The Genome of the North American Brown Bear or Grizzly: Ursus arctos ssp. horribilis

 , ,
, ,
Abstract
1. Introduction
2. Methods
3. Results and Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, C.R.; Waits, L.P.; Joyce, P. Phylogeography and mitochondrial diversity of extirpated brown bear (Ursus arctos) populations in the contiguous United States and Mexico. Mol. Ecol. 2006, 15, 4477–4485. [Google Scholar] [CrossRef] [PubMed]
- Gyug, L.; Hamilton, T.; Austin, M. Grizzly bear (Ursus Arctos). Accounts and Measures for Managing Identified Wildlife–Accounts V2004; Ministry of Water, Land and Air Protection British Columbia: National Library of Canada: BC, Canada, 2004. [Google Scholar]
- Jones, S.J.M.; Taylor, G.A.; Chan, S.; Warren, R.L.; Hammond, S.A.; Bilobram, S.; Mordecai, G.; Suttle, C.A.; Miller, K.M.; Schulze, A.; et al. The genome of the Beluga Whale (Delphinapterus leucas). Genes 2017, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.J.; Haulena, M.; Taylor, G.A.; Chan, M.; Bilobram, S.; Warren, R.L.; Hammond, S.A.; Mungall, K.L.; Choo, C.; Kirk, H.; et al. The genome of the Northern Sea Otter (Enhydra lutris kenyoni). Genes 2017, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.D.; Coombe, L.; Chu, J.; Warren, R.L.; Vandervalk, B.P.; Yeo, S.; Xue, Z.; Mohamadi, H.; Bohlmann, J.; Jones, S.J.M.; Birol, I. Tigmint: Correcting assembly errors using linked reads from large molecules. BMC Bioinform. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.; Coombe, L.; Warren, R.L.; Birol, I. ARCS: Scaffolding genome drafts with linked reads. Bioinformatics 2018, 34, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.L.; Yang, C.; Vandervalk, B.P.; Behsaz, B.; Lagman, A.; Jones, S.J.M.; Birol, I. LINKS: Scalable, alignment-free scaffolding of draft genomes with long reads. Gigascience 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Paulino, D.; Warren, R.L.; Vandervalk, B.P.; Raymond, A.; Jackman, S.D.; Birol, I. Sealer: A scalable gap-closing application for finishing draft genomes. BMC Bioinform. 2015, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Brown, G.R.; Hiatt, S.M.; Thibaud-Nissen, F.; Astashyn, A.; Ermolaeva, O.; Farrell, C.M.; Hart, J.; Landrum, M.J.; McGarvey, K.M.; et al. RefSeq: An update on mammalian reference sequences. Nucleic Acids Res. 2014, 42, D756–763. [Google Scholar] [CrossRef] [PubMed]
- Wurster-Hill, D.H.; Bush, M. The interrelationship of chromosome banding patterns in the giant panda (Ailuropoda melanoleuca), hybrid bear (Ursus middendorfi X Thalarctos maritimus), and other carnivores. Cytogenet. Cell Genet. 1980, 27, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Nash, W.G.; Wienberg, J.; Ferguson-Smith, M.A.; Menninger, J.C.; O’Brien, S.J. Comparative genomics: Tracking chromosome evolution in the family Ursidae using reciprocal chromosome painting. Cytogenet. Cell Genet. 1998, 83, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lorenzen, E.D.; Fumagalli, M.; Li, B.; Harris, K.; Xiong, Z.; Zhou, L.; Korneliussen, T.S.; Somel, M.; Babbitt, C.; et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 2014, 157, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Chu, J. Jupiter Plot: A Circos-Based Tool to Visualize Genome Assembly Consistency (Version 1.0). Zenodo. Available online: https://zenodo.org/record/1241235#.XA92q2hKiUk (accessed on 15 November 2018). [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]


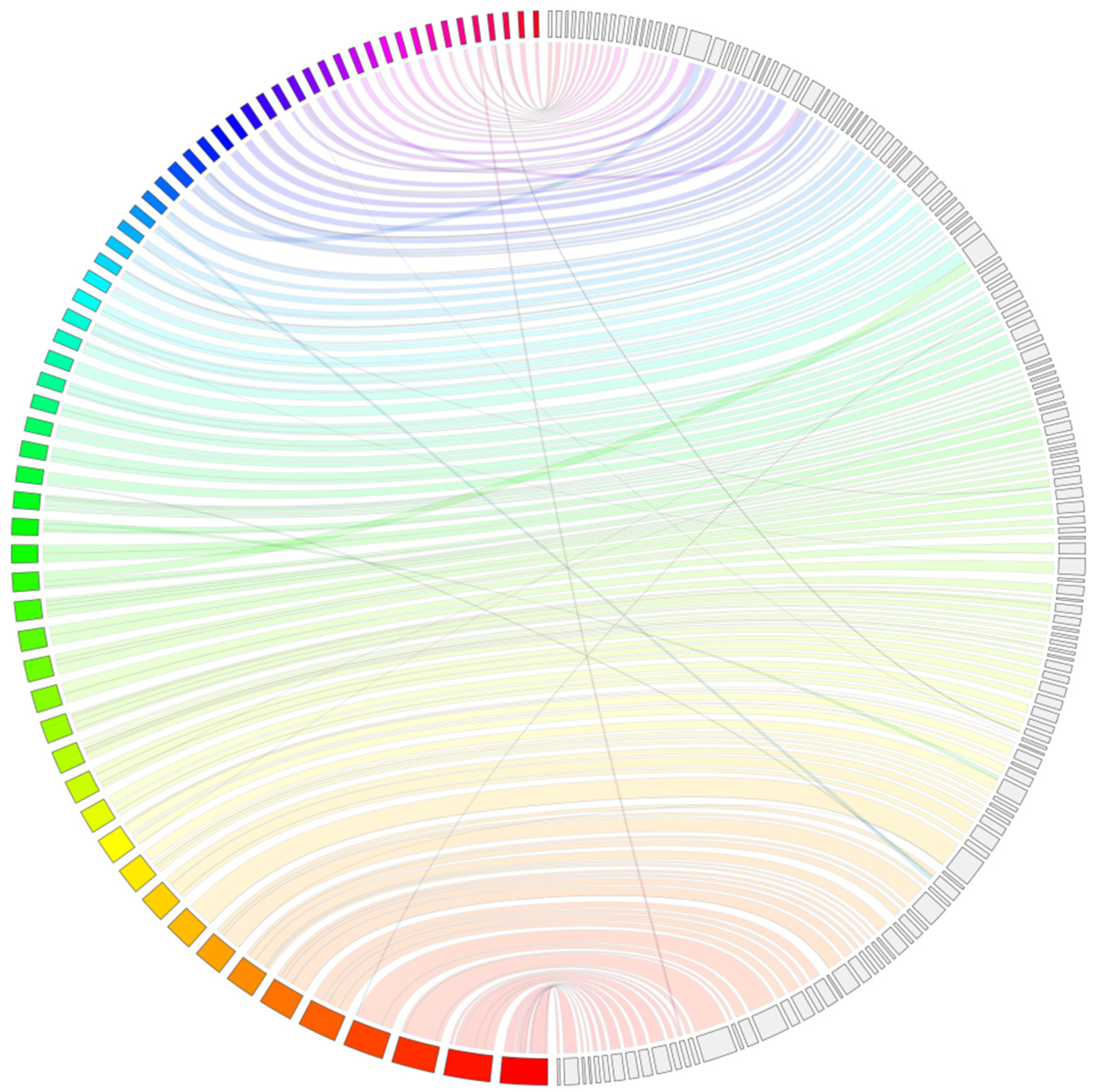{kind=link}
| Assembly | # of Scaffolds | Gaps within Scaffolds | Scaffold N50 (bp) | Longest Scaffold (bp) | BUSCO Complete Genes (of 4104) |
|---|---|---|---|---|---|
| Supernova | 8474 | 21,957 | 33.78 × 106 | 105.9 × 106 | 3943 (96.1%) |
| Tigmint | 8728 | 21,947 | 26.32 × 106 | 92.41 × 106 | 3943 (96.1%) |
| ARCS | 8679 | 21,996 | 27.77 × 106 | 92.41 × 106 | 3943 (96.1%) |
| LINKS1 | 8350 | 22,219 | 27.77 × 106 | 92.41 × 106 | 3943 (96.1%) |
| LINKS8 | 6673 | 23,947 | 36.71 × 106 | 92.42 × 106 | 3943 (96.1%) |
| Sealer | 6673 | 15,572 | 36.71 × 106 | 92.43 × 106 | 3943 (96.1%) |
| Assembly | # of Scaffolds | Scaffold N50 (bp) | Scaffold L50 | # of Contigs | Contig N50 (bp) | Contig L50 | BUSCO Complete Genes |
|---|---|---|---|---|---|---|---|
| Grizzly Bear | 6673 | 36.71 × 106 | 21 | 22,245 | 314 × 103 | 2191 | 3943 (96.1%) |
| Polar Bear | 23,819 | 15.94 × 106 | 46 | 134,162 | 46 × 103 | 14,124 | 3890 (94.7%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, G.A.; Kirk, H.; Coombe, L.; Jackman, S.D.; Chu, J.; Tse, K.; Cheng, D.; Chuah, E.; Pandoh, P.; Carlsen, R.; et al. The Genome of the North American Brown Bear or Grizzly: Ursus arctos ssp. horribilis. Genes 2018, 9, 598. https://doi.org/10.3390/genes9120598
Taylor GA, Kirk H, Coombe L, Jackman SD, Chu J, Tse K, Cheng D, Chuah E, Pandoh P, Carlsen R, et al. The Genome of the North American Brown Bear or Grizzly: Ursus arctos ssp. horribilis. Genes. 2018; 9(12):598. https://doi.org/10.3390/genes9120598
Chicago/Turabian StyleTaylor, Gregory A., Heather Kirk, Lauren Coombe, Shaun D. Jackman, Justin Chu, Kane Tse, Dean Cheng, Eric Chuah, Pawan Pandoh, Rebecca Carlsen, and et al. 2018. "The Genome of the North American Brown Bear or Grizzly: Ursus arctos ssp. horribilis" Genes 9, no. 12: 598. https://doi.org/10.3390/genes9120598
APA StyleTaylor, G. A., Kirk, H., Coombe, L., Jackman, S. D., Chu, J., Tse, K., Cheng, D., Chuah, E., Pandoh, P., Carlsen, R., Zhao, Y., Mungall, A. J., Moore, R., Birol, I., Franke, M., Marra, M. A., Dutton, C., & Jones, S. J. M. (2018). The Genome of the North American Brown Bear or Grizzly: Ursus arctos ssp. horribilis. Genes, 9(12), 598. https://doi.org/10.3390/genes9120598