Radial |
| 3 | 26 | c.904T > G | 8 | Radial |
| 4 | 8 | c.1047delT | 10 | Radial |
| 5 | 8 | c.1047delT | 10 | Radial |
| 6 | 50 | c.1377_1378 | 11 | Radial |
| 7 | 57 | c.2405_2406delAG | ORF15 | Radial |
| 8 | 76 | c.3092delA | ORF15 | Radial |
| 9 | 30 | c.2993_2997delAAGGG | ORF15 | Radial |
| 10 | 55 | c.2426_2427 del AG | ORF15 | Radial |
| 11 | 64 | c.2628_2629delGG | ORF15 | Radial |
| 12 | 39 | c.3178_3179delGA | ORF15 | Radial |
| 13 | 21 | c.2426_2427delAG | ORF15 | Radial |
| 14 | 26 | c.2650G > T | ORF15 | Radial |
| 15 | 60 | c.2405_2406delAG | ORF15 | Radial |
| 16 | 43 | c.215delA | 3 | Focal |
| 17 | 50 | c.408delT | 5 | Focal |
| 18 | 52 | c.581G > A | 6 | Focal |
| 19 | 64 | c.2426_2427delAG | ORF15 | Focal |
| 20 | 48 | c.2993_2997delAAGGG | ORF15 | Focal |
| 21 | 47 | c.1379T > A | 11 | Male |
| 22 | 60 | c.1571delA | 13 | Male |
| 23 | 21 | c.2405_2406delAG | ORF15 | Male |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanda, A.; Salvetti, A.P.; Clouston, P.; Downes, S.M.; MacLaren, R.E. Exploring the Variable Phenotypes of RPGR Carrier Females in Assessing Their Potential for Retinal Gene Therapy. Genes 2018, 9, 643. https://doi.org/10.3390/genes9120643
Nanda A, Salvetti AP, Clouston P, Downes SM, MacLaren RE. Exploring the Variable Phenotypes of RPGR Carrier Females in Assessing Their Potential for Retinal Gene Therapy. Genes. 2018; 9(12):643. https://doi.org/10.3390/genes9120643
Chicago/Turabian StyleNanda, Anika, Anna P. Salvetti, Penny Clouston, Susan M. Downes, and Robert E. MacLaren. 2018. "Exploring the Variable Phenotypes of RPGR Carrier Females in Assessing Their Potential for Retinal Gene Therapy" Genes 9, no. 12: 643. https://doi.org/10.3390/genes9120643
APA StyleNanda, A., Salvetti, A. P., Clouston, P., Downes, S. M., & MacLaren, R. E. (2018). Exploring the Variable Phenotypes of RPGR Carrier Females in Assessing Their Potential for Retinal Gene Therapy. Genes, 9(12), 643. https://doi.org/10.3390/genes9120643