The Complete Plastome Sequences of Eleven Capsicum Genotypes: Insights into DNA Variation and Molecular Evolution

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Chloroplast Isolation and DNA Extraction
2.3. Chloroplast DNA Sequencing and Genome Assembly
2.4. Genome Annotation and Analysis of Nucleotide Variability
2.5. Molecular Evolution Analysis on Protein-Coding Genes
2.6. Primer Design and PCR Amplification
3. Results
3.1. Chloroplast Genome Size and Organization
3.2. Sequence Variation within Capsicum Genotypes
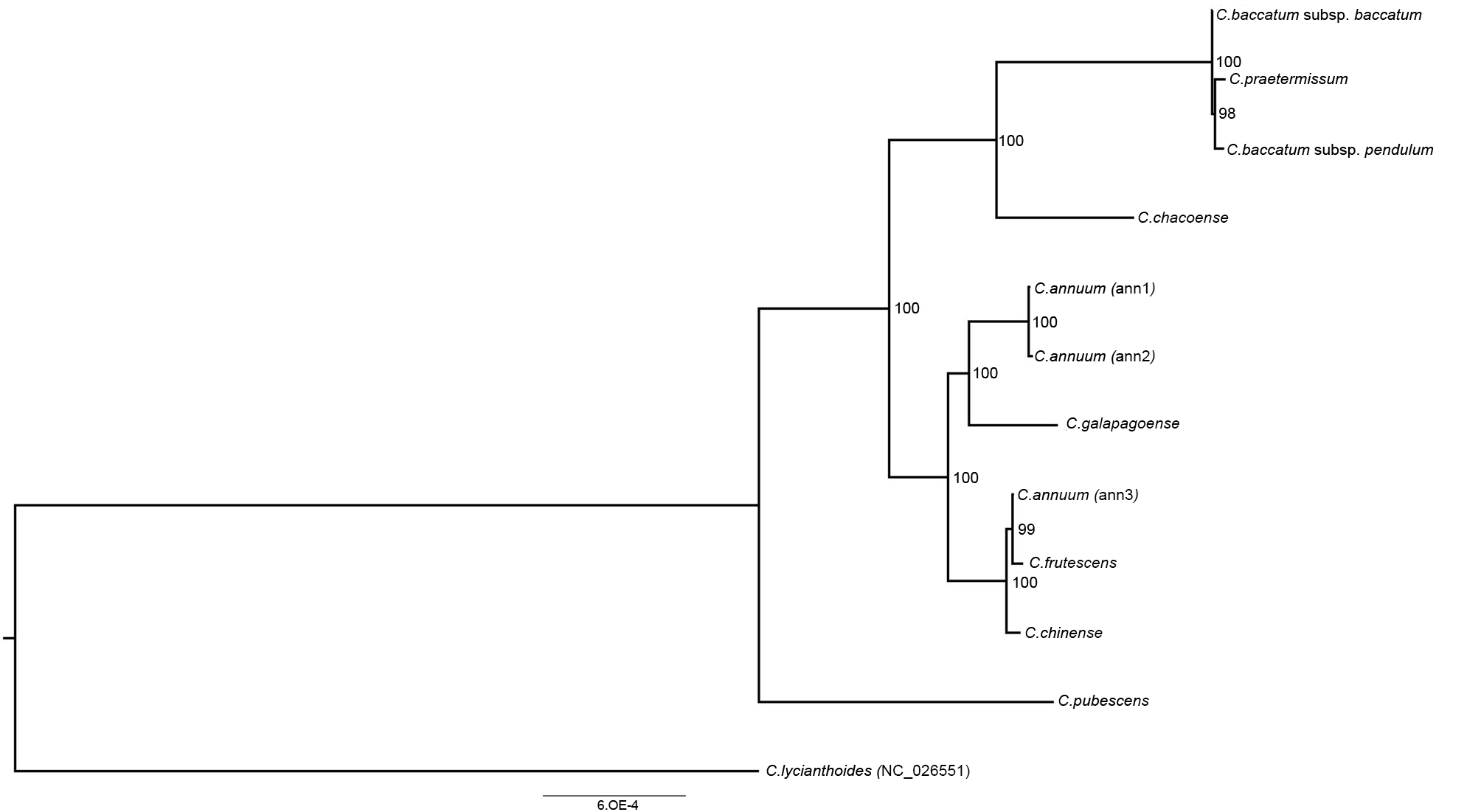
3.3. Phylogenetic Reconstruction and Molecular Evolution
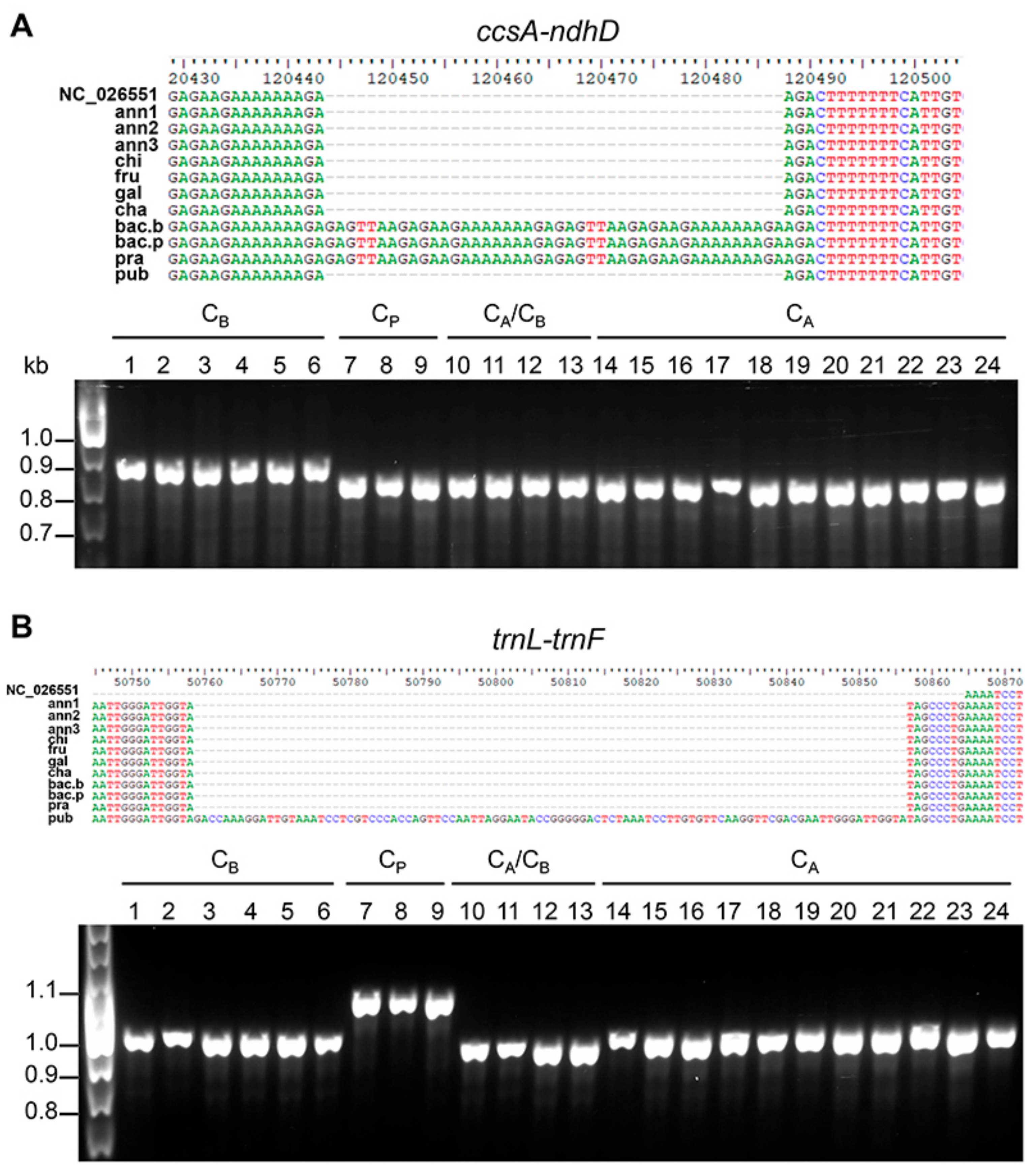
3.4. Chloroplast-Specific Molecular Markers for Capsicum spp.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; de Pamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in Angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Olmstead, R.G.; Bohs, L.; Migid, H.A.; Santiago-Valentin, E.; Garcia, V.F.; Collier, S.M. A molecular phylogeny of the Solanaceae. Taxon 2008, 57, 1159–1181. [Google Scholar]
- Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; Stevens, P.F.; et al. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Powell, W.; Morgante, M.; Andre, C.; McNicol, J.W.; Machray, G.C.; Doyle, J.J.; Tingey, S.V.; Rafalski, J.A. Hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr. Biol. 1995, 5, 1023–1029. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, Z.; Li, B.; Qin, C.; Wu, Z.; Trejo-Saavedra, D.L.; Luo, X.; Cui, J.; Rivera-Bustamante, R.F.; Li, S.; et al. A comprehensive characterization of simple sequence repeats in pepper genomes provides valuable resources for marker development in Capsicum. Sci. Rep. 2016, 6, 18919. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2016, 7, 1955. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, R.; Li, X. Comparative analysis of the complete chloroplast genome of four known Ziziphus species. Genes 2017, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Huang, J.-P.; Wu, C.-S.; Hsu, C.-Y.; Chaw, S.-M. Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome Biol. Evol. 2010, 2, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Cheng, T.; Lin, K.; Zhou, S. Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 2013, 5, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Curci, P.L.; De Paola, D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef] [PubMed]
- Rousseau-Gueutin, M.; Bellot, S.; Martin, G.E.; Boutte, J.; Chelaifa, H.; Lima, O.; Michon-Coudouel, S.; Naquin, D.; Salmon, A.; Ainouche, K.; et al. The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): Comparative analyses and molecular dating. Mol. Phylogenet. Evol. 2015, 93, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Gu, C.; Tembrock, L.R.; Zhang, D.; Ge, S. Characterization of the whole chloroplast genome of Chikusichloa mutica and its comparison with other rice tribe (Oryzeae) species. PLoS ONE 2017, 12, e0177553. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Q.; Drew, B.T.; Yang, J.-B.; Gao, L.-M.; Li, D.-Z. Comparative chloroplast genomes of eleven Schima (Theaceae) species: Insights into DNA barcoding and phylogeny. PLoS ONE 2017, 12, e0178026. [Google Scholar] [CrossRef] [PubMed]
- Carrizo García, C.; Barfuss, M.H.J.; Sehr, E.M.; Barboza, G.E.; Samuel, R.; Moscone, E.A.; Ehrendorfer, F. Phylogenetic relationships, diversification and expansion of chili peppers (Capsicum, Solanaceae). Ann. Bot. 2016, 118, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.A.; Ashrafi, H.; Reyes-Chin-Wo, S.; Yao, J.; Stoffel, K.; Truco, M.-J.; Kozik, A.; Michelmore, R.W.; Van Deynze, A. Characterization of Capsicum annuum genetic diversity and population structure based on parallel polymorphism discovery with a 30 k unigene pepper genechip. PLoS ONE 2013, 8, e56200. [Google Scholar] [CrossRef] [PubMed]
- Moscone, E.A.; Scaldaferro, M.A.; Grabiele, M.; Cecchini, N.M.; Sánchez García, Y.; Jarret, R.; Daviña, J.R.; Ducasse, D.A.; Barboza, G.E.; Ehrendorfer, F. The Evolution of Chili Peppers (Capsicum—Solanaceae): A Cytogenetic Perspective, 2007; International Society for Horticultural Science (ISHS): Leuven, Belgium, 2007; pp. 137–170. [Google Scholar]
- Perry, L.; Dickau, R.; Zarrillo, S.; Holst, I.; Pearsall, D.M.; Piperno, D.R.; Berman, M.J.; Cooke, R.G.; Rademaker, K.; Ranere, A.J.; et al. Starch fossils and the domestication and dispersal of chili peppers (Capsicum spp. L.) in the americas. Science 2007, 315, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Ince, A.G.; Karaca, M.; Onus, A.N. Genetic relationships within and between Capsicum species. Biochem. Genet. 2010, 48, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Walsh, B.M.; Hoot, S.B. Phylogenetic relationships of Capsicum (Solanaceae) using DNA sequences from two noncoding regions: The chloroplast atpB-rbcL spacer region and nuclear waxy introns. Int. J. Plant Sci. 2001, 162, 1409–1418. [Google Scholar] [CrossRef]
- Rai, V.P.; Kumar, R.; Kumar, S.; Rai, A.; Kumar, S.; Singh, M.; Singh, S.P.; Rai, A.B.; Paliwal, R. Genetic diversity in Capsicum germplasm based on microsatellite and random amplified microsatellite polymorphism markers. Physiol. Mol. Biol. Plants 2013, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Nicolaï, M.; Cantet, M.; Lefebvre, V.; Sage-Palloix, A.-M.; Palloix, A. Genotyping a large collection of pepper (Capsicum spp.) with SSR loci brings new evidence for the wild origin of cultivated C. annuum and the structuring of genetic diversity by human selection of cultivar types. Genet. Resour. Crop Evol. 2013, 60, 2375–2390. [Google Scholar] [CrossRef]
- Thul, S.T.; Darokar, M.P.; Shasany, A.K.; Khanuja, S.P.S. Molecular profiling for genetic variability in Capsicum species based on ISSR and RAPD markers. Mol. Biotechnol. 2012, 51, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.D.; Park, J.; Kim, J.; Song, W.; Hur, C.-G.; Lee, Y.-H.; Kang, B.-C. Complete sequencing and comparative analyses of the pepper (Capsicum annuum L.) plastome revealed high frequency of tandem repeats and large insertion/deletions on pepper plastome. Plant Cell Rep. 2011, 30, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Raveendar, S.; Na, Y.-W.; Lee, J.-R.; Shim, D.; Ma, K.-H.; Lee, S.-Y.; Chung, J.-W. The complete chloroplast genome of Capsicum annuum var. glabriusculum using illumina sequencing. Molecules 2015, 20, 13080–13088. [Google Scholar] [CrossRef] [PubMed]
- Raveendar, S.; Jeon, Y.-A.; Lee, J.-R.; Lee, G.-A.; Lee, K.J.; Cho, G.-T.; Ma, K.-H.; Lee, S.-Y.; Chung, J.-W. The complete chloroplast genome sequence of Korean landrace “Subicho” pepper (Capsicum annuum var. annuum). Plant Breed. Biotechnol. 2015, 3, 88–94. [Google Scholar] [CrossRef]
- Shim, D.; Raveendar, S.; Lee, J.-R.; Lee, G.-A.; Ro, N.-Y.; Jeon, Y.-A.; Cho, G.-T.; Lee, H.-S.; Ma, K.-H.; Chung, J.-W. The complete chloroplast genome of Capsicum frutescens (Solanaceae). Appl. Plant Sci. 2016, 4, 1600002. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-S.; Lee, J.; Lee, S.-C.; Yang, T.-J.; Yoon, J.B. The complete chloroplast genome sequence of Capsicum chinense jacq. (Solanaceae). Mitochondrial DNA B Resour. 2016, 1, 164–165. [Google Scholar] [CrossRef]
- Zeng, F.-C.; Gao, C.-W.; Gao, L.-Z. The complete chloroplast genome sequence of American bird pepper (Capsicum annuum var. glabriusculum). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Raveendar, S.; Lee, K.J.; Shin, M.-J.; Cho, G.-T.; Lee, J.-R.; Ma, K.-H.; Lee, G.-A.; Chung, J.-W. Complete chloroplast genome sequencing and genetic relationship analysis of Capsicum chinense jacq. Plant Breed. Biotechnol. 2017, 5, 261–268. [Google Scholar] [CrossRef]
- Rogalski, M.; do Nascimento Vieira, L.; Fraga, H.P.; Guerra, M.P. Plastid genomics in horticultural species: Importance and applications for plant population genetics, evolution, and biotechnology. Front. Plant Sci. 2015, 6, 586. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What can we do with 1000 plastid genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Kemble, R.J. A rapid, single leaf, nucleic acid assay for determining the cytoplasmic organelle complement of rapeseed and related Brassica species. Theor. Appl. Genet. 1987, 73, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Scotti, N.; Cardi, T.; Marechal Drouard, L. Mitochondrial DNA and RNA isolation from small amounts of potato tissue. Plant Mol. Biol. Rep. 2001, 19, 67. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with dogma. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar genome draw (ogdraw): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using clustalw and clustalx. Curr. Protoc. Bioinform. 2003. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. Snp-sites: Rapid efficient extraction of SNPs from multi-fasta alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef] [PubMed]
- Poliakov, A.; Foong, J.; Brudno, M.; Dubchak, I. Genomevista—An integrated software package for whole-genome alignment and visualization. Bioinformatics 2014, 30, 2654–2655. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. Dnasp v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Doron-Faigenboim, A.; Erez, E.; Martz, E.; Bacharach, E.; Pupko, T. Selecton 2007: Advanced models for detecting positive and purifying selection using a bayesian inference approach. Nucleic Acids Res. 2007, 35, W506–W511. [Google Scholar] [CrossRef] [PubMed]
- Kelchner, S.A. The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann. Mo. Bot. Gard. 2000, 87, 482–498. [Google Scholar] [CrossRef]
- Twyford, A.D.; Ness, R.W. Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 2017, 17, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Xue, Q.; Zhu, S.; Sun, J.; Liu, W.; Ding, X. The complete plastome sequences of four orchid species: Insights into the evolution of the orchidaceae and the utility of plastomic mutational hotspots. Front. Plant Sci. 2017, 8, 715. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, M.H.; Pang, A.W.C.; Bunikis, I.; Höijer, I.; Vinnere-Petterson, O.; Suh, A.; Wolf, J.B.W. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017, 27, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in Angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Rongwen, J.; Akkaya, M.; Bhagwat, A.; Lavi, U.; Cregan, P. The use of microsatellite DNA markers for soybean genotype identification. Theor. Appl. Genet. 1995, 90, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.B.; Linares, A.R.; Cavalli-Sforza, L.L.; Feldman, M.W. An evaluation of genetic distances for use with microsatellite loci. Genetics 1995, 139, 463–471. [Google Scholar] [PubMed]
- Wang, H.-L.; Yang, J.; Boykin, L.M.; Zhao, Q.-Y.; Wang, Y.-J.; Liu, S.-S.; Wang, X.-W. Developing conversed microsatellite markers and their implications in evolutionary analysis of the Bemisia tabaci complex. Sci. Rep. 2014, 4, 6351. [Google Scholar] [CrossRef] [PubMed]
- Diekmann, K.; Hodkinson, T.R.; Barth, S. New chloroplast microsatellite markers suitable for assessing genetic diversity of Lolium perenne and other related grass species. Ann. Bot. 2012, 110, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- De Santana Lopes, A.; Pacheco, T.G.; Nimz, T.; do Nascimento Vieira, L.; Guerra, M.P.; Nodari, R.O.; de Souza, E.M.; de Oliveira Pedrosa, F.; Rogalski, M. The complete plastome of macaw palm [Acrocomia aculeata (Jacq.) Lodd. ex Mart.] and extensive molecular analyses of the evolution of plastid genes in Arecaceae. Planta 2018, 247, 1011–1030. [Google Scholar] [CrossRef] [PubMed]
- Bock, D.G.; Andrew, R.L.; Rieseberg, L.H. On the adaptive value of cytoplasmic genomes in plants. Mol. Ecol. 2014, 23, 4899–4911. [Google Scholar] [CrossRef] [PubMed]
- Piot, A.; Hackel, J.; Christin, P.-A.; Besnard, G. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 2018, 247, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-M.; Wang, J.; Feng, L.; Liu, S.; Pang, H.; Qi, L.; Li, J.; Sun, Y.; Qiao, W.; Zhang, L.; et al. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 2017, 7, 1555. [Google Scholar]
- Givnish, T.J.; Spalink, D.; Ames, M.; Lyon, S.P.; Hunter, S.J.; Zuluaga, A.; Iles, W.J.; Clements, M.A.; Arroyo, M.T.; Leebens-Mack, J. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. R. Soc. B 2015, 282, 20151553. [Google Scholar] [CrossRef] [PubMed]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Jarret, R.L. DNA barcoding in a crop genebank: The Capsicum annuum species complex. Open Biol. J. 2008, 1, 35–42. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype Code | Species | Complex a | Germplasm Bank Identifier (ID) | Size (Base Pairs) | % GC | |||
|---|---|---|---|---|---|---|---|---|
| Total | LSC d | SSC d | IR d | |||||
| ann1 | C. annuum | CA | CGN21526 b | 157,052 | 87,380 | 17,882 | 25,895 | 37.71 |
| ann2 | C. annuum | CA | CAP319 c | 156,842 | 87,380 | 17,960 | 25,751 | 37.72 |
| ann3 | C. annuum | CA | CAP1546 c | 156,872 | 87,341 | 17,917 | 25,807 | 37.73 |
| chi | C. chinense | CA | CGN22099 b | 156,858 | 87,288 | 17,860 | 25,855 | 37.73 |
| fru | C. frutescens | CA | CGN22779 b | 156,836 | 87,359 | 17,911 | 25,783 | 37.72 |
| gal | C. galapagoense | CA | CGN22208 b | 157,029 | 87,366 | 17,941 | 25,861 | 37.69 |
| cha | C. chacoense | CA/CB | CGN22084 b | 156,841 | 87,346 | 17,893 | 25,801 | 37.72 |
| bac.b | C. baccatum subsp. baccatum | CB | CGN23261 b | 157,053 | 87,350 | 17,973 | 25,865 | 36.45 |
| bac.p | C. baccatum subsp. pendulum | CB | CGN21512 b | 157,144 | 87,351 | 17,973 | 25,910 | 37.66 |
| pra | C. praetermissum | CB | CGN20805 b | 157,056 | 87,351 | 17,973 | 25,866 | 37.66 |
| pub | C. pubescens | CP | CGN22108 b | 157,390 | 87,688 | 17,928 | 25,887 | 37.69 |
| Genotypes | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Marker | Region | ann1 | ann2 | ann3 | chi | fru | gal | cha | bac.b | bac.p | pra | pub | Notes |
| SNPa | |||||||||||||
| AAACC[A/G]TTTA | psbA | 0 b | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | Gain of a HpyCH4III restriction site |
| GAATT[C/A]TATC | rps16 intron | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | Loss of a EcoRI restriction site |
| ATATT[C/T]CCGA | atpI | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | Loss of a Hpy188III restriction site |
| TGCGA[G/T]ATCG | rps2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | Loss of a Sau3AI restriction site |
| TCTTG[C/A]ATAT | rpoB | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | Loss of a HpyCH4V restriction site |
| CCAGC[T/C]CCCC | atpB | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | Loss of a AluI restriction site |
| SSRc | |||||||||||||
| TTTC(A)nTCAT | psbK–psbI | 9 d | 9 | 9 | 9 | 9 | 9 | 10 | 10 | 10 | 10 | 2 | |
| TCTG(T)nCAAA | trnG–trnR | 12 | 12 | 12 | 12 | 12 | 12 | 11 | 11 | 11 | 11 | 10 | |
| AAT(ATAA)nAT | psaA–ycf3 | 4 | 4 | 4 | 4 | 4 | 4 | 3 | 2 | 2 | 2 | 3 | |
| CTTC(CT)nTATC | ycf3 intron | 5 | 5 | 5 | 5 | 5 | 5 | 4 | 5 | 5 | 5 | 5 | |
| TTTC(A)nGGTA | atpB–rbcL | 11 | 11 | 11 | 11 | 11 | 11 | 9 | 9 | 9 | 9 | 8 | |
| GTTA(T)nAGGT | rpl20–rps12 | 14 | 14 | 14 | 14 | 14 | 14 | 15 | 16 | 16 | 16 | 13 | |
| TAAC(T)nGTTG | rpl32–trnL | 6 | 6 | 6 | 6 | 6 | 9 | 6 | 6 | 6 | 6 | 6 | |
| TRe | |||||||||||||
| GGAT(TTATC…GCCTA)37AAGG | trnS–rps4 | 1 f | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | |
| AAGA(GAGTT…AAAGA)22AGAC | ccsA–ndhD | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 3 | 3 | 1 | |
| TTAA(TTGGT…TTGTT)30TAAG | ycf1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 1 | |
| TCTC(ATTGA…ATTGT)25ATTT | ycf2–trnI | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Agostino, N.; Tamburino, R.; Cantarella, C.; De Carluccio, V.; Sannino, L.; Cozzolino, S.; Cardi, T.; Scotti, N. The Complete Plastome Sequences of Eleven Capsicum Genotypes: Insights into DNA Variation and Molecular Evolution. Genes 2018, 9, 503. https://doi.org/10.3390/genes9100503
D’Agostino N, Tamburino R, Cantarella C, De Carluccio V, Sannino L, Cozzolino S, Cardi T, Scotti N. The Complete Plastome Sequences of Eleven Capsicum Genotypes: Insights into DNA Variation and Molecular Evolution. Genes. 2018; 9(10):503. https://doi.org/10.3390/genes9100503
Chicago/Turabian StyleD’Agostino, Nunzio, Rachele Tamburino, Concita Cantarella, Valentina De Carluccio, Lorenza Sannino, Salvatore Cozzolino, Teodoro Cardi, and Nunzia Scotti. 2018. "The Complete Plastome Sequences of Eleven Capsicum Genotypes: Insights into DNA Variation and Molecular Evolution" Genes 9, no. 10: 503. https://doi.org/10.3390/genes9100503
APA StyleD’Agostino, N., Tamburino, R., Cantarella, C., De Carluccio, V., Sannino, L., Cozzolino, S., Cardi, T., & Scotti, N. (2018). The Complete Plastome Sequences of Eleven Capsicum Genotypes: Insights into DNA Variation and Molecular Evolution. Genes, 9(10), 503. https://doi.org/10.3390/genes9100503