Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Intestinal Epithelium Architecture and Cellular Composition
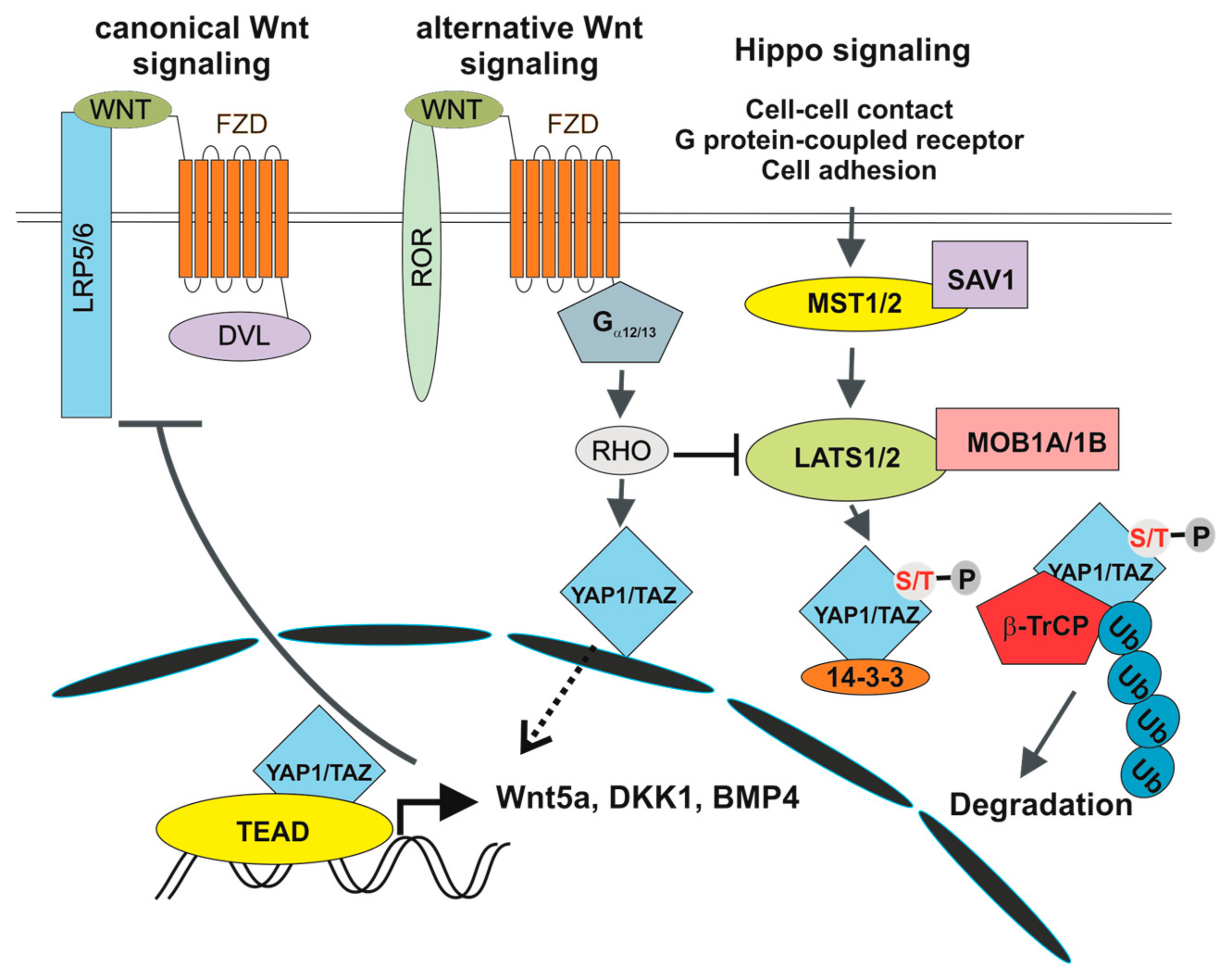
3. Wnt Signalling in the Intestine
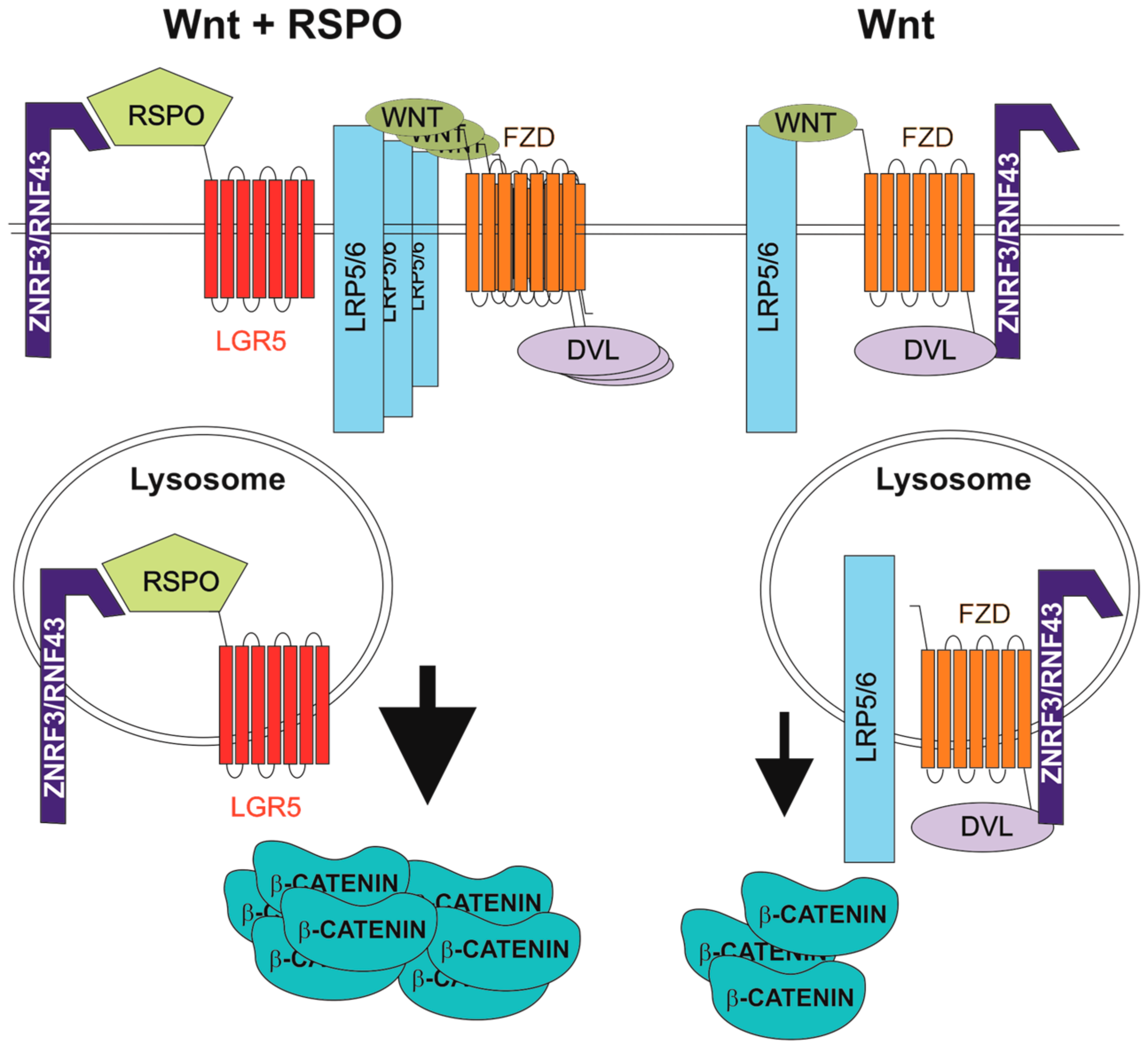
4. R-Spondins/Leucine-Rich-Repeat-Containing G-Protein-Coupled Receptor Signalling
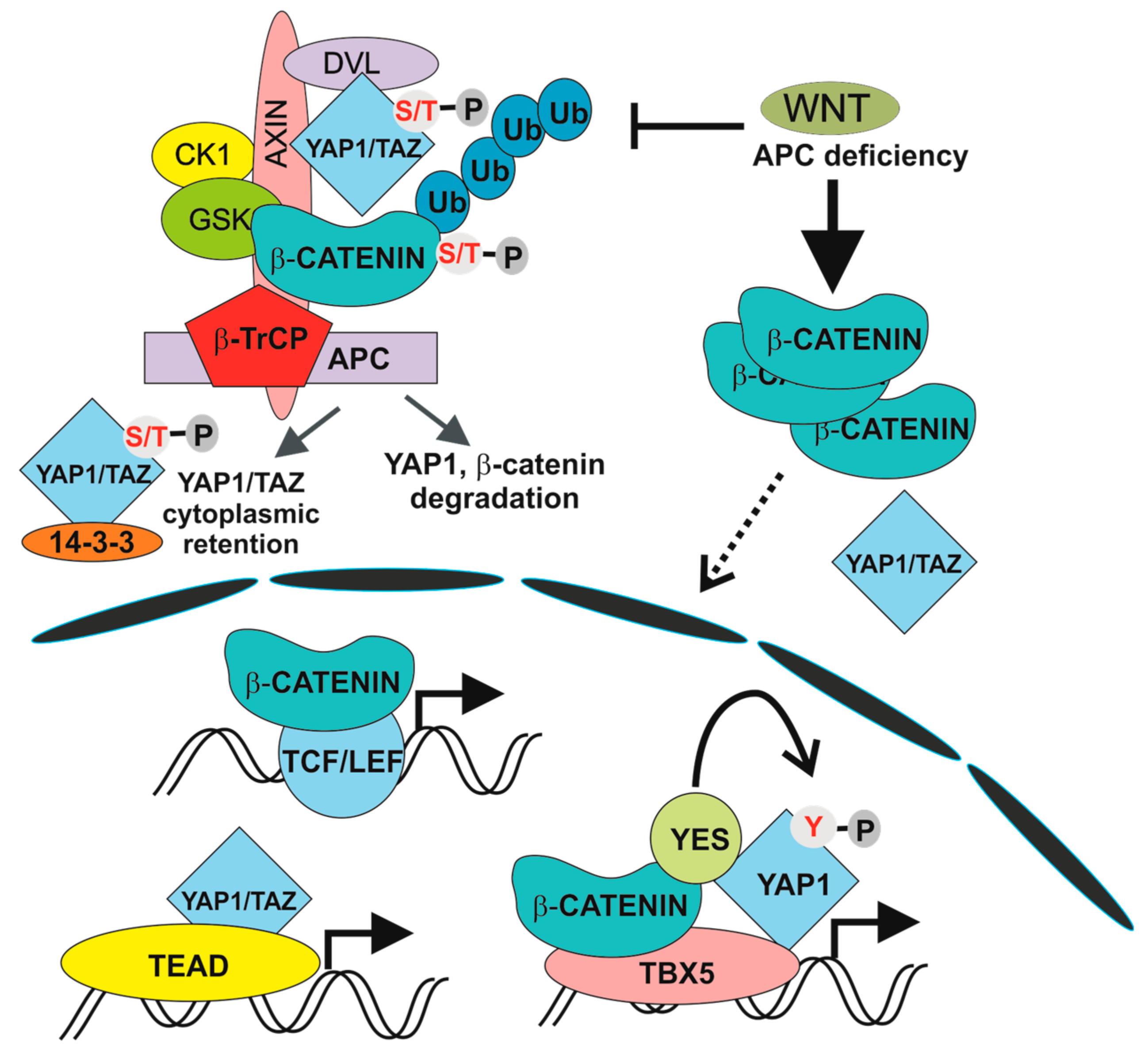
5. The Hippo Pathway
Acknowledgments
Conflicts of Interest
References
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Huber, O.; Korn, R.; McLaughlin, J.; Ohsugi, M.; Herrmann, B.G.; Kemler, R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 1996, 59, 3–10. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; vanWichen, D.; deWeger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signalling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single LGR5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as mediator of Wnt signalling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt signalling activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Marti, P.; et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, Z.; Ma, Y.; Yue, Z.; Lin, H.; Qu, G.; Huang, J.; Dai, W.; Li, C.; Zheng, C.; et al. Lgr4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat. Med. 2016, 22, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Stem cells. What is an adult stem cell? Science 2015, 350, 1319–1320. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Wagers, A.J. No place like home: Anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell Biol. 2008, 9, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Van der Flier, L.G.; Sabates-Bellver, J.; Oving, I.; Haegebarth, A.; De Palo, M.; Anti, M.; Van Gijn, M.E.; Suijkerbuijk, S.; Van de Wetering, M.; Marra, G.; et al. The intestinal Wnt/TCF signature. Gastroenterology 2007, 132, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene LGR5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; Haegebarth, A.; Stange, D.E.; van de Wetering, M.; Clevers, H. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology 2009, 137, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Stange, D.E.; Schepers, A.G.; van de Wetering, M.; Koo, B.K.; Itzkovitz, S.; Volckmann, R.; Kung, K.S.; Koster, J.; Radulescu, S.; et al. The LGR5 intestinal stem cell signature: Robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012, 31, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Fafilek, B.; Krausova, M.; Vojtechova, M.; Pospichalova, V.; Tumova, L.; Sloncova, E.; Huranova, M.; Stancikova, J.; Hlavata, A.; Svec, J.; et al. Troy, a tumour necrosis factor receptor family member, interacts with LGR5 to inhibit Wnt signalling in intestinal stem cells. Gastroenterology 2013, 144, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sachs, N.; Wiebrands, K.; Ellenbroek, S.I.; Fumagalli, A.; Lyubimova, A.; Begthel, H.; van den Born, M.; van Es, J.H.; Karthaus, W.R.; et al. Reg4+ deep crypt secretory cells function as epithelial niche for LGR5+ stem cells in colon. Proc. Natl. Acad. Sci. USA 2016, 113, E5399–E5407. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S.; Booth, C.; Pritchard, D.M. The intestinal epithelial stem cell: The mucosal governor. Int. J. Exp. Pathol. 1997, 78, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; de Sauvage, F.J. A reserve stem cell population in small intestine renders LGR5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Itzkovitz, S.; Lyubimova, A.; Blat, I.C.; Maynard, M.; van Es, J.; Lees, J.; Jacks, T.; Clevers, H.; van Oudenaarden, A. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat. Cell Biol. 2012, 14, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Asfaha, S.; Hayakawa, Y.; Muley, A.; Stokes, S.; Graham, T.A.; Ericksen, R.E.; Westphalen, C.B.; von Burstin, J.; Mastracci, T.L.; Worthley, D.L.; et al. Krt19+/LGR5− cells are radioresistant cancer-initiating stem cells in the colon and intestine. Cell Stem Cell 2015, 16, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; de Sauvage, F.; van Es, J.H.; et al. Replacement of lost LGR5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell 2016, 18, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Buczacki, S.J.; Zecchini, H.I.; Nicholson, A.M.; Russell, R.; Vermeulen, L.; Kemp, R.; Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing LGR5. Nature 2013, 495, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Meran, L.; Baulies, A.; Li, V.S.W. Intestinal stem cell niche: The extracellular matrix and cellular components. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for LGR5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Van Es, J.H.; Clevers, H. Redundant sources of wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 2012, 143, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Escudero, S.; Shivdasani, R.A. Intact function of LGR5 receptor-expressing intestinal stem cells in the absence of Paneth cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3932–3937. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, Z.; Greicius, G.; Madan, B.; Biechele, S.; Zhong, Z.; Zaribafzadeh, H.; Edison; Aliyev, J.; Wu, Y.; Bunte, R.; et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development 2014, 141, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, R.; Nusse, R. Towards an integrated view of Wnt signalling in development. Development 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R., 3rd; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Takada, R.; Satomi, Y.; Kurata, T.; Ueno, N.; Norioka, S.; Kondoh, H.; Takao, T.; Takada, S. Monounsaturated fatty acid modification of Wnt protein: Its role in Wnt secretion. Dev. Cell 2006, 11, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Doubravska, L.; Krausova, M.; Gradl, D.; Vojtechova, M.; Tumova, L.; Lukas, J.; Valenta, T.; Pospichalova, V.; Fafilek, B.; Plachy, J.; et al. Fatty acid modification of Wnt1 and Wnt3a at serine is prerequisite for lipidation at cysteine and is essential for Wnt signalling. Cell. Signal. 2011, 23, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Langton, P.F.; Kakugawa, S.; Vincent, J.P. Making, exporting and modulating Wnts. Trends Cell Biol. 2016, 26, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Gregorieff, A.; Pinto, D.; Begthel, H.; Destree, O.; Kielman, M.; Clevers, H. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology 2005, 129, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, D.J.; Phesse, T.J.; Barker, N.; Schwab, R.H.; Amin, N.; Malaterre, J.; Stange, D.E.; Nowell, C.J.; Currie, S.A.; Saw, J.T.; et al. Frizzled7 functions as a Wnt receptor in intestinal epithelial LGR5+ stem cells. Stem Cell Rep. 2015, 4, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ueno, K.; Hazama, S.; Mitomori, S.; Nishioka, M.; Suehiro, Y.; Hirata, H.; Oka, M.; Imai, K.; Dahiya, R.; Hinoda, Y. Down-regulation of frizzled-7 expression decreases survival, invasion and metastatic capabilities of colon cancer cells. Br. J. Cancer 2009, 101, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Hrckulak, D.; Kolar, M.; Strnad, H.; Korinek, V. TCF/LEF transcription factors: An update from the internet resources. Cancers 2016, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Tortelote, G.G.; Reis, R.R.; de Almeida Mendes, F.; Abreu, J.G. Complexity of the Wnt/beta-catenin pathway: Searching for an activation model. Cell. Signal. 2017, 40, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Davis, C.R.; Wang, H.T.; Chu, P.; Lee, M.; Yuan, J.; Nusse, R.; Kuo, C.J. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc. Natl. Acad. Sci. USA 2004, 101, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Nishisho, I.; Kinzler, K.W.; Vogelstein, B.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Nagase, H. Mutations of the adenomatous polyposis coli gene in familial polyposis coli patients and sporadic colorectal tumors. Princess Takamatsu Symp. 1991, 22, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Bright-Thomas, R.M.; Hargest, R. APC, beta-catenin and hTCF-4; an unholy trinity in the genesis of colorectal cancer. Eur. J. Surg. Oncol. 2003, 29, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, S.M.; Petty, E.M.; Stoffel, E.M.; Fearon, E.R. An AXIN2 mutant allele associated with predisposition to colorectal neoplasia has context-dependent effects on AXIN2 protein function. Neoplasia 2015, 17, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.S.; Dismuke, A.D.; Powell, A.E.; Carroll, K.H.; Wong, M.H. Wnt-reporter expression pattern in the mouse intestine during homeostasis. BMC Gastroenterol. 2008, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Jordens, I.; Mosa, M.H.; Basak, O.; Korving, J.; Tauriello, D.V.; de Punder, K.; Angers, S.; Peters, P.J.; Maurice, M.M.; et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 2016, 530, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Mlodzik, M. Planar cell polarity signaling: From fly development to human disease. Annu. Rev. Genet. 2008, 42, 517–540. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Mehdawi, L.M.; Prasad, C.P.; Ehrnstrom, R.; Andersson, T.; Sjolander, A. Non-canonical WNT5A signaling up-regulates the expression of the tumor suppressor 15-PGDH and induces differentiation of colon cancer cells. Mol. Oncol. 2016, 10, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Medegan, B.; Braun, D.P. Wnt9A induction linked to suppression of human colorectal cancer cell proliferation. Int. J. Mol. Sci. 2016, 17, 495. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.R.; Das, A.M.; Helvensteijn, W.; Franken, P.F.; Swagemakers, S.; van der Valk, M.A.; ten Hagen, T.L.; Kuipers, E.J.; van Veelen, W.; Smits, R. Wnt5a promotes human colon cancer cell migration and invasion but does not augment intestinal tumorigenesis in Apc1638N mice. Carcinogenesis 2013, 34, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.K.; Lee, J.S. Cellular signaling and biological functions of R-spondins. Cell. Signal. 2012, 24, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Yokota, C.; Semenov, M.V.; Doble, B.; Woodgett, J.; He, X. R-spondin1 is a high affinity ligand for LRP6 and induces LRP6 phosphorylation and beta-catenin signaling. J. Biol. Chem. 2007, 282, 15903–15911. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Ruffner, H.; Sprunger, J.; Charlat, O.; Leighton-Davies, J.; Grosshans, B.; Salathe, A.; Zietzling, S.; Beck, V.; Therier, M.; Isken, A.; et al. R-spondin potentiates Wnt/beta-catenin signaling through orphan receptors LGR4 and LGR5. PLoS ONE 2012, 7, e40976. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/LGR5/RNF43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zamponi, R.; Charlat, O.; Ramones, M.; Swalley, S.; Jiang, X.; Rivera, D.; Tschantz, W.; Lu, B.; Quinn, L.; et al. Interaction with both ZNRF3 and LGR4 is required for the signalling activity of R-spondin. EMBO Rep. 2013, 14, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Zebisch, M.; Xu, Y.; Krastev, C.; MacDonald, B.T.; Chen, M.; Gilbert, R.J.; He, X.; Jones, E.Y. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 2013, 4, 2787. [Google Scholar] [CrossRef] [PubMed]
- Loregger, A.; Grandl, M.; Mejias-Luque, R.; Allgauer, M.; Degenhart, K.; Haselmann, V.; Oikonomou, C.; Hatzis, P.; Janssen, K.P.; Nitsche, U.; et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated beta-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 2015, 8, ra90. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Yi, J.; Thomas, A.; Liu, Q. RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E1221–E1229. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Charlat, O.; Zamponi, R.; Yang, Y.; Cong, F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell 2015, 58, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of APC in vivo immediately perturbs Wnt signaling, differentiation and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.S.; Janda, C.Y.; Chang, J.; Zheng, G.X.Y.; Larkin, K.A.; Luca, V.C.; Chia, L.A.; Mah, A.T.; Han, A.; Terry, J.M.; et al. Non-equivalence of Wnt and R-spondin ligands during LGR5+ intestinal stem-cell self-renewal. Nature 2017, 545, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Janda, C.Y.; Dang, L.T.; You, C.; Chang, J.; de Lau, W.; Zhong, Z.A.; Yan, K.S.; Marecic, O.; Siepe, D.; Li, X.; et al. Surrogate Wnt agonists that phenocopy canonical Wnt and beta-catenin signalling. Nature 2017, 545, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Wu, F.J.; Kudo, M.; Hsiao, C.L.; Hsueh, A.J.; Luo, C.W. A naturally occurring LGR4 splice variant encodes a soluble antagonist useful for demonstrating the gonadal roles of LGR4 in mammals. PLoS ONE 2014, 9, e106804. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.Y.; Liang, S.G.; Hsueh, A.J. Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine-rich repeats and a G protein-coupled, seven-transmembrane region. Mol. Endocrinol. 1998, 12, 1830–1845. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.; Wang, R.; Bailey, W.; Xie, G.; Chen, F.; Caskey, C.T.; Liu, Q. Identification and cloning of an orphan G protein-coupled receptor of the glycoprotein hormone receptor subfamily. Biochem. Biophys. Res. Commun. 1998, 247, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Jaks, V.; Barker, N.; Kasper, M.; van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgard, R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 2008, 40, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Brenot, A.; Lawson, D.A.; Linnemann, J.R.; Van Kappel, E.C.; Wong, K.C.; de Sauvage, F.; Klein, O.D.; Werb, Z. Lgr5-expressing cells are sufficient and necessary for postnatal mammary gland organogenesis. Cell Rep. 2013, 3, 70–78. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Ciampricotti, M.; Michalak, E.M.; Tan, D.W.; Speksnijder, E.N.; Hau, C.S.; Clevers, H.; Barker, N.; Jonkers, J. Developmental stage-specific contribution of LGR5+ cells to basal and luminal epithelial lineages in the postnatal mammary gland. J. Pathol. 2012, 228, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.K.; Li, Y.; Redding, K.M.; Iwatsuki, K.; Margolskee, R.F.; Jiang, P. Lgr5-EGFP marks taste bud stem/progenitor cells in posterior tongue. Stem Cells 2013, 31, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Rookmaaker, M.B.; Kujala, P.; Ng, A.; Leushacke, M.; Snippert, H.; van de Wetering, M.; Tan, S.; Van Es, J.H.; Huch, M.; et al. Lgr5+ve stem/progenitor cells contribute to nephron formation during kidney development. Cell Rep. 2012, 2, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Mazerbourg, S.; Bouley, D.M.; Luo, C.W.; Kawamura, K.; Kuwabara, Y.; Baribault, H.; Tian, H.; Hsueh, A.J. Neonatal lethality of LGR5 null mice is associated with ankyloglossia and gastrointestinal distension. Mol. Cell. Biol. 2004, 24, 9736–9743. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; van Es, J.H.; Barker, N.; van de Wetering, M.; van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 2010, 327, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.A.; Mitchell, K.J.; Goodrich, L.V.; Lu, X.; Pinson, K.; Scherz, P.; Skarnes, W.C.; Tessier-Lavigne, M. Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature 2001, 410, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Mazerbourg, S.; Bouley, D.M.; Sudo, S.; Klein, C.A.; Zhang, J.V.; Kawamura, K.; Goodrich, L.V.; Rayburn, H.; Tessier-Lavigne, M.; Hsueh, A.J. Leucine-rich repeat-containing, G protein-coupled receptor 4 null mice exhibit intrauterine growth retardation associated with embryonic and perinatal lethality. Mol. Endocrinol. 2004, 18, 2241–2254. [Google Scholar] [CrossRef] [PubMed]
- Pawaputanon Na Mahasarakham, C.; Ezura, Y.; Kawasaki, M.; Smriti, A.; Moriya, S.; Yamada, T.; Izu, Y.; Nifuji, A.; Nishimori, K.; Izumi, Y.; et al. BMP-2 enhances Lgr4 gene expression in osteoblastic cells. J. Cell. Physiol. 2016, 231, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, W.; Guo, C.A.; Han, N.; Pan, J.F.; Fei, T.; Yan, Z.Q. Stat3 upregulates leucine-rich repeat-containing G protein-coupled receptor 4 expression in osteosarcoma cells. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhou, W.; Zhou, X.; Li, D.; Weng, J.; Yi, Z.; Cho, S.G.; Li, C.; Yi, T.; Wu, X.; et al. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 2009, 136, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, A.; Norozi, F.; Shahrabi, S.; Shahjahani, M.; Saki, N. Wnt/beta-catenin signaling in bone marrow niche. Cell Tissue Res. 2016, 363, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; San Martin, J.; Dansey, R. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Styrkarsdottir, U.; Thorleifsson, G.; Sulem, P.; Gudbjartsson, D.F.; Sigurdsson, A.; Jonasdottir, A.; Oddsson, A.; Helgason, A.; Magnusson, O.T.; Walters, G.B.; et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 2013, 497, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ning, T.; Shi, J.; Chen, M.; Ding, L.; Huang, Y.; Kauderer, S.; Xu, M.; Cui, B.; Bi, Y.; et al. Association of a gain-of-function variant in LGR4 with central obesity. Obesity (Silver Spring) 2017, 25, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, R.; Wang, F.; Hong, J.; Li, X.; Chen, M.; Ke, Y.; Zhang, X.; Ma, Q.; Wang, R.; et al. Ablation of LGR4 promotes energy expenditure by driving white-to-brown fat switch. Nat. Cell Biol. 2013, 15, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Reddy, P.; Cheng, Y.; Luo, C.W.; Hsiao, C.L.; Hsueh, A.J. Multi-functional norrin is a ligand for the LGR4 receptor. J. Cell Sci. 2013, 126, 2060–2068. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Zhao, J.; Andarmani, S.; Kakitani, M.; Oshima, T.; Binnerts, M.E.; Abo, A.; Tomizuka, K.; Funk, W.D. R-spondin proteins: A novel link to beta-catenin activation. Cell Cycle 2006, 5, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Tomizuka, K.; Horikoshi, K.; Kitada, R.; Sugawara, Y.; Iba, Y.; Kojima, A.; Yoshitome, A.; Yamawaki, K.; Amagai, M.; Inoue, A.; et al. R-spondin1 plays an essential role in ovarian development through positively regulating Wnt-4 signaling. Hum. Mol. Genet. 2008, 17, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Yamada, W.; Nagao, K.; Horikoshi, K.; Fujikura, A.; Ikeda, E.; Inagaki, Y.; Kakitani, M.; Tomizuka, K.; Miyazaki, H.; Suda, T.; et al. Craniofacial malformation in R-spondin2 knockout mice. Biochem. Biophys. Res. Commun. 2009, 381, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Mieda, M.; Ikeda, T.; Hamada, Y.; Nakamura, H.; Okamoto, H. R-spondin3 is required for mouse placental development. Dev. Biol. 2007, 301, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Kazanskaya, O.; Ohkawara, B.; Heroult, M.; Wu, W.; Maltry, N.; Augustin, H.G.; Niehrs, C. The Wnt signaling regulator R-spondin 3 promotes angioblast and vascular development. Development 2008, 135, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Bruchle, N.O.; Frank, J.; Frank, V.; Senderek, J.; Akar, A.; Koc, E.; Rigopoulos, D.; van Steensel, M.; Zerres, K.; Bergmann, C. RSPO4 is the major gene in autosomal-recessive anonychia and mutations cluster in the furin-like cysteine-rich domains of the Wnt signaling ligand R-spondin 4. J. Investig. Dermatol. 2008, 128, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Schatoff, E.M.; Murphy, C.; Zafra, M.P.; Wilkinson, J.E.; Elemento, O.; Dow, L.E. R-spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat. Commun. 2017, 8, 15945. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.Y.; Lee, T.L.; Hao, S.S.; Mahooti, S.; Baird, S.M.; Donoghue, D.J.; Haas, M. Visualization of early prostatic adenocarcinoma as a stem cell disease. Oncotarget 2016, 7, 76159–76168. [Google Scholar] [CrossRef] [PubMed]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Yue, J.; Wang, J.; Zhang, L.; Fan, R.; Zhang, H.; Zhang, Q. GPCR48/LGR4 promotes tumorigenesis of prostate cancer via PI3K/Akt signaling pathway. Med. Oncol. 2015, 32, 49. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The Hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Hariharan, I.K. The Hippo pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011288. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Hong, W. The emerging role of the Hippo pathway in cell contact inhibition, organ size control and cancer development in mammals. Cancer Cell 2008, 13, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Konsavage, W.M., Jr.; Yochum, G.S. Intersection of Hippo/YAP and Wnt/beta-catenin signaling pathways. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. Taz: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. Tead mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.K.; Sullivan, A.J.; Medina, R.; Ito, Y.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 2004, 23, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the Hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; McNeill, H.; et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 2010, 18, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Imajo, M.; Miyatake, K.; Iimura, A.; Miyamoto, A.; Nishida, E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signalling. EMBO J. 2012, 31, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Maitra, A.; Anders, R.A.; Taketo, M.M.; Pan, D. Beta-catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015, 29, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Beta-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Oudhoff, M.J.; Braam, M.J.S.; Freeman, S.A.; Wong, D.; Rattray, D.G.; Wang, J.; Antignano, F.; Snyder, K.; Refaeli, I.; Hughes, M.R.; et al. SETD7 controls intestinal regeneration and tumorigenesis by regulating Wnt/beta-catenin and Hippo/YAP signaling. Dev. Cell 2016, 37, 47–57. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kriz, V.; Korinek, V. Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells. Genes 2018, 9, 20. https://doi.org/10.3390/genes9010020
Kriz V, Korinek V. Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells. Genes. 2018; 9(1):20. https://doi.org/10.3390/genes9010020
Chicago/Turabian StyleKriz, Vitezslav, and Vladimir Korinek. 2018. "Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells" Genes 9, no. 1: 20. https://doi.org/10.3390/genes9010020
APA StyleKriz, V., & Korinek, V. (2018). Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells. Genes, 9(1), 20. https://doi.org/10.3390/genes9010020