De Novo Hepatic Transcriptome Assembly and Systems Level Analysis of Three Species of Dietary Fish, Sardinops sagax, Scomber japonicus, and Pleuronichthys verticalis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish Sampling
2.2. RNA Extraction
2.3. High-Throughput Sequencing
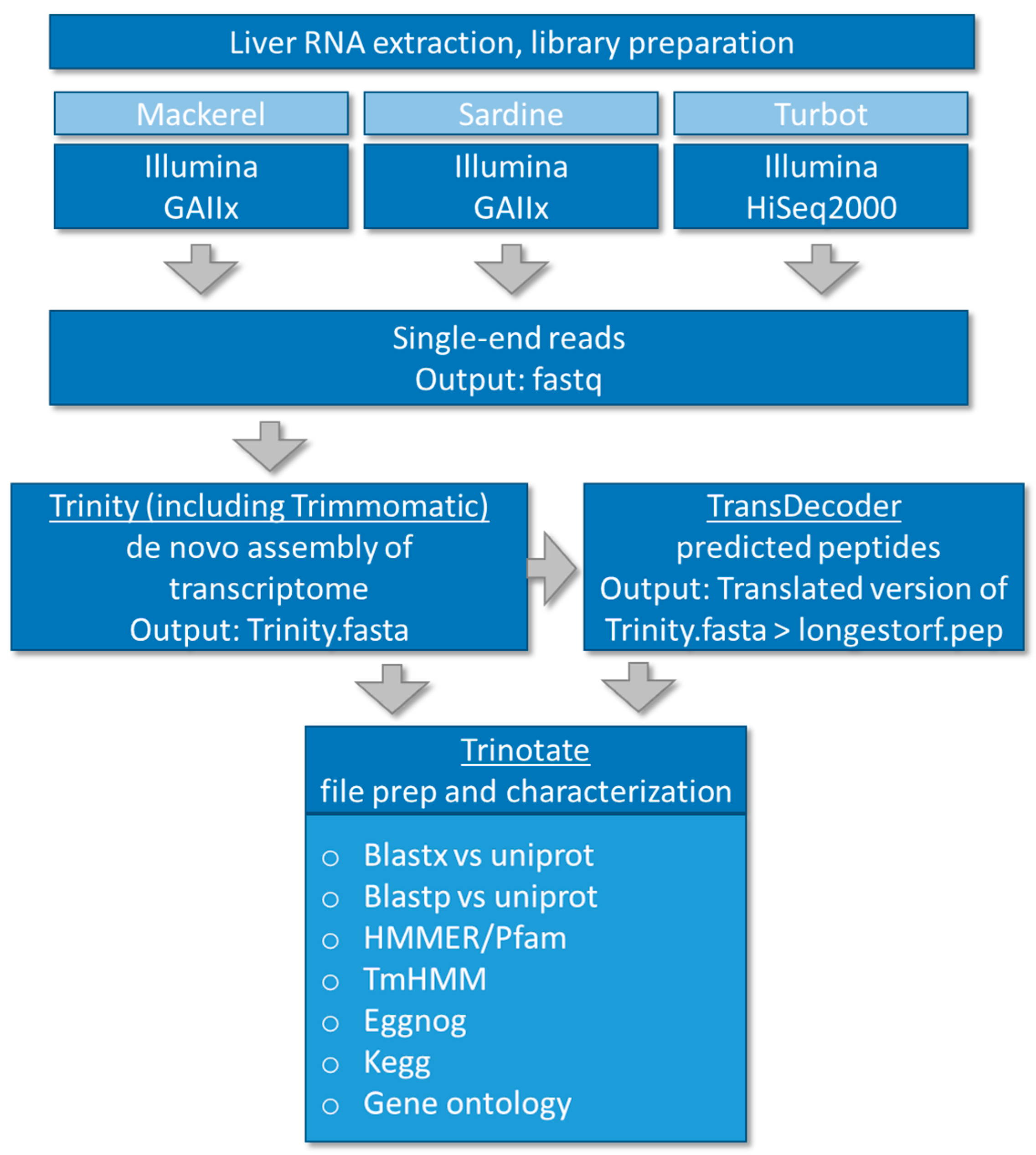
2.4. De Novo Assembly and Transcriptome Annotation
2.5. Systems Level Analysis
3. Results
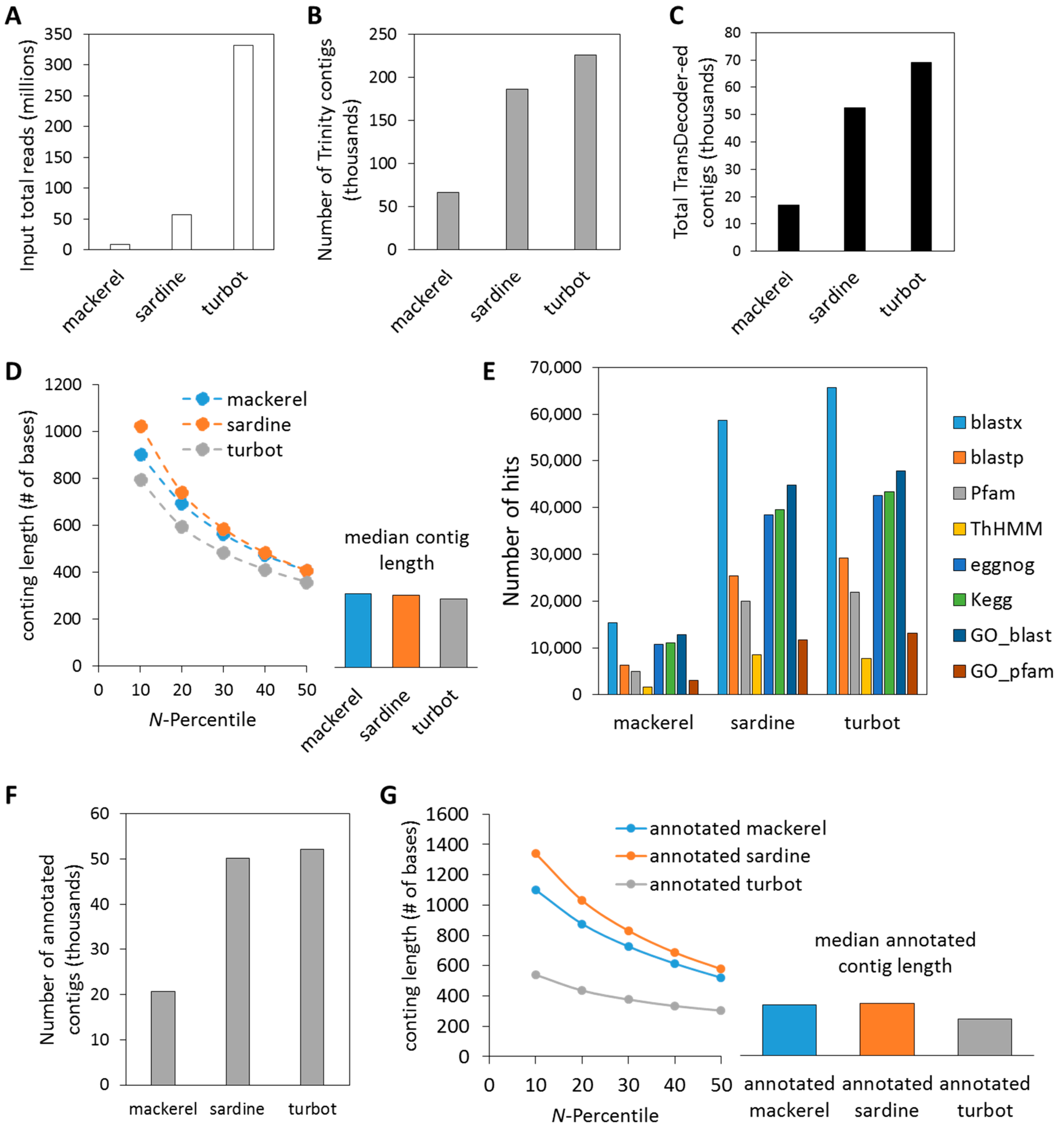
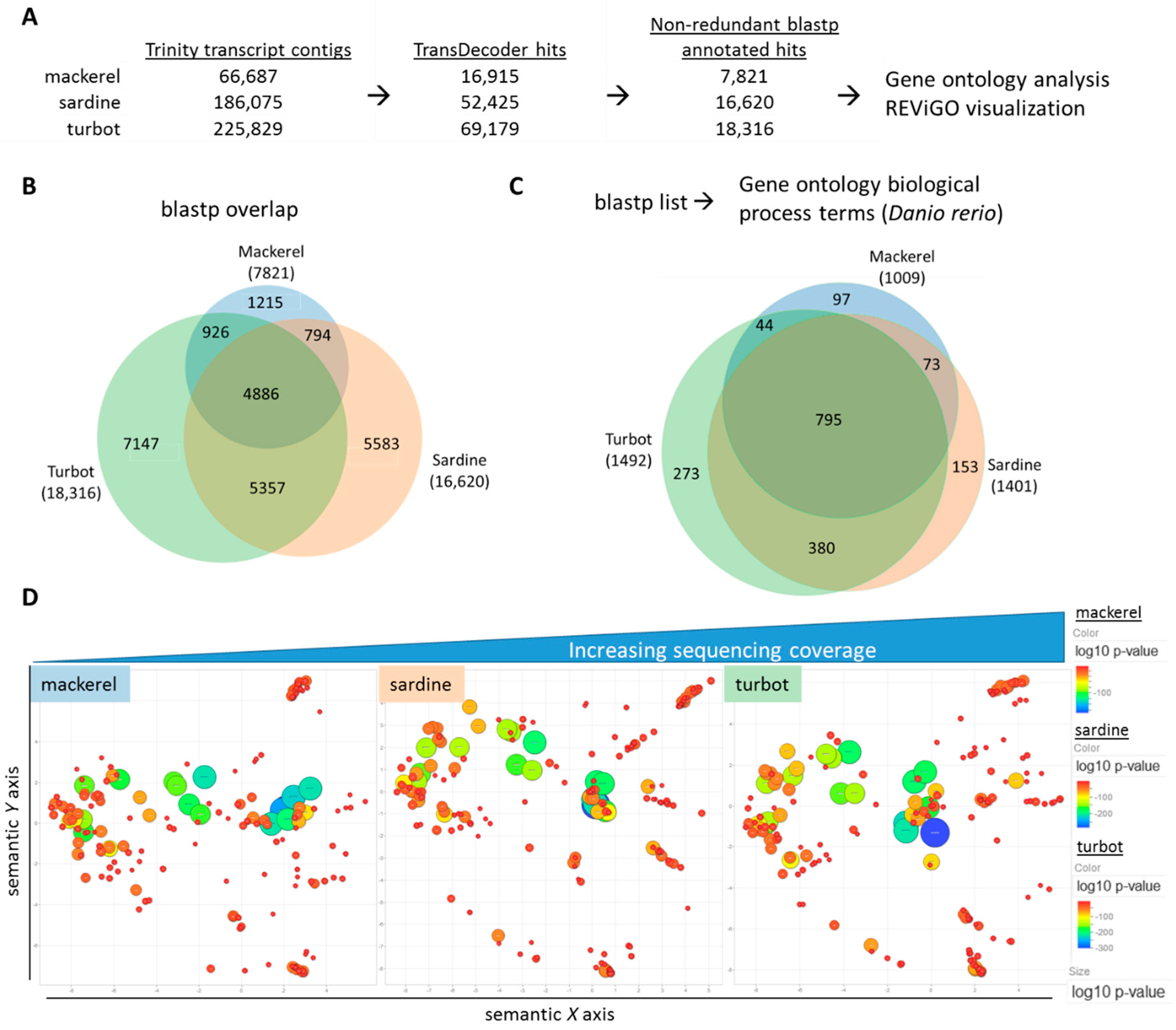
3.1. De Novo Assembly of Transcriptomes Using Trinity
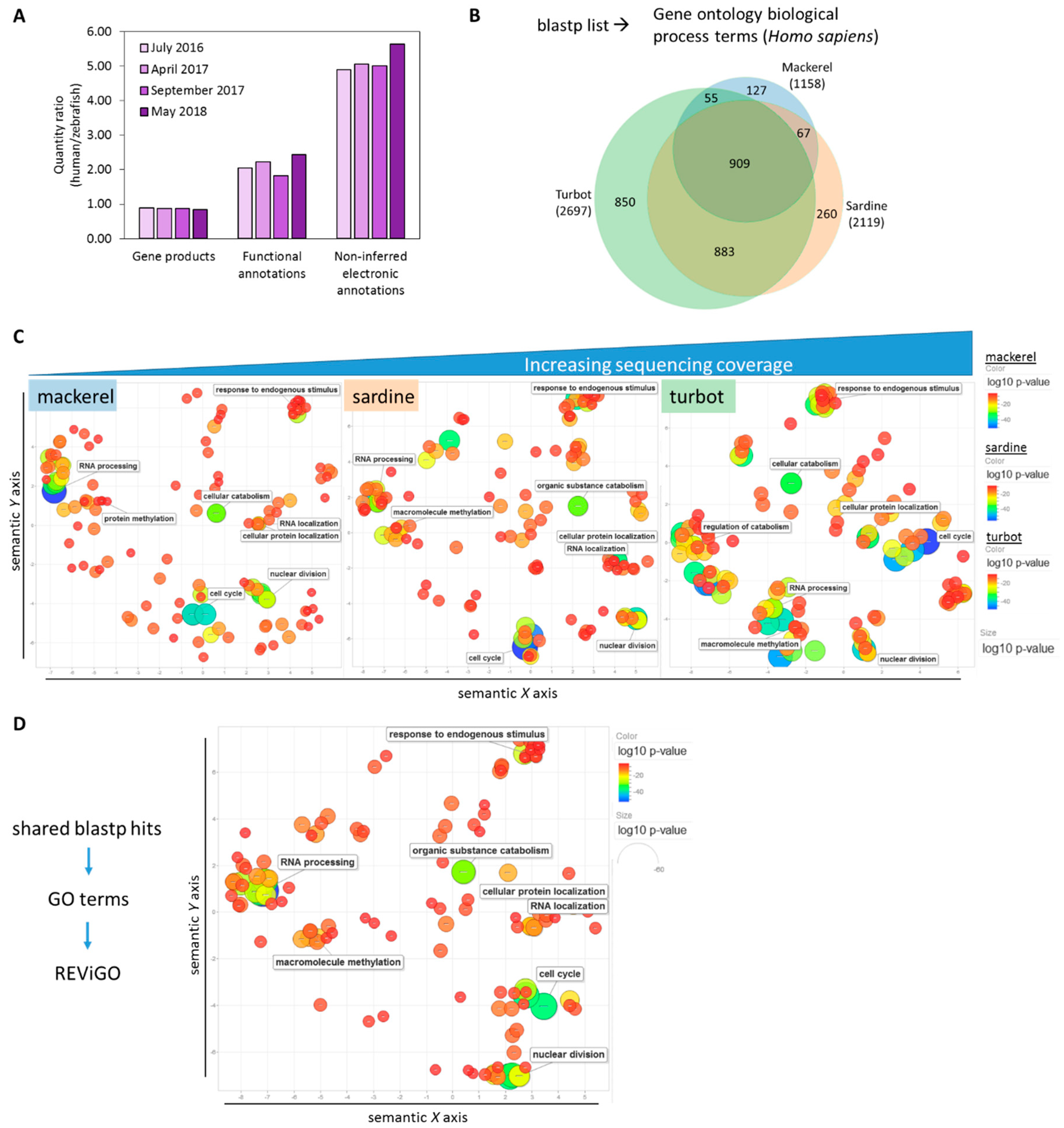
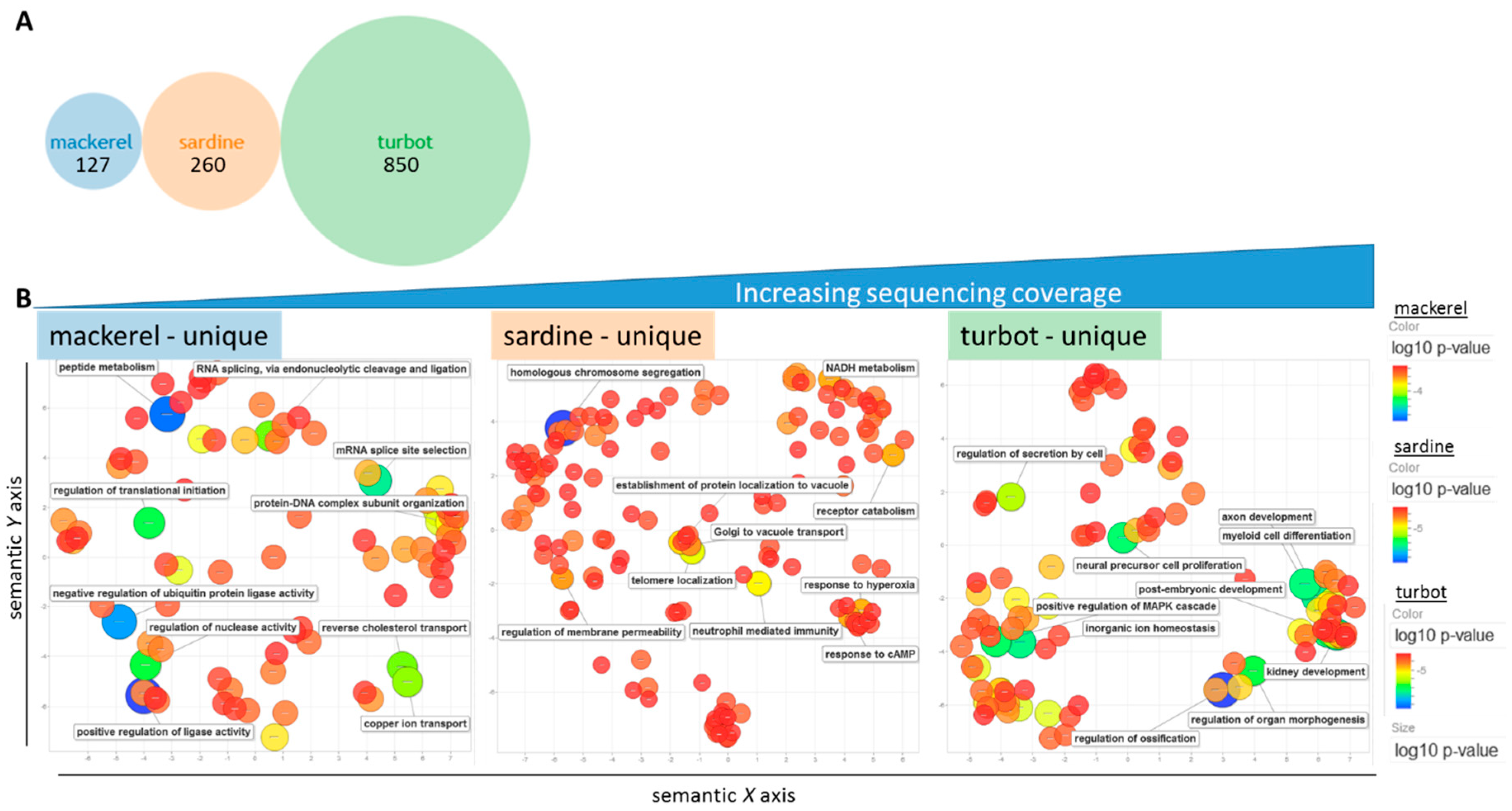
3.2. Functional Analysis of De Novo Hepatic Transcriptomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aloo, P.A. Health problems associated with consumption of fish and the role of aquatic environments in the transmission of human diseases. Afr. J. Health Sci. 2000, 7, 107–113. [Google Scholar] [PubMed]
- Boltana, S.; Sanhueza, N.; Aguilar, A.; Gallardo-Escarate, C.; Arriagada, G.; Valdes, J.A.; Soto, D.; Quinones, R.A. Influences of thermal environment on fish growth. Ecol. Evol. 2017, 7, 6814–6825. [Google Scholar] [CrossRef] [PubMed]
- Essington, T.E.; Beaudreau, A.H.; Wiedenmann, J. Fishing through marine food webs. Proc. Natl. Acad. Sci. USA 2006, 103, 3171–3175. [Google Scholar] [CrossRef] [PubMed]
- Glomset, J.A. Fish, fatty acids, and human health. N. Engl. J. Med. 1985, 312, 1253–1254. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Ashburn, S.M.; Razzouk, L.; Smith, D.A. Fish oils, coronary heart disease, and the environment. Am. J. Public Health 2013, 103, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.D.; Randall Hughes, A.; Hultgren, K.M.; Miner, B.G.; Sorte, C.J.; Thornber, C.S.; Rodriguez, L.F.; Tomanek, L.; Williams, S.L. The impacts of climate change in coastal marine systems. Ecol. Lett. 2006, 9, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.; Andersen, J.S.; Kaewmak, T.; Somsiri, T.; Dalsgaard, A. Impact of integrated fish farming on antimicrobial resistance in a pond environment. Appl. Environ. Microbiol. 2002, 68, 6036–6042. [Google Scholar] [CrossRef] [PubMed]
- Szuwalski, C.S.; Hilborn, R. Environment drives forage fish productivity. Proc. Natl. Acad. Sci. USA 2015, 112, E3314–E3315. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; White, P.A. The mutagenic hazards of aquatic sediments: A review. Mutat. Res. 2004, 567, 151–225. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Grant, S.B.; Surbeck, C.Q.; DiGiacomo, P.M.; Nezlin, N.P.; Jiang, S. Coastal water quality impact of stormwater runoff from an urban watershed in southern California. Environ. Sci. Technol. 2005, 39, 5940–5953. [Google Scholar] [CrossRef] [PubMed]
- Given, S.; Pendleton, L.H.; Boehm, A.B. Regional public health cost estimates of contaminated coastal waters: A case study of gastroenteritis at southern California beaches. Environ. Sci. Technol. 2006, 40, 4851–4858. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Noble, R.; Chu, W. Human adenoviruses and coliphages in urban runoff-impacted coastal waters of southern California. Appl. Environ. Microbiol. 2001, 67, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Sercu, B.; Van De Werfhorst, L.C.; Murray, J.; Holden, P.A. Storm drains are sources of human fecal pollution during dry weather in three urban southern California watersheds. Environ. Sci. Technol. 2009, 43, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Parnell, P.E.; Groce, A.K.; Stebbins, T.D.; Dayton, P.K. Discriminating sources of PCB contamination in fish on the coastal shelf off San Diego, California (USA). Mar. Pollut. Bull. 2008, 56, 1992–2002. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C. Does breast cancer start in the womb? Basic Clin. Pharmacol. Toxicol. 2008, 102, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Grun, F.; Blumberg, B. Minireview: The case for obesogens. Mol. Endocrinol. 2009, 23, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.; Lowry, M.S.; Yochem, P.K.; Stewart, B.S.; Heath, C.B. Seasonal and annual variability in the diet of California sea lions, Zalophus californianus, at San Nicolas Island, California, 1981–1986. Fish. Bull 1991, 89, 331–336. [Google Scholar]
- LLefebvre, K.; Silver, M.; Coale, S.; Tjeerdema, R. Domoic acid in planktivorous fish in relation to toxic Pseudo-nitzschia cell densities. Mar. Biol. 2002, 140, 625–631. [Google Scholar] [CrossRef]
- Klinger, D.H.; Dale, J.J.; Machado, B.E.; Incardona, J.P.; Farwell, C.J.; Block, B.A. Exposure to Deepwater Horizon weathered crude oil increases routine metabolic demand in chub mackerel, Scomber japonicus. Mar. Pollut. Bull. 2015, 98, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.; Roney, J.; Kleppel, G. Fish food-habits along a pollution gradient. Calif. Fish Game 1985, 71, 28–39. [Google Scholar]
- Miller, D.J.; Lea, R.N. Guide to the Coastal Marine Fishes of California; UCANR Publications: Oakland, CA, USA, 1972. [Google Scholar]
- Baker, M.E.; Ruggeri, B.; Sprague, L.J.; Eckhardt-Ludka, C.; Lapira, J.; Wick, I.; Soverchia, L.; Ubaldi, M.; Polzonetti-Magni, A.M.; Vidal-Dorsch, D.; et al. Analysis of endocrine disruption in southern California coastal fish using an aquatic multispecies microarray. Environ. Health Perspect. 2009, 117, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J.; American Heart Association; Nutrition, C. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 2002, 106, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E.; Hardiman, G. Transcriptional analysis of endocrine disruption using zebrafish and massively parallel sequencing. J. Mol. Endocrinol. 2014, 52, R241–R256. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Dorsch, D.E.; Bay, S.M.; Ribecco, C.; Sprague, L.J.; Angert, M.; Ludka, C.; Ricciardelli, E.; Carnevali, O.; Greenstein, D.J.; Schlenk, D.; et al. Genomic and phenotypic response of hornyhead turbot exposed to municipal wastewater effluents. Aquat. Toxicol. 2013, 140–141, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E.; Sprague, L.J.; Ribecco, C.; Ruggeri, B.; Lekmine, N.; Ludka, C.; Wick, I.; Soverchia, L.; Ubaldi, M.; Šášik, R.; et al. Application of a targeted endocrine q-PCR panel to monitor the effects of pollution in southern California flatfish. Endocr. Disrupt. 2014, 2, e969598. [Google Scholar] [CrossRef]
- Xu, E.G.; Mager, E.M.; Grosell, M.; Pasparakis, C.; Schlenker, L.S.; Stieglitz, J.D.; Benetti, D.; Hazard, E.S.; Courtney, S.M.; Diamante, G.; et al. Time- and oil-dependent transcriptomic and physiological responses to deepwater horizon oil in mahi-mahi (Coryphaena hippurus) embryos andlarvae. Environ. Sci. Technol. 2016, 50, 7842–7851. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.G.; Khursigara, A.J.; Magnuson, J.; Hazard, E.S.; Hardiman, G.; Esbaugh, A.J.; Roberts, A.P.; Schlenk, D. Larval red drum (Sciaenops ocellatus) sublethal exposure to weathered deepwater horizon crude oil: Developmental and transcriptomic consequences. Environ. Sci. Technol. 2017, 51, 10162–10172. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.G.; Mager, E.M.; Grosell, M.; Stieglitz, J.D.; Hazard, E.S.; Hardiman, G.; Schlenk, D. Developmental transcriptomic analyses for mechanistic insights into critical pathways involved in embryogenesis of pelagic mahi-mahi (Coryphaena hippurus). PLoS ONE 2017, 12, e0180454. [Google Scholar] [CrossRef] [PubMed]
- Miracle, A.L.; Ankley, G.T. Ecotoxicogenomics: Linkages between exposure and effects in assessing risks of aquatic contaminants to fish. Reprod. Toxicol. 2005, 19, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E.; Vidal-Dorsch, D.E.; Ribecco, C.; Sprague, L.J.; Angert, M.; Lekmine, N.; Ludka, C.; Martella, A.; Ricciardelli, E.; Bay, S.M.; et al. Molecular analysis of endocrine disruption in hornyhead turbot at wastewater outfalls in southern california using a second generation multi-species microarray. PLoS ONE 2013, 8, e75553. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- The UniProt, C. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- The Gene Ontology, C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2017. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simao, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef] [PubMed]
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011, 27, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Venny Oliveros, J. An interactive Tool for Comparing Lists with Venn’s Diagrams. 2007. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 25 October 2018).
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Huff, M.; da Silveira, W.A.; Carnevali, O.; Renaud, L.; Hardiman, G. Systems analysis of the liver transcriptome in adult male zebrafish exposed to the plasticizer (2-ethylhexyl) phthalate (DEHP). Sci. Rep. 2018, 8, 2118. [Google Scholar] [CrossRef] [PubMed]
- Compeau, P.E.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.Y.; Wang, Y.; Kong, Y.M.; Luo, D.; Li, X.; Hao, P. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: A comparative study. BMC Bioinform. 2011, 12 (Suppl. 14), S2. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B.; et al. Comparison of the two major classes of assembly algorithms: Overlap-layout-consensus and de-bruijn-graph. Brief Funct. Genom. 2012, 11, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Sudhagar, A.; Kumar, G.; El-Matbouli, M. Transcriptome analysis based on RNA-Seq in understanding pathogenic mechanisms of diseases and the immune system of fish: A comprehensive review. Int. J. Mol. Sci. 2018, 19, 245. [Google Scholar] [CrossRef] [PubMed]
- Uren Webster, T.M.; Shears, J.A.; Moore, K.; Santos, E.M. Identification of conserved hepatic transcriptomic responses to 17β-estradiol using high-throughput sequencing in brown trout. Physiol. Genom. 2015, 47, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Oomen, R.A.; Hutchings, J.A. Variation in spawning time promotes genetic variability in population responses to environmental change in a marine fish. Conserv. Physiol. 2015, 3, cov027. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.A.; Byrne, M. Acclimatization and adaptive capacity of marine species in a changing ocean. Adv. Mar. Biol. 2016, 74, 69–116. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Zeng, Z.; Shi, T. Comparative study of de novo assembly and genome-guided assembly strategies for transcriptome reconstruction based on RNA-Seq. Sci. China Life Sci. 2013, 56, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, A.; Pech, N.; Martin, J.F.; McCairns, R.J.S.; Mevy, J.P.; Chappaz, R.; Gilles, A. Challenges and advances for transcriptome assembly in non-model species. PLoS ONE 2017, 12, e0185020. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, M.; Matschiner, M.; Torresen, O.K.; Jakobsen, K.S.; Jentoft, S. Whole genome sequencing data and de novo draft assemblies for 66 teleost species. Sci. Data 2017, 4, 160132. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, M.; Matschiner, M.; Torresen, O.K.; Star, B.; Snipen, L.G.; Hansen, T.F.; Baalsrud, H.T.; Nederbragt, A.J.; Hanel, R.; Salzburger, W.; et al. Evolution of the immune system influences speciation rates in teleost fishes. Nat. Genet. 2016, 48, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, M.; Yurchenko, A.A.; Augley, J.J.; Adams, C.E.; Herzyk, P.; Elmer, K.R. De novo transcriptome assembly, annotation and comparison of four ecological and evolutionary model salmonid fish species. BMC Genom. 2018, 19, 32. [Google Scholar] [CrossRef]
- Kroon, F.; Streten, C.; Harries, S. A protocol for identifying suitable biomarkers to assess fish health: A systematic review. PLoS ONE 2017, 12, e0174762. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Liu, S.; Zhou, T.; Tian, C.; Bao, L.; Dunham, R.; Liu, Z. Comparative genome analysis of 52 fish species suggests differential associations of repetitive elements with their living aquatic environments. BMC Genom. 2018, 19, 141. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richards, D.J.; Renaud, L.; Agarwal, N.; Starr Hazard, E.; Hyde, J.; Hardiman, G. De Novo Hepatic Transcriptome Assembly and Systems Level Analysis of Three Species of Dietary Fish, Sardinops sagax, Scomber japonicus, and Pleuronichthys verticalis. Genes 2018, 9, 521. https://doi.org/10.3390/genes9110521
Richards DJ, Renaud L, Agarwal N, Starr Hazard E, Hyde J, Hardiman G. De Novo Hepatic Transcriptome Assembly and Systems Level Analysis of Three Species of Dietary Fish, Sardinops sagax, Scomber japonicus, and Pleuronichthys verticalis. Genes. 2018; 9(11):521. https://doi.org/10.3390/genes9110521
Chicago/Turabian StyleRichards, Dylan J., Ludivine Renaud, Nisha Agarwal, E. Starr Hazard, John Hyde, and Gary Hardiman. 2018. "De Novo Hepatic Transcriptome Assembly and Systems Level Analysis of Three Species of Dietary Fish, Sardinops sagax, Scomber japonicus, and Pleuronichthys verticalis" Genes 9, no. 11: 521. https://doi.org/10.3390/genes9110521
APA StyleRichards, D. J., Renaud, L., Agarwal, N., Starr Hazard, E., Hyde, J., & Hardiman, G. (2018). De Novo Hepatic Transcriptome Assembly and Systems Level Analysis of Three Species of Dietary Fish, Sardinops sagax, Scomber japonicus, and Pleuronichthys verticalis. Genes, 9(11), 521. https://doi.org/10.3390/genes9110521