Modular Proteoglycan Perlecan/HSPG2: Mutations, Phenotypes, and Functions



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
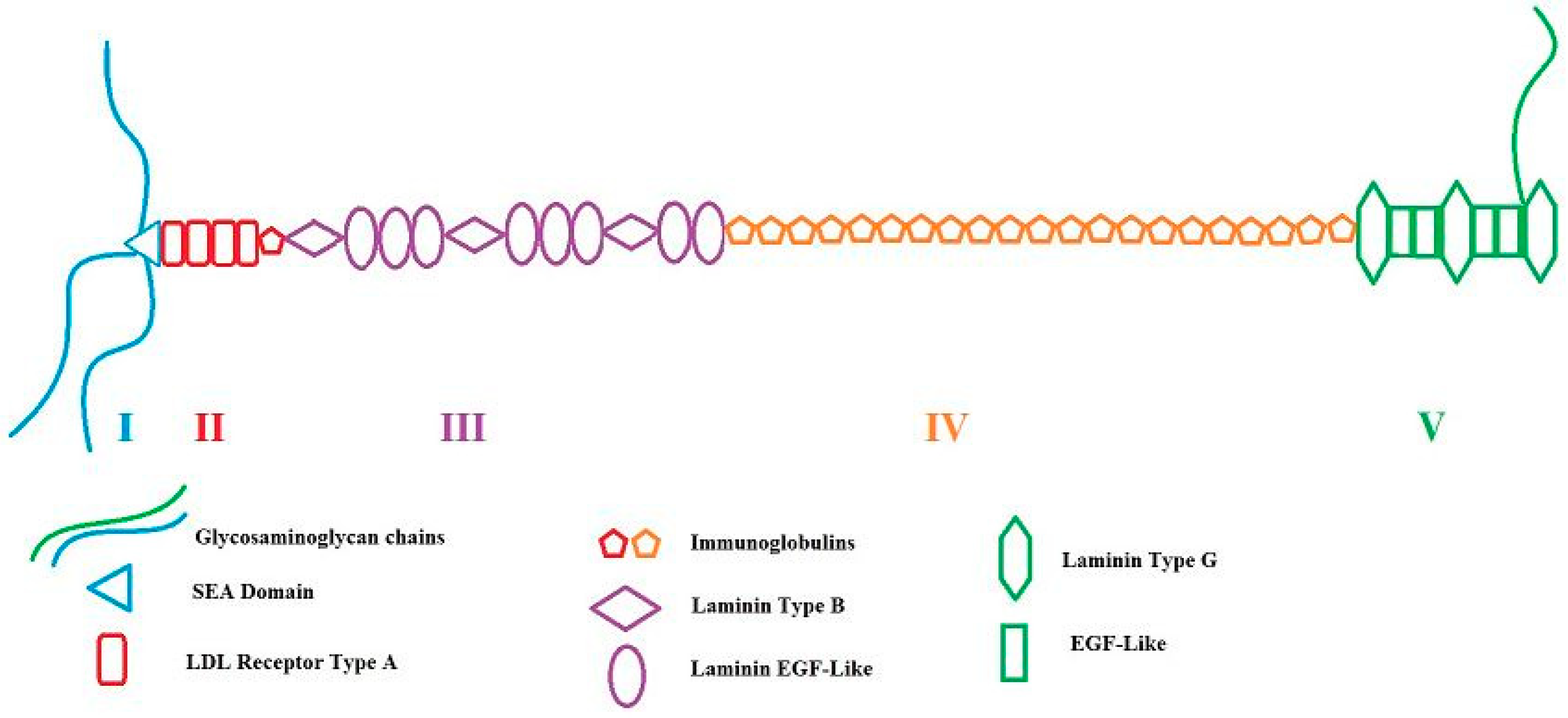
2. HSPG2/Perlecan: Gene and Proteoglycan
3. Conservation of HSPG2
4. HSPG2 Variants and Homologues
5. Developmental Expression of Perlecan
6. HSPG2 Associated Skeletal Defects
7. Mechanisms Underlying Perlecan Developmental Defects
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nicole, S.; Davoine, C.-S.; Topaloglu, H.; Cattolico, L.; Barral, D.; Beighton, P.; Hamida, C.B.; Hammouda, H.; Cruaud, C.; White, P.S.; et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat. Genet. 2000, 26, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Arikawa-Hirasawa, E.; Wilcox, W.R.; Le, A.H.; Silverman, N.; Govindraj, P.; Hassell, J.R.; Yamada, Y. Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat. Genet. 2001, 27, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Ito, M.; Nakata, T.; Noguchi, Y.; Okuno, T.; Ohkawara, B.; Masuda, A.; Goto, T.; Adachi, M.; Osaka, H.; et al. A missense mutation in domain III in HSPG2 in Schwartz-Jampel syndrome compromises secretion of perlecan into the extracellular space. Neuromuscul. Disord. 2015, 25, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Bauché, S.; Boerio, D.; Davoine, C.-S.; Bernard, V.; Stum, M.; Bureau, C.; Fardeau, M.; Romero, N.B.; Fontaine, B.; Koenig, J.; et al. Peripheral nerve hyperexcitability with preterminal nerve and neuromuscular junction remodeling is a hallmark of Schwartz-Jampel syndrome. Neuromuscul. Disord. 2013, 23, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Stum, M.; Davoine, C.-S.; Vicart, S.; Guillot-Noël, L.; Topaloglu, H.; Carod-Artal, F.J.; Kayserili, H.; Hentati, F.; Merlini, L.; Urtizberea, J.A.; et al. Spectrum of HSPG2 (Perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum. Mutat. 2006, 27, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Arikawa-Hirasawa, E.; Le, A.H.; Nishino, I.; Nonaka, I.; Ho, N.C.; Francomano, C.A.; Govindraj, P.; Hassell, J.R.; Devaney, J.M.; Spranger, J.; et al. Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am. J. Hum. Genet. 2002, 70, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Rieubland, C.; Jacquemont, S.; Mittaz, L.; Osterheld, M.-C.; Vial, Y.; Superti-Furga, A.; Unger, S.; Bonafé, L. Phenotypic and molecular characterization of a novel case of dyssegmental dysplasia, Silverman-Handmaker type. Eur. J. Med. Genet. 2010, 53, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Das Bhowmik, A.; Dalal, A.; Matta, D.; Kandadai, R.M.; Kanikannan, M.A.; Aggarwal, S. Identification of a novel splice site HSPG2 mutation and prenatal diagnosis in Schwartz Jampel Syndrome type 1 using whole exome sequencing. Neuromuscul. Disord. 2016, 26, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.D.; Sasaki, T.; Aszodi, A.; Jacenko, O. Reduced perlecan in mice results in chondrodysplasia resembling Schwartz-Jampel syndrome. Hum. Mol. Genet. 2007, 16, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Pierce, M.; Datta, M.W. Perlecan signaling: Helping hedgehog stimulate prostate cancer growth. Int. J. Biochem. Cell Biol. 2006, 38, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Conde-Knape, K. Heparan sulfate proteoglycans in experimental models of diabetes: A role for perlecan in diabetes complications. Diabetes Metab. Res. Rev. 2001, 17, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wang, X.L.; Wilcken, D.E. Genetic polymorphism of heparan sulfate proteoglycan (Perlecan, HSPG2), lipid profiles and coronary artery disease in the Australian population. Atherosclerosis 2000, 148, 125–129. [Google Scholar] [CrossRef]
- Lepelletier, F.-X.; Mann, D.M.A.; Robinson, A.C.; Pinteaux, E.; Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Farach-Carson, M.C.; Carson, D.D. Perlecan—A multifunctional extracellular proteoglycan scaffold. Glycobiology 2007, 17, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Wintle, R.F.; Kisilevsky, R.; Noonan, D.; Duncan, A.M.V. In situ hybridization to human chromosome 1 of a cDNA probe for the gene encoding the basement membrane heparan sulfate proteoglycan (HSPG). Cytogenet. Genome Res. 1990, 54, 60–61. [Google Scholar] [CrossRef] [PubMed]
- Dodge, G.R.; Kovalszky, I.; Chu, M.L.; Hassell, J.R.; McBride, O.W.; Yi, H.F. Heparan sulfate proteoglycan of human colon: Partial molecular cloning, cellular expression, and mapping of the gene (HSPG2) to the short arm of human chromosome 1. Genomics 1991, 10, 673–680. Available online: http://www.ncbi.nlm.nih.gov/pubmed/1679749 (accessed on 7 September 2018). [CrossRef]
- Kallunki, P.; Eddy, R.L.; Byers, M.G.; Kestilä, M.; Shows, T.B.; Tryggvason, K. Cloning of human heparan sulfate proteoglycan core protein, assignment of the gene (HSPG2) to 1p36.1→p35 and identification of a BamHI restriction fragment length polymorphism. Genomics 1991, 11, 389–396. [Google Scholar] [CrossRef]
- Cohen, I.R.; Grassel, S.; Murdoch, A.D.; Iozzo, R.V. Structural Characterization of the Complete Human Perlecan Gene and Its Promoter (Heparan Sulfate/Proteoglycan/Basement Membrane). 1993. Available online: http://www.pnas.org/content/pnas/90/21/10404.full.pdf (accessed on 7 September 2018).
- Murdoch, A.D.; Dodge, G.R.; Cohen, I.; Tuan, R.S.; Iozzo, R.V. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growt. J. Biol. Chem. 1992, 267, 8544–8557. Available online: http://www.jbc.org/content/267/12/8544.short (accessed on 12 February 2016). [PubMed]
- Graham, L.D.; Whitelock, J.M.; Underwood, P.A. Expression of human perlecan domain I as a recombinant heparan sulfate proteoglycan with 20-kDa glycosaminoglycan chains. Biochem. Biophys. Res. Commun. 1999, 256, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Hassell, J.R.; Robeyt, P.G.; Barracht, H.-J.; Wilczekt, J.; Rennardt, S.I.; Martint, G.R. Isolation of a Heparan Sulfate-Containing Proteoglycan from Basement Membrane (Glycosaminoglyeans/Transplantable Tumor/Extraceliular Matrix/Lamina Lucida/Anionic Component). 1980. Available online: http://www.pnas.org/content/pnas/77/8/4494.full.pdf (accessed on 7 September 2018).
- Paulsson, M.; Yurchenco, P.D.; Ruben, G.C.; Engel, J.; Timpl, R. Structure of low density heparan sulfate proteoglycan isolated from a mouse tumor basement membrane. J. Mol. Biol. 1987, 197, 297–313. [Google Scholar] [CrossRef]
- Dolan, M.; Horchar, T.; Rigatti, B.; Hassell, J.R. Identification of Sites in Domain I of Perlecan That Regulate Heparan Sulfate Synthesis. J. Biol. Chem. 1997, 272, 4316–4322. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Patthy, L. The SEA module: A new extracellular domain associated with O-glycosylation. Protein Sci. 1995, 4, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Whitelock, J.M.; Graham, L.D.; Melrose, J.; Murdoch, A.D.; Iozzo, R.V.; Anne Underwood, P. Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 1999, 18, 163–178. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, J.; Cao, R.; Morita, H.; Soininen, R.; Chan, K.M.; Liu, B.; Cao, Y.; Tryggvason, K. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004, 64, 4699–4702. [Google Scholar] [CrossRef] [PubMed]
- Whitelock, J.M.; Murdoch, A.D.; Iozzo, R.V.; Underwood, P.A. The Degradation of Human Endothelial Cell-derived Perlecan and Release of Bound Basic Fibroblast Growth Factor by Stromelysin, Collagenase, Plasmin, and Heparanases. J. Biol. Chem. 1996, 271, 10079–10086. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Yang, Y.; Kelly, T.; Macleod, V.; Dai, Y.; Theus, A. Enzymatic Remodeling of Heparan Sulfate Proteoglycans within the Tumor Microenvironment: Growth Regulation and the Prospect of New Cancer Therapies. J. Cell. Biochem. 2005, 96, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Costell, M.; Sasaki, T.; Mann, K.; Yamada, Y.; Timpl, R. Structural characterization of recombinant domain II of the basement membrane proteoglycan perlecan. FEBS Lett. 1996, 396, 127–131. [Google Scholar] [CrossRef]
- Kamimura, K.; Ueno, K.; Nakagawa, J.; Hamada, R.; Saitoe, M.; Maeda, N. Perlecan regulates bidirectional Wnt signaling at the Drosophila neuromuscular junction. J. Cell Biol. 2013, 200, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Mann, K.; Battistutta, R.; Wiedemann, H.; Timpl, R. Structural properties of recombinant domain III-3 of perlecan containing a globular domain inserted into an epidermal-growth-factor-like motif. Eur. J. Biochem. 1995, 231, 551–556. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7649154 (accessed on 12 February 2016). [CrossRef] [PubMed]
- Smith, S.M.-L.; West, L.A.; Hassell, J.R. The core protein of growth plate perlecan binds FGF-18 and alters its mitogenic effect on chondrocytes. Arch. Biochem. Biophys. 2007, 468, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Hopf, M.; Göhring, W.; Kohfeldt, E.; Yamada, Y.; Timpl, R. Recombinant domain IV of perlecan binds to nidogens, laminin-nidogen complex, fibronectin, fibulin-2 and heparin. Eur. J. Biochem. 1999, 259, 917–925. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10092882 (accessed on 8 September 2016). [CrossRef] [PubMed]
- Improta, S.; Politou, A.S.; Pastore, A. Immunoglobulin-like modules from titin I-band: Extensible components of muscle elasticity. Structure 1996, 4, 323–337. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8805538 (accessed on 12 February 2016). [CrossRef]
- Wijeratne, S.S.; Martinez, J.R.; Grindel, B.J.; Frey, E.W.; Li, J.; Wang, L.; Farach-Carson, M.C.; Kiang, C.H. Single molecule force measurements of perlecan/HSPG2: A key component of the osteocyte pericellular matrix. Matrix Biol. 2015, 50, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Sweeney, S.M.; San Antonio, J.D.; Fu, J.; Iozzo, R.V. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 2003, 278, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, J.J.; Whitelock, J.M.; Iozzo, R.V. Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. 2009, 28, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Douglass, S.; Goyal, A.; Iozzo, R.V. The role of perlecan and endorepellin in the control of tumor angiogenesis and endothelial cell autophagy. Connect. Tissue Res. 2015, 56, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, A.; Bix, G. Investigating the role of perlecan domain V in post-ischemic cerebral angiogenesis. Methods Mol. Biol. 2014, 1135, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Grindel, B.J.; Martinez, J.R.; Tellman, T.V.; Harrington, D.A.; Zafar, H.; Nakhleh, L.; Chung, L.W.; Farach-Carson, M.C. Matrilysin/MMP-7 Cleavage of Perlecan/HSPG2 Complexed with Semaphorin 3A Supports FAK-Mediated Stromal Invasion by Prostate Cancer Cells. Sci. Rep. 2018, 8, 7262. [Google Scholar] [CrossRef] [PubMed]
- Knox, S.; Fosang, A.J.; Last, K.; Melrose, J.; Whitelock, J. Perlecan from human epithelial cells is a hybrid heparan/chondroitin/keratan sulfate proteoglycan. FEBS Lett. 2005, 579, 5019–5023. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.P.; Trumble, T.N.; Plaas, A.H.K.; Sandy, J.D.; Romano, M.; Hernandez, J.; Merritt, K.A. Exercise and injury increase chondroitin sulfate chain length and decrease hyaluronan chain length in synovial fluid. Osteoarthr. Cartil. 2007, 15, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.J.; Gardner, D.L. Changes with age in the glycosaminoglycans of human articular cartilage. Ann. Rheum. Dis. 1979, 38, 371–377. Available online: http://www.ncbi.nlm.nih.gov/pubmed/496451 (accessed on 2 November 2018). [CrossRef] [PubMed]
- Knox, S.; Merry, C.; Stringer, S.; Melrose, J.; Whitelock, J. Not All Perlecans Are Created Equal. J. Biol. Chem. 2002, 277, 14657–14665. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.R.; Kassir, E.; Spurlin, J.; Martinez, J.; Putnam, N.H.; Farach-Carson, M.C. Evolution of the Perlecan/HSPG2 Gene and Its Activation in Regenerating Nematostella vectensis. PLoS ONE 2015, 10, e0124578. [Google Scholar] [CrossRef] [PubMed]
- Farach-Carson, M.C.; Warren, C.R.; Harrington, D.A.; Carson, D.D. Border patrol: Insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol. 2014, 34, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.R.; Grindel, B.J.; Hubka, K.M.; Dodge, G.R.; Farach-Carson, M.C. Perlecan/HSPG2: Signaling role of domain IV in chondrocyte clustering with implications for Schwartz-Jampel Syndrome. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, S.; Horchar, T.; Jefferson, B.; Laurie, G.W.; Hassell, J.R. Recombinant Domain III of Perlecan Promotes Cell Attachment through Its RGDS Sequence. J. Biol. Chem. 1995, 270, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Begovic, E.; Chapman, J.; Putnam, N.H.; Hellsten, U.; Kawashima, T.; Kuo, A.; Mitros, T.; Salamov, A.; Carpenter, M.L.; et al. The Trichoplax genome and the nature of placozoans. Nature 2008, 454, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Srivastava, M.; Hellsten, U.; Dirks, B.; Chapman, J.; Salamov, A.; Terry, A.; Shapiro, H.; Lindquist, E.; Kapitonov, V.V.; et al. Sea Anemone Genome Reveals Ancestral Eumetazoan Gene Repertoire and Genomic Organization. Science 2007, 317, 86–94. [Google Scholar] [CrossRef] [PubMed]
- King, N.; Westbrook, M.J.; Young, S.L.; Kuo, A.; Abedin, M.; Chapman, J.; Fairclough, S.; Hellsten, U.; Isogai, Y.; Letunic, I.; et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 2008, 451, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.F.; Pang, K.; Schnitzler, C.E.; Nguyen, A.-D.; Moreland, R.T.; Simmons, D.K.; Koch, B.J.; Francis, W.R.; Havlak, P.; Smith, S.A.; et al. The Genome of the Ctenophore Mnemiopsis leidyi and Its Implications for Cell Type Evolution. Science 2013, 342, 1242592. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Chen, Z.; de Mendoza, A.; Sebé-Pedrós, A.; Brown, M.W.; Kramer, E.; Carr, M.; Kerner, P.; Vervoort, M.; Sánchez-Pons, N.; et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nat. Commun. 2013, 4, 2325. [Google Scholar] [CrossRef] [PubMed]
- Tucker, R.P.; Shibata, B.; Blankenship, T.N. Ultrastructure of the mesoglea of the sea anemone Nematostella vectensis (Edwardsiidae). Invertebr. Biol. 2011, 130, 11–24. [Google Scholar] [CrossRef]
- Ringrose, J.H.; van den Toorn, H.W.P.; Eitel, M.; Post, H.; Neerincx, P.; Schierwater, B.; Altelaar, A.M.; Heck, A.J. Deep proteome profiling of Trichoplax adhaerens reveals remarkable features at the origin of metazoan multicellularity. Nat. Commun. 2013, 4, 1408. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Jung, M.; Cheng, B.; Whitelock, J.M. Transcriptional complexity of the HSPG2 gene in the human mast cell line, HMC-1. Matrix Biol. 2014, 35, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Lord, M.S.; Cheng, B.; Lyons, J.G.; Alkhouri, H.; Hughes, J.M.; McCarthy, S.J.; Iozzo, R.V.; Whitelock, J.M. Mast cells produce novel shorter forms of perlecan that contain functional endorepellin: A role in angiogenesis and wound healing. J. Biol. Chem. 2013, 288, 3289–3304. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, T.M.; Williams, B.D.; Mullen, G.P.; Moerman, D.G. Products of the unc-52 gene in Caenorhabditis elegans are homologous to the core protein of the mammalian basement membrane heparan sulfate proteoglycan. Genes Dev. 1993, 7, 1471–1484. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8393416 (accessed on 27 October 2015). [CrossRef] [PubMed]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1213120&tool=pmcentrez&rendertype=abstract (accessed on 18 July 2014). [PubMed]
- Mackenzie, J.M., Jr.; Garcea, R.L.; Zengel, J.M.; Epstein, H.F. Muscle development in Caenorhabditis elegans: Mutants exhibiting retarded sarcomere construction. Cell 1978, 15, 751–762. [Google Scholar] [CrossRef]
- Merz, D.C.; Alves, G.; Kawano, T.; Zheng, H.; Culotti, J.G. UNC-52/perlecan affects gonadal leader cell migrations in C. elegans hermaphrodites through alterations in growth factor signaling. Dev. Biol. 2003, 256, 173–186. Available online: http://www.ncbi.nlm.nih.gov/pubmed/12654300 (accessed on 27 October 2015). [CrossRef]
- Gilchrist, E.J.; Moerman, D.G. Mutations in the sup-38 gene of Caenorhabditis elegans suppress muscle-attachment defects in unc-52 mutants. Genetics 1992, 132, 431–442. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1205147&tool=pmcentrez&rendertype=abstract (accessed on 27 October 2015). [PubMed]
- Jafari, G.; Burghoorn, J.; Kawano, T.; Mathew, M.; Mörck, C.; Axäng, C.; Ailion, M.; Thomas, J.H.; Culotti, J.G.; Swoboda, P.; et al. Genetics of extracellular matrix remodeling during organ growth using the Caenorhabditis elegans pharynx model. Genetics 2010, 186, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Spartz, A.K.; Herman, R.K.; Shaw, J.E. SMU-2 and SMU-1, Caenorhabditis elegans homologs of mammalian spliceosome-associated proteins RED and fSAP57, work together to affect splice site choice. Mol. Cell. Biol. 2004, 24, 6811–6823. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, E.A.; Herman, R.K.; Rogalski, T.M.; Mullen, G.P.; Moerman, D.G.; Shaw, J.E. The mec-8 gene of C. elegans encodes a protein with two RNA recognition motifs and regulates alternative splicing of unc-52 transcripts. Development 1996, 122, 1601–1610. [Google Scholar] [PubMed]
- Kabat, J.L.; Barberan-Soler, S.; Zahler, A.M. HRP-2, the Caenorhabditis elegans homolog of mammalian heterogeneous nuclear ribonucleoproteins Q and R is an alternative splicing factor that binds to UCUAUC splicing regulatory elements. J. Biol. Chem. 2009, 284, 28490–28497. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Zhu, Y.; Jiang, X.; Li, Y.; Zhu, M.; Dong, M.; Huang, Z.; Wang, C.; Labouesse, M.; Zhang, H. CCAR-1 affects hemidesmosome biogenesis by regulating unc-52/perlecan alternative splicing in the C. elegans epidermis. J. Cell Sci. 2018, 131, jcs214379. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Kankel, D.R. l(1)trol and l(1)devl, loci affecting the development of the adult central nervous system in Drosophila melanogaster. Genetics 1992, 130, 523–537. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1204870&tool=pmcentrez&rendertype=abstract (accessed on 28 October 2015). [PubMed]
- Cho, J.Y.; Chak, K.; Andreone, B.J.; Wooley, J.R.; Kolodkin, A.L. The extracellular matrix proteoglycan perlecan facilitates transmembrane semaphorin-mediated repulsive guidance. Genes Dev. 2012, 26, 2222–2235. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, M.; Liu, T.; Banerjee, U.; Hartenstein, V. The proteoglycan Trol controls the architecture of the extracellular matrix and balances proliferation and differentiation of blood progenitors in the Drosophila lymph gland. Dev Biol. 2013, 384, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; French, M.M.; Julian, J.; Paria, B.C.; Dey, S.K.; Carson, D.D. Expression of Heparan Sulfate Proteoglycan (Perlecan) in the Mouse Blastocyst Is Regulated during Normal and Delayed Implantation. Dev Biol. 1997, 184, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Carson, D.D.; Tang, J.-P.; Julian, J. Heparan Sulfate Proteoglycan (Perlecan) Expression by Mouse Embryos during Acquisition of Attachment Competence. Dev Biol. 1993, 155, 97–106. [Google Scholar] [CrossRef] [PubMed]
- French, M.M.; Smith, S.E.; Akanbi, K.; Sanford, T.; Hecht, J.; Farach-Carson, M.C.; Carson, D.D. Expression of the heparan sulfate proteoglycan, perlecan, during mouse embryogenesis and perlecan chondrogenic activity in vitro. J. Cell Biol. 1999, 145, 1103–1115. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2133131&tool=pmcentrez&rendertype=abstract (accessed on 4 October 2015). [CrossRef] [PubMed]
- Handler, M.; Yurchenco, P.D.; Iozzo, R.V. Developmental expression of perlecan during murine embryogenesis. Dev. Dyn. 1997, 210, 130–145. [Google Scholar] [CrossRef]
- Arikawa-Hirasawa, E.; Wilcox, W.R.; Yamada, Y. Dyssegmental dysplasia, Silverman-Handmaker type: Unexpected role of perlecan in cartilage development. Am. J. Med. Genet. 2001, 106, 254–257. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11891676 (accessed on 12 February 2016). [CrossRef] [PubMed]
- Stum, M.; Girard, E.; Bangratz, M.; Bernard, V.; Herbin, M.; Vignaud, A.; Ferry, A.; Davoine, C.S.; Echaniz-Laguna, A.; René, F.; et al. Evidence of a dosage effect and a physiological endplate acetylcholinesterase deficiency in the first mouse models mimicking Schwartz-Jampel syndrome neuromyotonia. Hum. Mol. Genet. 2008, 17, 3166–3179. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Jampel, R.S. Congenital blepharophimosis associated with a unique generalized myopathy. Arch. Ophthalmol. 1962, 68, 52–57. Available online: http://www.ncbi.nlm.nih.gov/pubmed/13909723 (accessed on 12 February 2016). [CrossRef] [PubMed]
- Mereu, T.R.; Porter, I.H.; Hug, G. Myotonia, shortness of stature, and hip dysplasia. Schwartz-Jampel syndrome. Am. J. Dis. Child. 1969, 117, 470–478. Available online: http://www.ncbi.nlm.nih.gov/pubmed/5773418 (accessed on 12 February 2016). [CrossRef] [PubMed]
- Handmaker, S.D.; Campbell, J.A.; Robinson, L.D.; Chinwah, O.; Gorlin, R.J. Dyssegmental dwarfism: A new syndrome of lethal dwarfism. Birth Defects Orig. Artic. Ser. 1977, 13, 79–90. Available online: http://www.ncbi.nlm.nih.gov/pubmed/922143 (accessed on 12 February 2016). [PubMed]
- Costell, M. Perlecan Maintains the Integrity of Cartilage and Some Basement Membranes. J. Cell Biol. 1999, 147, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Arikawa-Hirasawa, E.; Watanabe, H.; Takami, H.; Hassell, J.R.; Yamada, Y. Perlecan is essential for cartilage and cephalic development. Nat. Genet. 1999, 23, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Sasse, P.; Malan, D.; Fleischmann, M.; Roell, W.; Gustafsson, E.; Bostani, T.; Fan, Y.; Kolbe, T.; Breitbach, M.; Addicks, K.; et al. Perlecan is critical for heart stability. Cardiovasc. Res. 2008, 80, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Alicknavitch, M.; D’Souza, S.S.; Daikoku, T.; Kirn-Safran, C.B.; Marchetti, D.; Carson, D.D.; Farach-Carson, M.C. Heparanase expression and activity influences chondrogenic and osteogenic processes during endochondral bone formation. Bone 2008, 43, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Ishijima, M.; Suzuki, N.; Hozumi, K.; Matsunobu, T.; Kosaki, K.; Kaneko, H.; Hassell, J.R.; Arikawa-Hirasawa, E.; Yamada, Y. Perlecan modulates VEGF signaling and is essential for vascularization in endochondral bone formation. Matrix Biol. 2012, 31, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.A.; Lepori-Bui, N.; Fomin, P.V.; Sloofman, L.G.; Zhou, X.; Farach-Carson, M.C.; Wang, L.; Kirn-Safran, C.B. Deficiency in perlecan/HSPG2 during bone development enhances osteogenesis and decreases quality of adult bone in mice. Calcif. Tissue Int. 2014, 95, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.R.; Modla, S.; Grindel, B.J.; Czymmek, K.J.; Kirn-Safran, C.B.; Wang, L.; Duncan, R.L.; Farach-Carson, M.C. Perlecan/Hspg2 deficiency alters the pericellular space of the lacunocanalicular system surrounding osteocytic processes in cortical bone. J. Bone Miner. Res. 2011, 26, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Lai, X.; Price, C.; Thompson, W.R.; Li, W.; Quabili, T.R.; Tseng, W.J.; Liu, X.S.; Zhang, H.; Pan, J.; et al. Perlecan-containing pericellular matrix regulates solute transport and mechanosensing within the osteocyte lacunar-canalicular system. J. Bone Miner. Res. 2014, 29, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, R.E.; Defrate, L.E.; Guilak, F. A biomechanical role for perlecan in the pericellular matrix of articular cartilage. Matrix Biol. 2012, 31, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, R.E.; Sanchez-Adams, J.; Guilak, F. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol. 2014, 39, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Zelenski, N.A.; Leddy, H.A.; Sanchez-Adams, J.; Zhang, J.; Bonaldo, P.; Liedtke, W.; Guilak, F. Type VI Collagen Regulates Pericellular Matrix Properties, Chondrocyte Swelling, and Mechanotransduction in Mouse Articular Cartilage. Arthritis Rheumatol. 2015, 67, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.J.; Shu, C.C.; Lord, M.S.; Little, C.B.; Whitelock, J.M.; Melrose, J. Pericellular colocalisation and interactive properties of type VI collagen and perlecan in the intervertebral disc. Eur. Cells Mater. 2016, 32, 40–57. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27377666 (accessed on 2 November 2018). [CrossRef]
- Xu, Z.; Ichikawa, N.; Kosaki, K.; Yamada, Y.; Sasaki, T.; Sakai, L.Y.; Kurosawa, H.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan deficiency causes muscle hypertrophy, a decrease in myostatin expression, and changes in muscle fiber composition. Matrix Biol. 2010, 29, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Xu, Z.; Furuya, N.; Nonaka, R.; Yamada, Y.; Arikawa-Hirasawa, E. Perlecan inhibits autophagy to maintain muscle homeostasis in mouse soleus muscle. Matrix Biol. 2015, 48, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T.; Antonarakis, S.E. Mutation Nomenclature Extensions and Suggestions to Describe Complex Mutations: A Discussion. 2000. Available online: http://www.dmd.nl/mutnomen (accessed on 2 November 2018).
- Ladhani, N.N.N.; Chitayat, D.; Nezarati, M.M.; Laureane, M.C.; Keating, S.; Silver, R.J.; Unger, S.; Velsher, L.; Sirkin, W.; Toi, A.; et al. Dyssegmental dysplasia, Silverman-Handmaker type: Prenatal ultrasound findings and molecular analysis. Prenat. Diagn. 2013, 33, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Clarke, D.; Al Ahmad, A.; Kahle, M.; Parham, C.; Auckland, L.; Shaw, C.; Fidanboylu, M.; Orr, A.W.; Ogunshola, O.; et al. Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J. Clin. Investig. 2011, 121, 3005–3023. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.; Sasaki, T.; Gohring, W.; Yamada, Y.; Timpl, R. The C-Terminal Domain V of Perlecan Promotes β1 Integrin-Mediated Cell Adhesion, Binds Heparin, Nidogen and Fibulin-2 and Can be Modified by Glycosaminoglycans. Eur. J. Biochem. 1997, 250, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Kimbell, L.M.; Ohno, K.; Engel, A.G.; Rotundo, R.L. C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J. Biol. Chem. 2004, 279, 10997–11005. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, J.R.; Dhawan, A.; Farach-Carson, M.C. Modular Proteoglycan Perlecan/HSPG2: Mutations, Phenotypes, and Functions. Genes 2018, 9, 556. https://doi.org/10.3390/genes9110556
Martinez JR, Dhawan A, Farach-Carson MC. Modular Proteoglycan Perlecan/HSPG2: Mutations, Phenotypes, and Functions. Genes. 2018; 9(11):556. https://doi.org/10.3390/genes9110556
Chicago/Turabian StyleMartinez, Jerahme R., Akash Dhawan, and Mary C. Farach-Carson. 2018. "Modular Proteoglycan Perlecan/HSPG2: Mutations, Phenotypes, and Functions" Genes 9, no. 11: 556. https://doi.org/10.3390/genes9110556
APA StyleMartinez, J. R., Dhawan, A., & Farach-Carson, M. C. (2018). Modular Proteoglycan Perlecan/HSPG2: Mutations, Phenotypes, and Functions. Genes, 9(11), 556. https://doi.org/10.3390/genes9110556