A Rare Form of Retinal Dystrophy Caused by Hypomorphic Nonsense Mutations in CEP290

Abstract
:1. Introduction
2. Material and Methods
2.1. Clinical Examination
2.2. Genetic Testing
2.3. Sequence Analysis of the CEP290 Gene
2.4. Analysis of Hypomorphic Character of the c.4723A>T Variant
3. Results
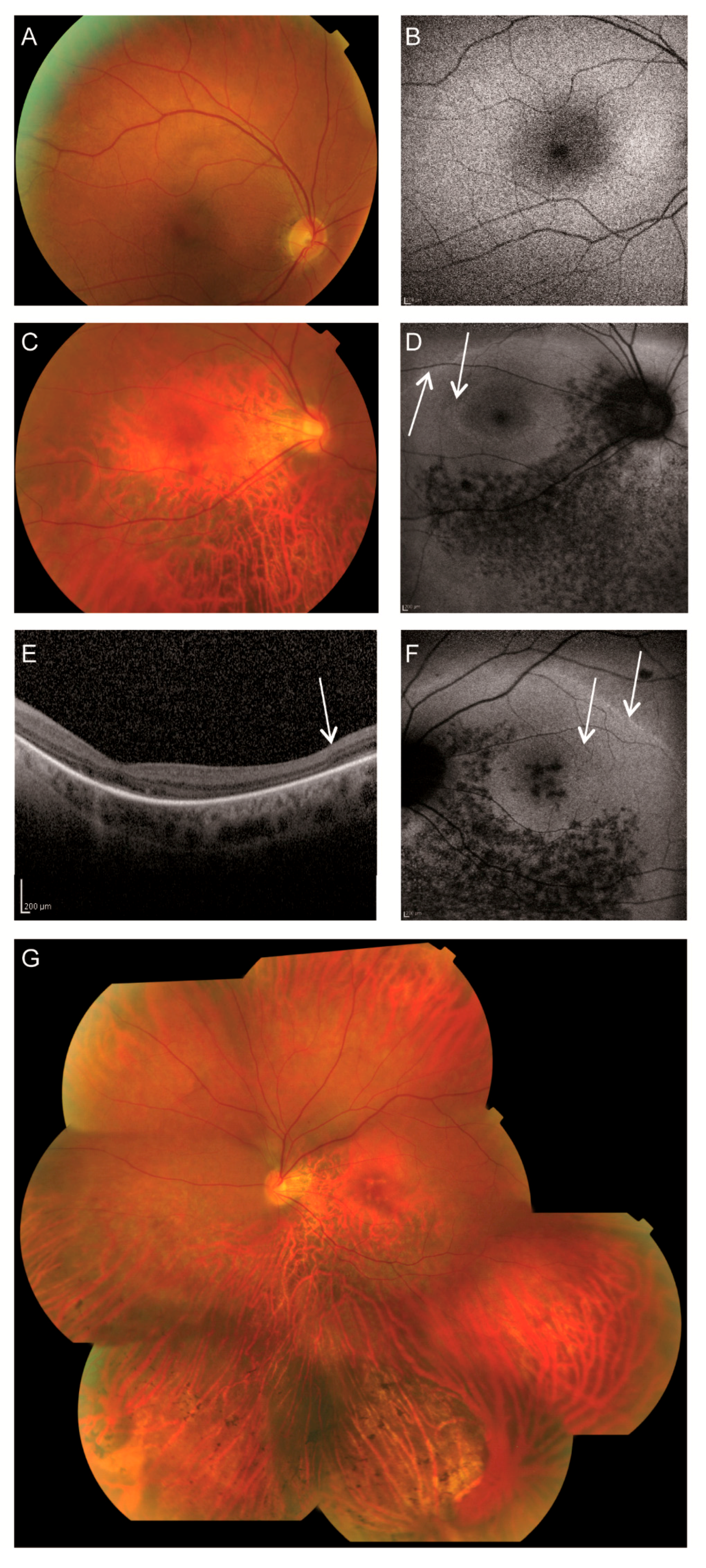
3.1. Clinical Findings
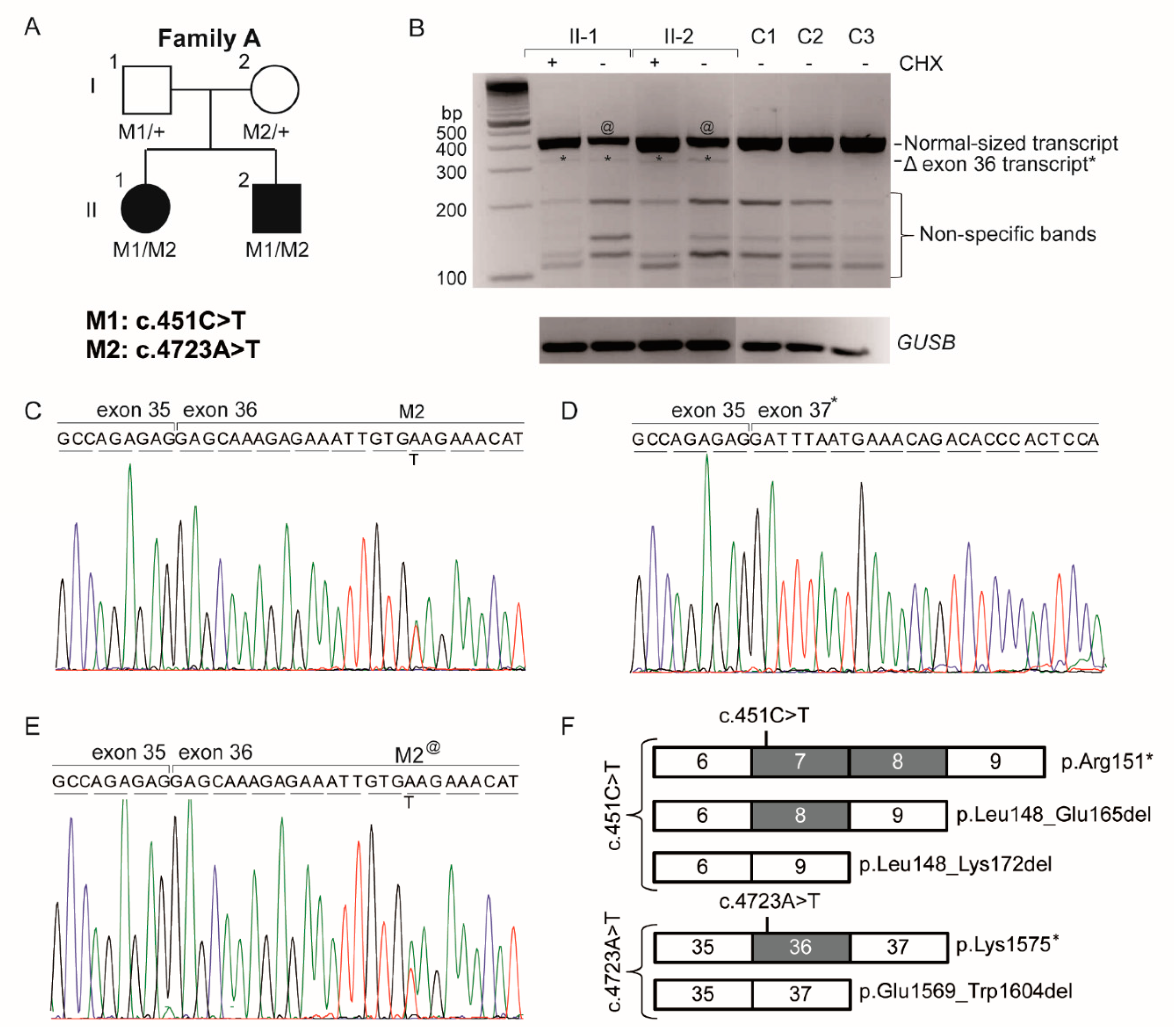
3.2. Genetic Analysis
3.3. Analysis of the Hypomorphic Character of c.4723A>T
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Craige, B.; Tsao, C.C.; Diener, D.R.; Hou, Y.; Lechtreck, K.F.; Rosenbaum, J.L.; Witman, G.B. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 2010, 190, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Barbelanne, M.; Song, J.; Ahmadzai, M.; Tsang, W.Y. Pathogenic NPHP5 mutations impair protein interaction with Cep290, a prerequisite for ciliogenesis. Hum. Mol. Genet. 2013, 22, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J.; et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Khanna, H.; Hawes, N.; Jimeno, D.; He, S.; Lillo, C.; Parapuram, S.K.; Cheng, H.; Scott, A.; Hurd, R.E.; et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 2006, 15, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; Pott, J.W.; Collin, R.W.J.; Kroes, H.Y.; Verheij, J.B.; Blokland, E.A.; de Castro Miro, M.; Hoyng, C.B.; Klaver, C.C.W.; Koenekoop, R.K.; et al. A novel nonsense mutation in CEP290 induces exon skipping and leads to a relatively mild retinal phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3646–3652. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Baala, L.; Audollent, S.; Martinovic, J.; Ozilou, C.; Babron, M.C.; Sivanandamoorthy, S.; Saunier, S.; Salomon, R.; Gonzales, M.; Rattenberry, E.; et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 2007, 81, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; De Baere, E. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Delphin, N.; Hanein, S.; Gerber, S.; Dufier, J.L.; Roche, O.; Defoort-Dhellemmes, S.; Dollfus, H.; Fazzi, E.; Munnich, A.; et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum. Mutat. 2007, 28, 416. [Google Scholar] [CrossRef] [PubMed]
- Van Lith, G.H.M. General cone dysfunction without achromatopsia. In 10th ISCERG Symposium; Documenta Ophthalmologica Proceedings Series; Springer: Dordrecht, The Netherlands, 1973; pp. 175–180. [Google Scholar]
- Vincent, A.; Wright, T.; Billingsley, G.; Westall, C.; Heon, E. Oligocone trichromacy is part of the spectrum of CNGA3-related cone system disorders. Ophthalmic Genet. 2011, 32, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Christoffersen, N.L.; Sander, B.; Edmund, C.; Larsen, M.; Grau, T.; Wissinger, B.; Kohl, S.; Rosenberg, T. Oligocone trichromacy: Clinical and molecular genetic investigations. Investig. Ophthalmol. Vis. Sci. 2010, 51, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Baumann, B.; Kohl, S.; Zrenner, E.; Jorgensen, A.L.; Wissinger, B. Variant phenotypes of incomplete achromatopsia in two cousins with GNAT2 gene mutations. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4256–4262. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Holder, G.E.; Bradshaw, K.; Hunt, D.M.; Mollon, J.D.; Moore, A.T. Oligocone trichromacy: A rare and unusual cone dysfunction syndrome. Br. J. Ophthalmol. 2004, 88, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.; Drumare, I.; Bouacha, I.; Puech, B.; Defoort-Dhellemmes, S. Long-term follow-up of two patients with oligocone trichromacy. Doc. Ophthalmol. 2015, 131, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M. ISCEV standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; van Genderen, M.M.; van Schooneveld, M.J.; Visser, L.; Riemslag, F.C.; Keunen, J.E.; Bakker, B.; Zonneveld, M.N.; den Hollander, A.I.; Cremers, F.P.; et al. A homozygous frameshift mutation in LRAT causes retinitis punctata albescens. Ophthalmology 2012, 119, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Frank, V.; den Hollander, A.I.; Bruchle, N.O.; Zonneveld, M.N.; Nurnberg, G.; Becker, C.; Du Bois, G.; Kendziorra, H.; Roosing, S.; Senderek, J.; et al. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat. 2008, 29, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- ExAC. Available online: http://exac.broadinstitute.org/ (accessed on 29 June 2017).
- IGSR: The International Genome Sample Resource; 1000 Genomes. Available online: http://www.1000genomes.org/ (accessed on 29 June 2017).
- Michaelides, M.; Rha, J.; Dees, E.W.; Baraas, R.C.; Wagner-Schuman, M.L.; Mollon, J.D.; Dubis, A.M.; Andersen, M.K.; Rosenberg, T.; Larsen, M.; et al. Integrity of the cone photoreceptor mosaic in oligocone trichromacy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4757–4764. [Google Scholar] [CrossRef] [PubMed]
- Hofer, H.; Carroll, J.; Neitz, J.; Neitz, M.; Williams, D.R. Organization of the human trichromatic cone mosaic. J. Neurosci. 2005, 25, 9669–9679. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.; Chong, N.H.; Robson, A.G.; Holder, G.E.; Moore, A.T.; Bird, A.C. Fundus autofluorescence in patients with Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2747–2752. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Tran, H.V.; Vaclavik, V.; Borruat, F.X.; Schorderet, D.F.; Munier, F.L. Double concentric autofluorescence ring in NR2E3-p.G56R -linked autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4754–4764. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Krishnaswami, S.R.; Gleeson, J.G. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum. Mol. Genet. 2008, 17, 3796–3805. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peranen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Aleman, T.S.; Jacobson, S.G.; Khanna, H.; Sumaroka, A.; Aguirre, G.K.; Schwartz, S.B.; Windsor, E.A.; He, S.; Chang, B.; et al. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: Implications for therapy of Leber congenital amaurosis. Hum. Mutat. 2007, 28, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, S.; Shimauchi-Matsukawa, Y.; Tachibanaki, S.; Kawamura, S. Highly efficient retinal metabolism in cones. Proc. Natl. Acad. Sci. USA 2008, 105, 16051–16056. [Google Scholar] [CrossRef] [PubMed]
- Stearns, G.; Evangelista, M.; Fadool, J.M.; Brockerhoff, S.E. A mutation in the cone-specific pde6 gene causes rapid cone photoreceptor degeneration in zebrafish. J. Neurosci. 2007, 27, 13866–13874. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, S.; Michalakis, S.; Claes, E.; Seeliger, M.W.; Humphries, P.; Biel, M.; Feigenspan, A. Synaptic plasticity in cnga3(−/−) mice: Cone bipolar cells react on the missing cone input and form ectopic synapses with rods. J. Neurosci. 2006, 26, 5248–5255. [Google Scholar] [CrossRef] [PubMed]
- Boudard, D.L.; Tanimoto, N.; Huber, G.; Beck, S.C.; Seeliger, M.W.; Hicks, D. Cone loss is delayed relative to rod loss during induced retinal degeneration in the diurnal cone-rich rodent Arvicanthis ansorgei. Neuroscience 2010, 169, 1815–1830. [Google Scholar] [CrossRef] [PubMed]
- Camacho, E.T.; Colon Velez, M.A.; Hernandez, D.J.; Rodriguez Bernier, U.; Van Laarhoven, J.; Wirkus, S. A mathematical model for photoreceptor interactions. J. Theor. Biol. 2010, 267, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Freed, M.A.; Sterling, P. Microcircuitry of the dark-adapted cat retina: Functional architecture of the rod-cone network. J. Neurosci. 1986, 6, 3505–3517. [Google Scholar] [PubMed]
- Bloomfield, S.A.; Dacheux, R.F. Rod vision: Pathways and processing in the mammalian retina. Prog. Retin. Eye Res. 2001, 20, 351–384. [Google Scholar] [CrossRef]
- Organisciak, D.T.; Darrow, R.M.; Barsalou, L.; Kutty, R.K.; Wiggert, B. Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Investig. Ophthalmol. Vis. Sci. 2003, 44, 486–492. [Google Scholar] [CrossRef]
- Naash, M.L.; Peachey, N.S.; Li, Z.Y.; Gryczan, C.C.; Goto, Y.; Blanks, J.; Milam, A.H.; Ripps, H. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Investig. Ophthalmol. Vis. Sci. 1996, 37, 775–782. [Google Scholar]
- Novarino, G.; Akizu, N.; Gleeson, J.G. Modeling human disease in humans: The ciliopathies. Cell 2011, 147, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Drivas, T.G.; Wojno, A.P.; Tucker, B.A.; Stone, E.M.; Bennett, J. Basal exon skipping and genetic pleiotropy: A predictive model of disease pathogenesis. Sci. Transl. Med. 2015, 7, 291ra297. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Casteels, I.; Meire, F.; De Jaegere, S.; Hooghe, S.; van Regemorter, N.; Van Esch, H.; Matuleviciene, A.; Nunes, L.; Meersschaut, V.; et al. Genetic screening of LCA in Belgium: Predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum. Mutat. 2010, 31, E1709–E1766. [Google Scholar] [CrossRef] [PubMed]
- Walia, S.; Fishman, G.A.; Jacobson, S.G.; Aleman, T.S.; Koenekoop, R.K.; Traboulsi, E.I.; Weleber, R.G.; Pennesi, M.E.; Heon, E.; Drack, A.; et al. Visual acuity in patients with Leber’s congenital amaurosis and early childhood-onset retinitis pigmentosa. Ophthalmology 2010, 117, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Barrano, G.; Silhavy, J.L.; Marsh, S.E.; Travaglini, L.; Bielas, S.L.; Amorini, M.; Zablocka, D.; Kayserili, H.; Al-Gazali, L.; et al. CEP290 mutations are frequently identified in the oculo-renal form of joubert syndrome-related disorders. Am. J. Hum. Genet. 2007, 81, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Halbritter, J.; Diaz, K.; Chaki, M.; Porath, J.D.; Tarrier, B.; Fu, C.; Innis, J.L.; Allen, S.J.; Lyons, R.H.; Stefanidis, C.J.; et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012, 49, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M. Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LVIX Edward Jackson memorial lecture. Am. J. Ophthalmol. 2007, 144, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Papon, J.F.; Perrault, I.; Coste, A.; Louis, B.; Gerard, X.; Hanein, S.; Fares-Taie, L.; Gerber, S.; Defoort-Dhellemmes, S.; Vojtek, A.M.; et al. Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis with CEP290 mutations. J. Med. Genet. 2010, 47, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Patient ID | II-1 | II-2 |
|---|---|---|
| Sex | Female | Male |
| Age at diagnosis | 3 | 2 |
| Age recent examination | 46 | 45 |
| Nystagmus | Present | Present |
| Visual acuity RE | 20/100 | 20/80 |
| Visual acuity LE | 20/100 | 20/160 |
| Refraction | RE: +1.25–2.25 × 19; LE: −0.25–3.00 × 170 | RE: −14.25–1.75 × 5; LE: −10.25–2.25 × 155 |
| Lens | Clear | Clear |
| Fundus | Pink optic discs, mildly attenuated arterioles, coarse-grained aspect RPE posterior pole and mid-periphery superior quadrants, faintly recognizable foveola reflex BE with subtle indication for bull’s eye-like maculopathy, RPE atrophy in far periphery of superior quadrants, RPE atrophy in mid- and far periphery of inferior quadrants with scarce bone-spicule pigmentations | Pink, myopic optic discs, mildly attenuated arterioles, subtle RPE alterations macula RE, ring-shaped atrophy surrounding the fovea LE, faintly recognizable foveola reflex BE, perimacular RPE atrophy, mild RPE changes superior quadrants, RPE atrophy inferior quadrants with bone-spicule pigmentations |
| Fundus autofluorescence | Relatively normal | Double hyperautofluorescent ring, hypoautofluorescent spots along and inferior to the inferior vascular arcade BE, and in macula LE |
| OCT | Failed | No discernible photoreceptor complexes at the macula, but present at the peripheral part of the scan. |
| Color vision (Panel D-15) | Saturated: normal Desaturated: minor errors RE, multiple errors LE, no specific axis | RE: de- and saturated: multiple errors mainly in tritan axis LE: failed |
| Visual field (Goldmann) | Radius < 100 | Altitudinal defect BE, partially including the center RE, central scotoma LE |
| ERG Dark adapted | Mildly reduced isolated rod responses with significantly reduced ‘mixed’ responses | Significantly reduced isolated rod and ‘mixed’ responses |
| ERG Light adapted | Non-recordable | Non-recordable |
| Miscellaneous | Anorexia, depressions, followed by the diagnosis of schizofrenia at age 29 | None |
| First Allele | Second Allele | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample ID | Diagnosis | DNA Variant | Predicted Protein Variant | Predicted Proteins Based on RNA Study | DNA Variant | Predicted Protein Variant | Predicted Proteins Based on RNA Study | Ref. |
| Compound heterozygous | ||||||||
| Family A | OT | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] # | c.451C>T | p.(Arg151*) | p.[Arg151*, Leu148_Glu165del, Leu148_Lys172del] | This study |
| 809 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.1709C>G | p.(Ser570*) | ND | [10] |
| LCA-6 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.2991+1655A>G | p.(Cys998*) | p.[Cys998*, =] $ | [41] |
| LCA-7 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.2991+1655A>G | p.(Cys998*) | p.[Cys998*, =] | [41] |
| LCA-8 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.2991+1655A>G | p.(Cys998*) | p.[Cys998*, =] | [41] |
| LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.2991+1655A>G | p.(Cys998*) | p.[Cys998*, =] | [42] | |
| LCA-24 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4696G>C | p.(Ala1556Pro) | ND | [41] |
| COR031/CORS1 | CORS | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4393C>T | p.(Arg1465*) | ND | [10,43] |
| SLS-2 | SLSN | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4393C>T | p.(Arg1465*) | ND | [41] |
| SLS-3 | SLSN | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4393C>T | p.(Arg1465*) | ND | [41] |
| F283-21 | SLSN | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.1984C>T | p.(Gln662*) | ND | [44] |
| A3100-21 | SLSN | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.1987A>T | p.(Lys663*) | ND | [44] |
| A1210-21 | SLSN | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.3802C>T | p.(Gln1268*) | ND | [44] |
| F118-21 | SLSN | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4452_4455delAGAA | p.(Lys1484Asnfs*4) | ND | [44] |
| A1712-21 | SLSN | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.1189+1A>G | p.(?) | ND | [44] |
| Homozygous | ||||||||
| 1 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [46] |
| 2 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [46] |
| 738 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [10] |
| 848 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [10] |
| 258 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [10] |
| 419 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [10] |
| LEP | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [10] |
| LCA-25 | LCA | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [41] |
| 623 | JBTS+retina | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | c.4723A>T | p.(Lys1575*) | p.[Lys1575*, Glu1569_Trp1604del] | [10] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roosing, S.; Cremers, F.P.M.; Riemslag, F.C.C.; Zonneveld-Vrieling, M.N.; Talsma, H.E.; Klessens-Godfroy, F.J.M.; Den Hollander, A.I.; Van den Born, L.I. A Rare Form of Retinal Dystrophy Caused by Hypomorphic Nonsense Mutations in CEP290. Genes 2017, 8, 208. https://doi.org/10.3390/genes8080208
Roosing S, Cremers FPM, Riemslag FCC, Zonneveld-Vrieling MN, Talsma HE, Klessens-Godfroy FJM, Den Hollander AI, Van den Born LI. A Rare Form of Retinal Dystrophy Caused by Hypomorphic Nonsense Mutations in CEP290. Genes. 2017; 8(8):208. https://doi.org/10.3390/genes8080208
Chicago/Turabian StyleRoosing, Susanne, Frans P. M. Cremers, Frans C. C. Riemslag, Marijke N. Zonneveld-Vrieling, Herman E. Talsma, Francoise J. M. Klessens-Godfroy, Anneke I. Den Hollander, and L. Ingeborgh Van den Born. 2017. "A Rare Form of Retinal Dystrophy Caused by Hypomorphic Nonsense Mutations in CEP290" Genes 8, no. 8: 208. https://doi.org/10.3390/genes8080208
APA StyleRoosing, S., Cremers, F. P. M., Riemslag, F. C. C., Zonneveld-Vrieling, M. N., Talsma, H. E., Klessens-Godfroy, F. J. M., Den Hollander, A. I., & Van den Born, L. I. (2017). A Rare Form of Retinal Dystrophy Caused by Hypomorphic Nonsense Mutations in CEP290. Genes, 8(8), 208. https://doi.org/10.3390/genes8080208