Advances in Genomic Profiling and Analysis of 3D Chromatin Structure and Interaction

 , ,
, , 
Abstract
1. Introduction
2. Nuclear Organization and Functional Elements
2.1. The Structure and Functional Units of Chromosome
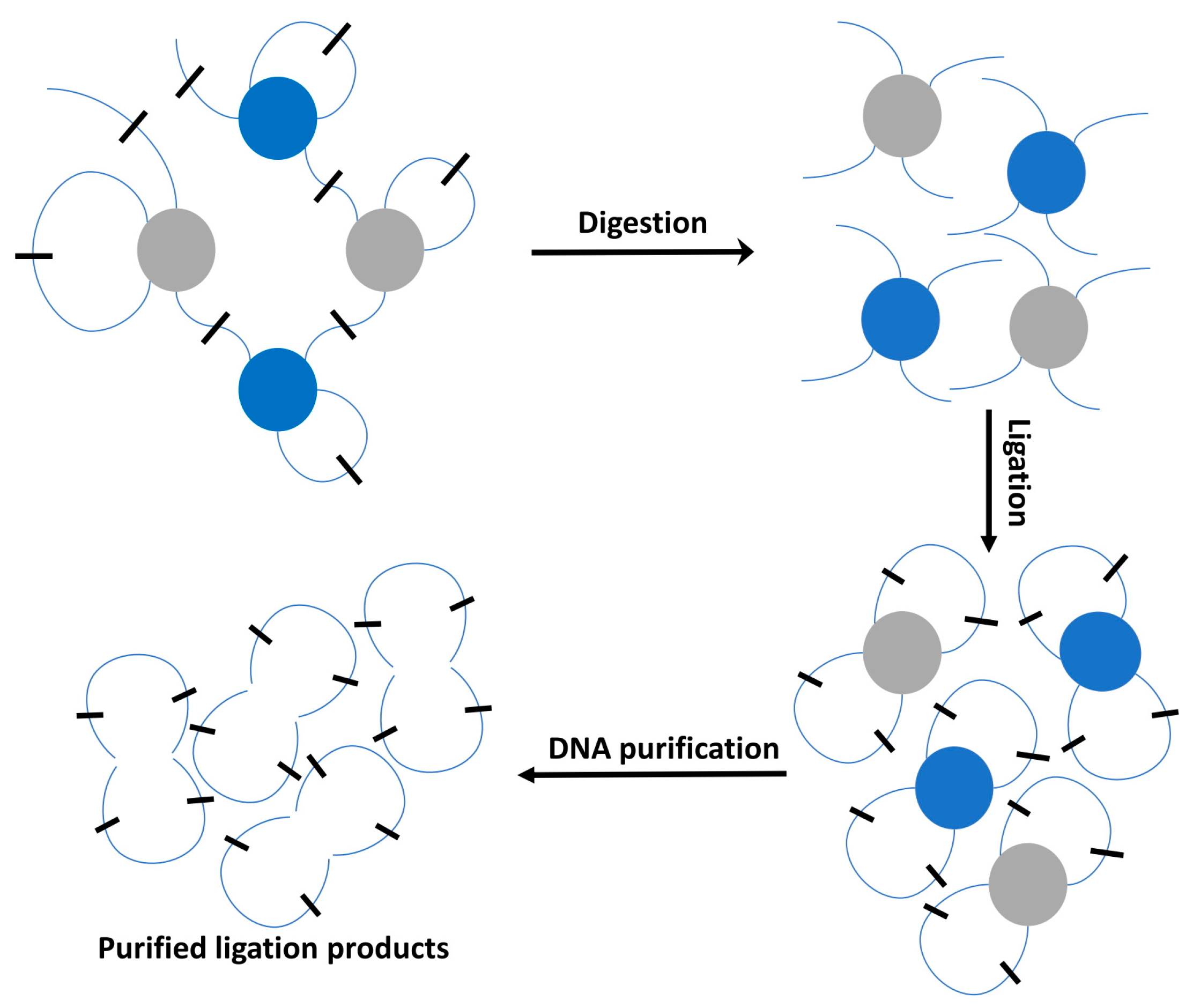
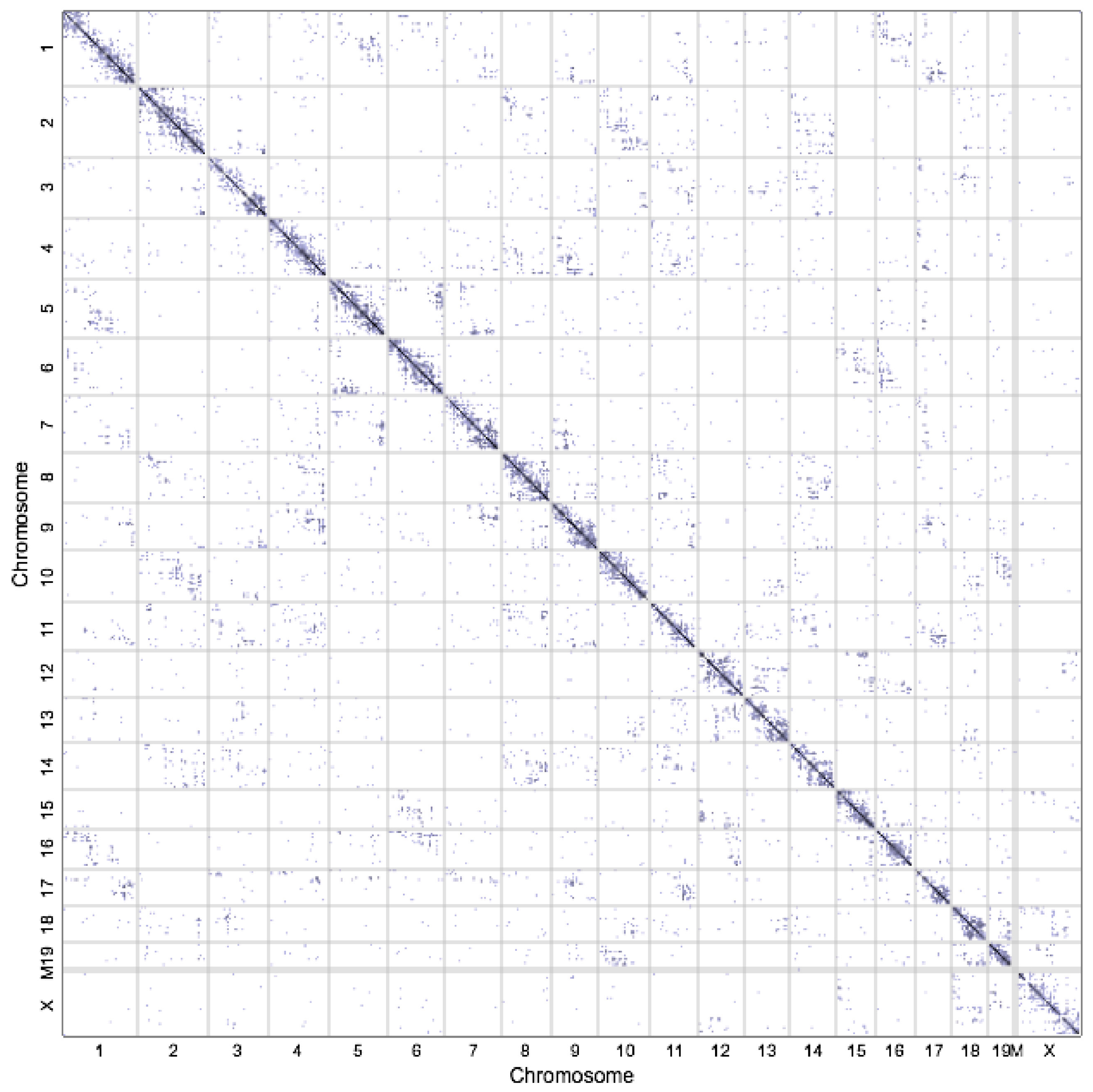
2.2. High-Throughput Profiling 3D Techniques
3. Advances in Statistical and Computational Analyses
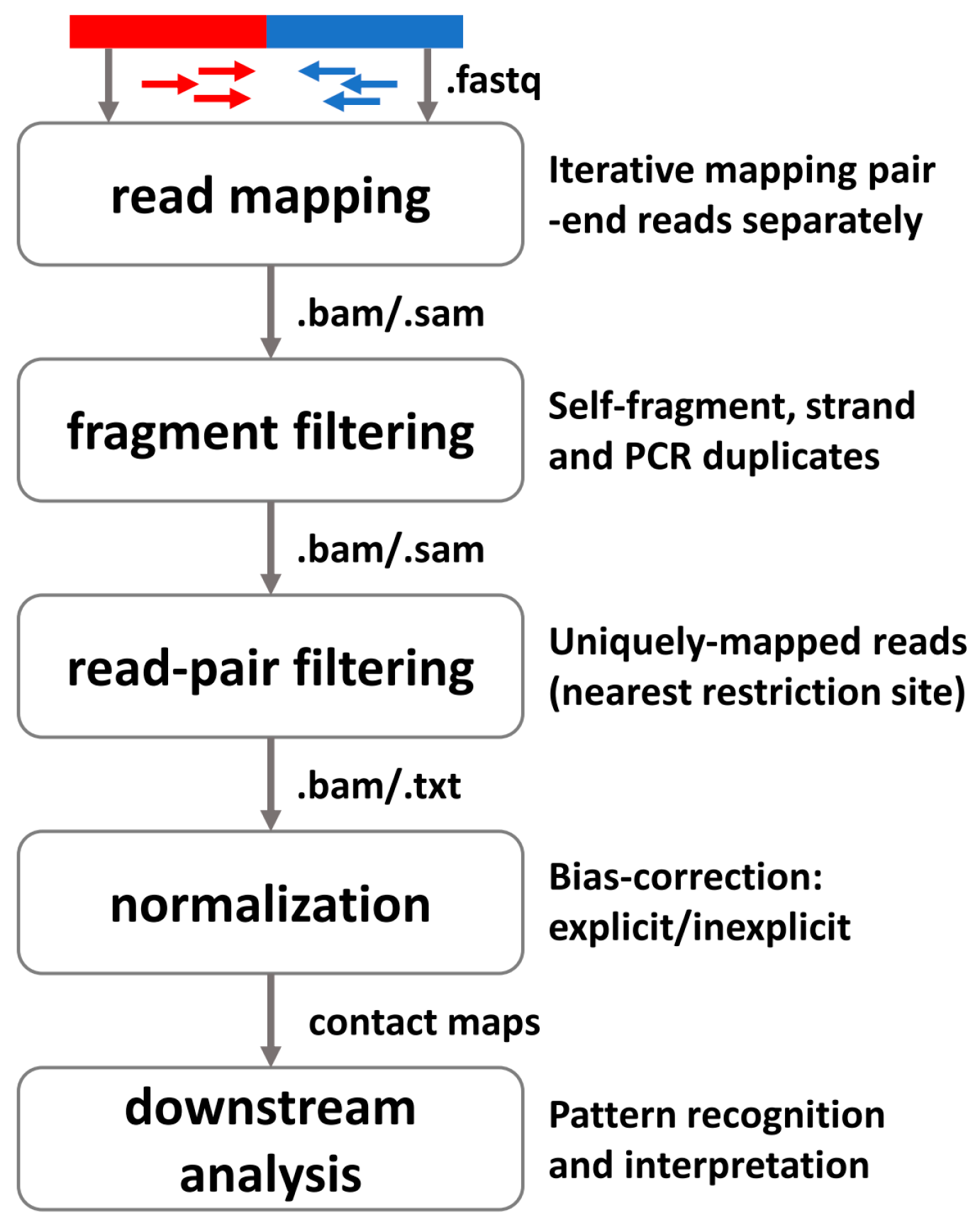
3.1. General Pipeline for Preprocessing High-Throughput Profiling 3D Data
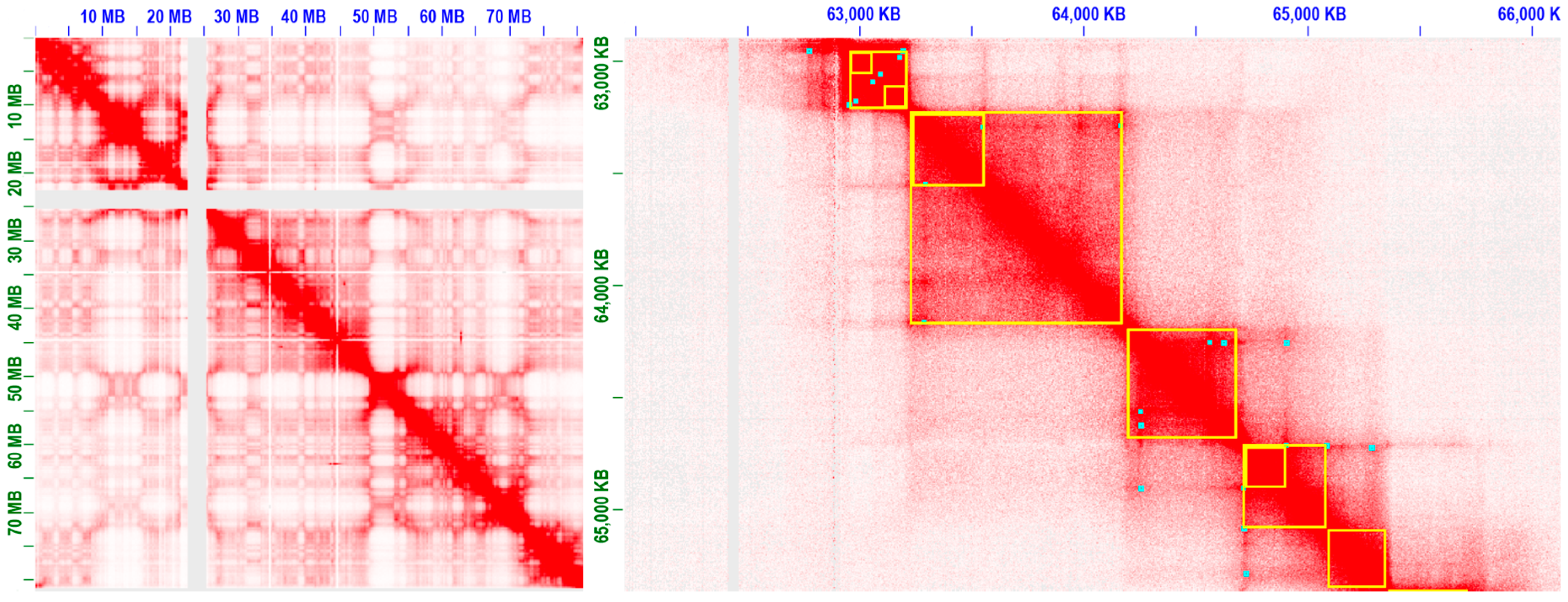
3.2. Progresses in Modeling and Analyse of 3D Chromatin Interactions
3.3. Processes in Inferring 3D Spatial Structure
4. Advances in Single Cell Hi-C Computational Analyses
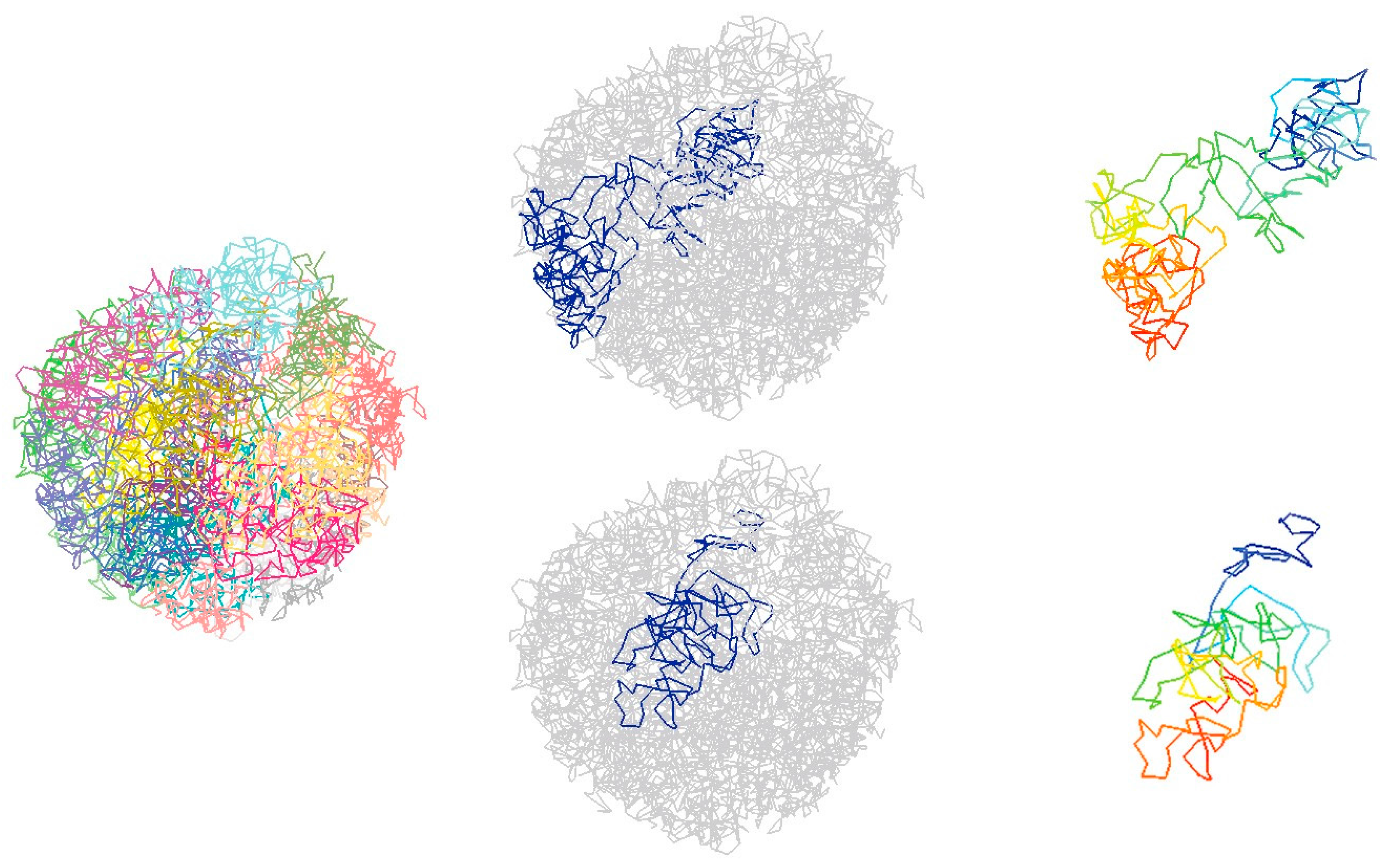
4.1. Computational Methods to Infer Chromatin 3D Structure from Single-Cell Hi-C Data
4.2. Challenges for Single Cell Hi-C Computational Analyses
5. Conclusions and Future Perspectives
5.1 Advances in 3D Chromatin Structure Interaction in Diseases
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Giniger, E.; Varnum, S.M.; Ptashne, M. Specific DNA binding of GAL4, a positive regulatory protein of yeast. Cell 1985, 40, 767–774. [Google Scholar] [CrossRef]
- Rusk, N. When chia pets meet Hi-C. Nat. Meth. 2009, 6, 863. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Feuerborn, A.; Cook, P.R. Why the activity of a gene depends on its neighbors. Trends Genet. 2015, 31, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Ahituv, N. Gene regulatory elements, major drivers of human disease. Ann. Rev. Genom. Hum. Genet. 2017, 18, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Pombo, A.; Dillon, N. Three-dimensional genome architecture: Players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015, 16, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, D.; Leleu, M.; Schorderet, P.; Joye, E.; Chabaud, F.; Duboule, D. Temporal dynamics and developmental memory of 3D chromatin architecture at hox gene loci. eLife 2014, 3, e02557. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y. Chromatin architecture reorganization during stem cell differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef] [PubMed]
- Tolhuis, B.; Palstra, R.-J.; Splinter, E.; Grosveld, F.; de Laat, W. Looping and interaction between hypersensitive sites in the active β-globin locus. Mol. Cell 2002, 10, 1453–1465. [Google Scholar] [CrossRef]
- Lan, X.; Witt, H.; Katsumura, K.; Ye, Z.; Wang, Q.; Bresnick, E.H.; Farnham, P.J.; Jin, V.X. Integration of Hi-C and CHIP-seq data reveals distinct types of chromatin linkages. Nucleic Acids Res. 2012, 40, 7690–7704. [Google Scholar] [CrossRef] [PubMed]
- Ktistaki, E.; Garefalaki, A.; Williams, A.; Andrews, S.R.; Bell, D.M.; Foster, K.E.; Spilianakis, C.G.; Flavell, R.A.; Kosyakova, N.; Trifonov, V.; et al. CD8 locus nuclear dynamics during thymocyte development. J. Immunol. 2010, 184, 5686–5695. [Google Scholar] [CrossRef] [PubMed]
- Markova, E.N.; Kantidze, O.L.; Razin, S.V. Transcriptional regulation and spatial organisation of the human AML1/RUNX1 gene. J. Cell. Biochem. 2011, 112, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Blackledge, N.P.; Ott, C.J.; Gillen, A.E.; Harris, A. An insulator element 3′ to the CFTR gene binds CTCF and reveals an active chromatin hub in primary cells. Nucleic Acids Res. 2009, 37, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Ghavi-Helm, Y.; Klein, F.A.; Pakozdi, T.; Ciglar, L.; Noordermeer, D.; Huber, W. Enhancer loops appear stable during development and are associated with paused polymerase. Nature 2014, 512, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yue, F.; McCleary, D.F.; Ye, Z.; Edsall, L.; Kuan, S.; Wagner, U.; Dixon, J.; Lee, L.; Lobanenkov, V.V.; et al. A map of the cis-regulatory sequences in the mouse genome. Nature 2012, 488, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Symmons, O.; Uslu, V.V.; Tsujimura, T.; Ruf, S.; Nassari, S.; Schwarzer, W.; Ettwiller, L.; Spitz, F. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014, 24, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.M.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Cremins, J.E.; Sauria, M.E.G.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.K.; Ong, C.-T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013, 153, 1281–1295. [Google Scholar]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Beagan, J.A.; Duong, M.T.; Titus, K.R.; Zhou, L.; Cao, Z.; Ma, J.; Lachanski, C.V.; Gillis, D.R.; Phillips-Cremins, J.E. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017, 27, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.-A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.; Cusanovich, D.A.; Hause, R.J.; Ma, W.; Qiu, R.; Deng, X.; Blau, C.A.; Disteche, C.M.; Noble, W.S.; Shendure, J.; et al. Mapping 3D genome architecture through in situ dnase Hi-C. Nat. Protoc. 2016, 11, 2104–2121. [Google Scholar] [CrossRef] [PubMed]
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; de Wit, E.; van Steensel, B.; de Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 2006, 38, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome conformation capture carbon copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Stadhouders, R.; Kolovos, P.; Brouwer, R.; Zuin, J.; van den Heuvel, A.; Kockx, C.; Palstra, R.-J.; Wendt, K.S.; Grosveld, F.; van Ijcken, W.; et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat. Protoc. 2013, 8, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hirao, A. Maintenance of genomic integrity in hematopoietic stem cells. Int. J. Hematol. 2011, 93, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.R.; Roberts, N.; McGowan, S.; Hay, D.; Giannoulatou, E.; Lynch, M.; De Gobbi, M.; Taylor, S.; Gibbons, R.; Higgs, D.R. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. 2014, 46, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Kalhor, R.; Tjong, H.; Jayathilaka, N.; Alber, F.; Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 2012, 30, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Kolovos, P.; van de Werken, H.J.; Kepper, N.; Zuin, J.; Brouwer, R.W.; Kockx, C.E.; Wendt, K.S.; van IJcken, W.F.; Grosveld, F.; Knoch, T.A. Targeted chromatin capture (T2C): A novel high resolution high throughput method to detect genomic interactions and regulatory elements. Epigenet. Chromatin 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Nicodemi, M.; Pombo, A. Models of chromosome structure. Curr. Opin. Cell Biol. 2014, 28. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.-M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Li, W.; Tjong, H.; Hao, S.; Zhou, Y.; Li, Q.; Chen, L.; Zhu, B.; Alber, F.; Jasmine Zhou, X. Mining 3D genome structure populations identifies major factors governing the stability of regulatory communities. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Whalen, S.; Truty, R.M.; Pollard, K.S. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016, 48, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Molitor, J.; Mallm, J.P.; Rippe, K.; Erdel, F. Retrieving chromatin patterns from deep sequencing data using correlation functions. Biophys. J. 2017, 112, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Bortle, K.V.; Corces, V.G. Nuclear organization and genome function. Ann. Rev. Cell Dev. Biol. 2012, 28, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Pancaldi, V.; Carrillo-de-Santa-Pau, E.; Javierre, B.M.; Juan, D.; Fraser, P.; Spivakov, M.; Valencia, A.; Rico, D. Integrating epigenomic data and 3D genomic structure with a new measure of chromatin assortativity. Genome Biol. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Michor, F. A computational strategy to adjust for copy number in tumor Hi-C data. Bioinformatics 2016, 32, 3695–3701. [Google Scholar] [CrossRef] [PubMed]
- Imakaev, M.; Fudenberg, G.; McCord, R.P.; Naumova, N.; Goloborodko, A.; Lajoie, B.R.; Dekker, J.; Mirny, L.A. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 2012, 9, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Bhardwaj, V.; Villaveces, J.; Arrigoni, L.; Gruening, B.A.; Lam, K.C.; Habermann, B.; Akhtar, A.; Manke, T. High-resolution TADs reveal DNA sequences underlying genome organization in flies. bioRxiv 2017. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.-J.; Vert, J.-P. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Serra, F.; Baù, D.; Filion, G.; Marti-Renom, M.A. Structural features of the fly chromatin colors revealed by automatic three-dimensional modeling. bioRxiv 2016. [Google Scholar] [CrossRef]
- Wingett, S.; Ewels, P.; Furlan-Magaril, M.; Nagano, T.; Schoenfelder, S.; Fraser, P.; Andrews, S. HiCUP: Pipeline for mapping and processing Hi-C data. F1000 Res. 2015, 4, 1310. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.W.; Grob, S.; Grossniklaus, U. HiCdat: A fast and easy-to-use Hi-C data analysis tool. BMC Bioinform. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.-C.; Lin, C.-F.; Valladares, O.; Malamon, J.; Kuksa, P.P.; Zheng, Q.; Gregory, B.D.; Wang, L.-S. Hippie: A high-throughput identification pipeline for promoter interacting enhancer elements. Bioinformatics 2015, 31, 1290–1292. [Google Scholar] [CrossRef] [PubMed]
- Castellano, G.; Le Dily, F.; Hermoso Pulido, A.; Beato, M.; Roma, G. Hi-Cpipe: A pipeline for high-throughput chromosome capture. bioRxiv 2015. [Google Scholar] [CrossRef]
- Weinreb, C.; Raphael, B.J. Identification of hierarchical chromatin domains. Bioinformatics 2016, 32, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Shi, Y.; Dai, C.; Tjong, H.; Gong, K.; Alber, F.; Zhou, X.J. Topdom: An efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016, 44, e70. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Mirny, L. The 3D genome as moderator of chromosomal communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Gorkin, D.U.; Leung, D.; Ren, B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 2014, 14, 762–775. [Google Scholar]
- Zhu, Y.; Chen, Z.; Zhang, K.; Wang, M.; Medovoy, D.; Whitaker, J.W. Constructing 3D interaction maps from 1D epigenomes. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ay, F.; Noble, W. Analysis methods for studying the 3D architecture of the genome. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Sekelja, M.; Paulsen, J.; Collas, P. 4D nucleomes in single cells: What can computational modeling reveal about spatial chromatin conformation? Genome Biol. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.M.; Hendy, M.D.; Pichugina, T.; Wake, G.C.; Langowski, J. The statistical-mechanics of chromosome conformation capture. Nucleus 2013, 4, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, G.; Toh, K.-C.; Sung, W.-K. 3D chromosome modeling with semi-definite programming and Hi-C data. J. Comput. Biol. 2013, 20, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Deng, K.; Qin, Z.; Dixon, J.; Selvaraj, S.; Fang, J.; Ren, B.; Liu, J.S. Bayesian inference of spatial organizations of chromosomes. PLoS Comput. Biol. 2013, 9, e1002893. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Naumova, N.; Imakaev, M.; Fudenberg, G.; Zhan, Y.; Lajoie, B.R.; Mirny, L.A.; Dekker, J. Organization of the mitotic chromosome. Science 2013, 342, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, F.J.; Disteche, C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Li, B.; Conneely, K.N.; Wu, H.; Hu, M.; Ayyala, D.; Park, Y.; Jin, V.X.; Zhang, F.; Zhang, H.; et al. Statistical challenges in analyzing methylation and long-range chromosomal interaction data. Stat. Biosci. 2016, 8, 284–309. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Lubling, Y.; Yaffe, E.; Wingett, S.W.; Dean, W.; Tanay, A.; Fraser, P. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat. Protoc. 2015, 10, 1986–2003. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.; Gramstad, O.; Collas, P. Manifold based optimization for single-cell 3D genome reconstruction. PLoS Comput. Biol. 2015, 11, e1004396. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-cell epigenomics: Powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhou, Y.; Wang, C.-M.; Huang, T.H.M.; Jin, V.X. Integration of DNA methylation and gene transcription across nineteen cell types reveals cell type-specific and genomic region-dependent regulatory patterns. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [PubMed]
- Barutcu, A.R.; Lajoie, B.R.; McCord, R.P.; Tye, C.E.; Hong, D.; Messier, T.L.; Browne, G.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; et al. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.-Y.; Hsu, H.-K.; Lan, X.; Juan, L.; Yan, P.S.; Labanowska, J.; Heerema, N.; Hsiao, T.-H.; Chiu, Y.-C.; Chen, Y.; et al. Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer. Cancer Cell 2013, 24, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stutz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 2017, 49, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Ganeshan, B.; Irshad, S.; Lawler, K.; Eisenblätter, M.; Milewicz, H.; Rodriguez-Justo, M.; Miles, K.; Ellis, P.; Groves, A.; et al. The use of molecular imaging combined with genomic techniques to understand the heterogeneity in cancer metastasis. Br. J. Radiol. 2014, 87. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Mai, S. The use of 3D telomere FISH for the characterization of the nuclear architecture in EBV-positive hodgkin’s lymphoma. In Epstein Barr Virus: Methods and Protocols; Minarovits, J., Niller, H.H., Eds.; Springer: New York, NY, USA, 2017; pp. 93–104. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Sequencing Reads Aligner | Feature | Programing Language |
|---|---|---|---|
| HiCapp [44] | Bowtie2 | Correct copy number bias | Shell |
| hiclib [45] | Bowtie2 | Iterative | Python |
| HiCExplorer [46] | Bwa, Bowtie2, Hisat2 | Check inter chromosomal fraction of reads | Python |
| HiC-Pro [47] | Bowtie2 | Trimming of reads | Python, R |
| TADbit [48] | GEM | Iterative | Python |
| HiCUP [49] | Bowtie, Bowtie2 | Pre-truncation | Perl, R |
| HiC-Box [50] | Bowtie2 | Correct contact maps for systematic biases | Python |
| HiCdat [51] | Subread | Analyze larger structural features | C++, R |
| HIPPIE [52] | BWA | Extract enhancer-target gene relationships | Python, Perl, R |
| HiC-inspector [53] | Bowtie | Focus on mapping and filtering | Perl, R |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.; Cheng, X.; Xi, Y.; Chen, Z.; Zhou, Y.; Jin, V.X. Advances in Genomic Profiling and Analysis of 3D Chromatin Structure and Interaction. Genes 2017, 8, 223. https://doi.org/10.3390/genes8090223
Tang B, Cheng X, Xi Y, Chen Z, Zhou Y, Jin VX. Advances in Genomic Profiling and Analysis of 3D Chromatin Structure and Interaction. Genes. 2017; 8(9):223. https://doi.org/10.3390/genes8090223
Chicago/Turabian StyleTang, Binhua, Xiaolong Cheng, Yunlong Xi, Zixin Chen, Yufan Zhou, and Victor X. Jin. 2017. "Advances in Genomic Profiling and Analysis of 3D Chromatin Structure and Interaction" Genes 8, no. 9: 223. https://doi.org/10.3390/genes8090223
APA StyleTang, B., Cheng, X., Xi, Y., Chen, Z., Zhou, Y., & Jin, V. X. (2017). Advances in Genomic Profiling and Analysis of 3D Chromatin Structure and Interaction. Genes, 8(9), 223. https://doi.org/10.3390/genes8090223