EYS Mutations Causing Autosomal Recessive Retinitis Pigmentosa: Changes of Retinal Structure and Function with Disease Progression

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Genetic Analysis
2.3. Psychophysics
2.4. Imaging
3. Results
3.1. Clinical Characteristics of the EYS Patients in this Study
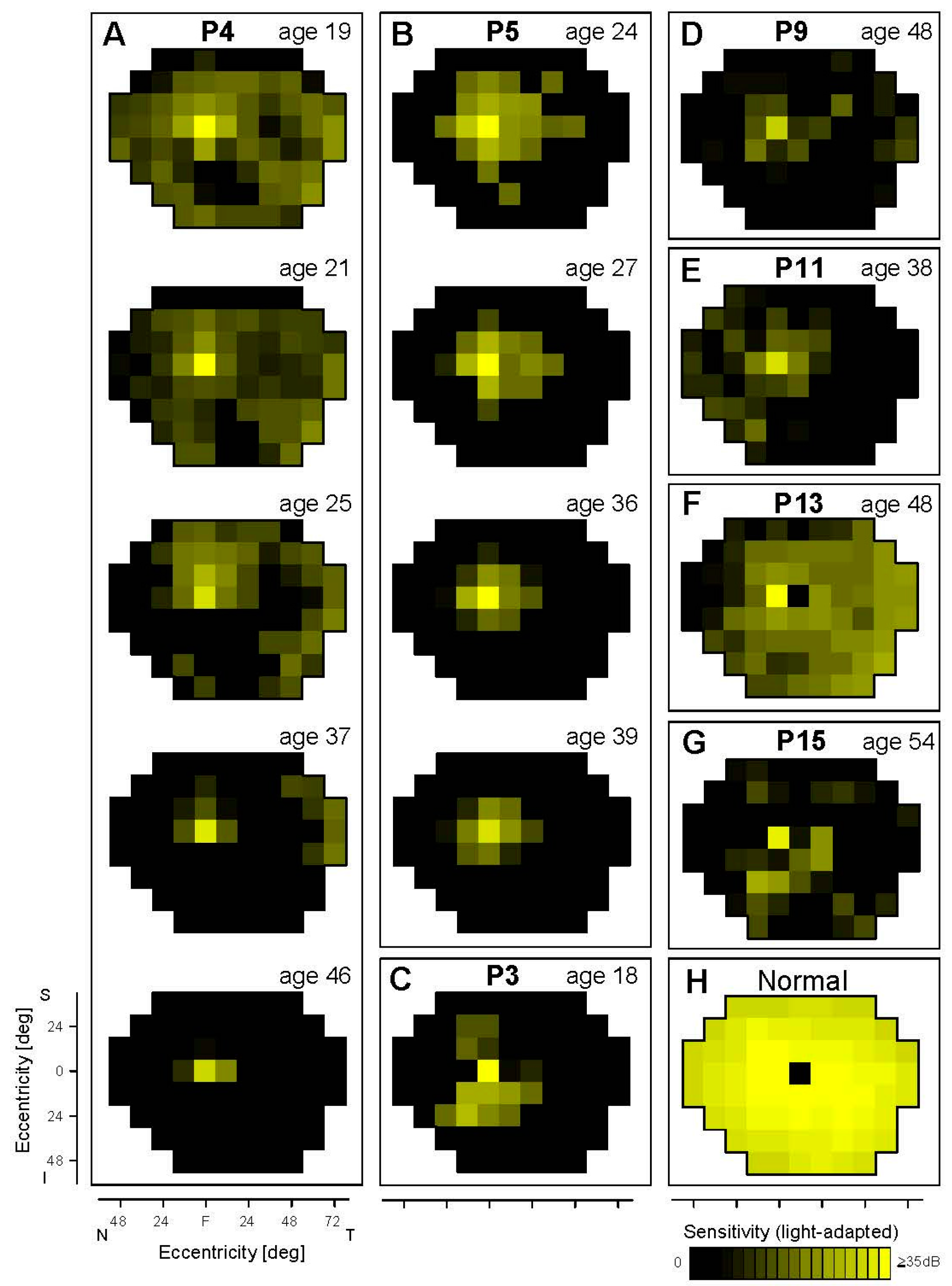
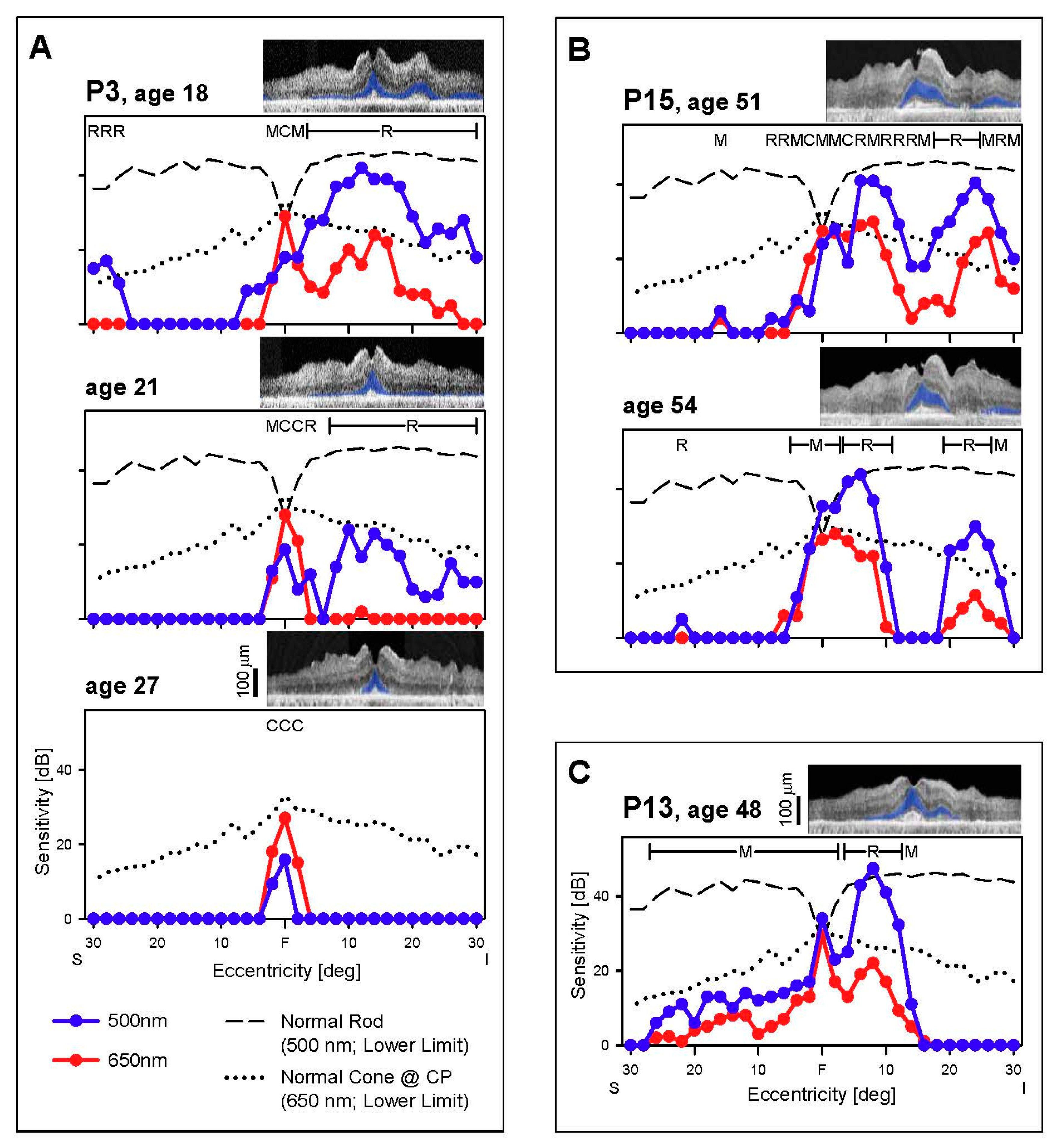
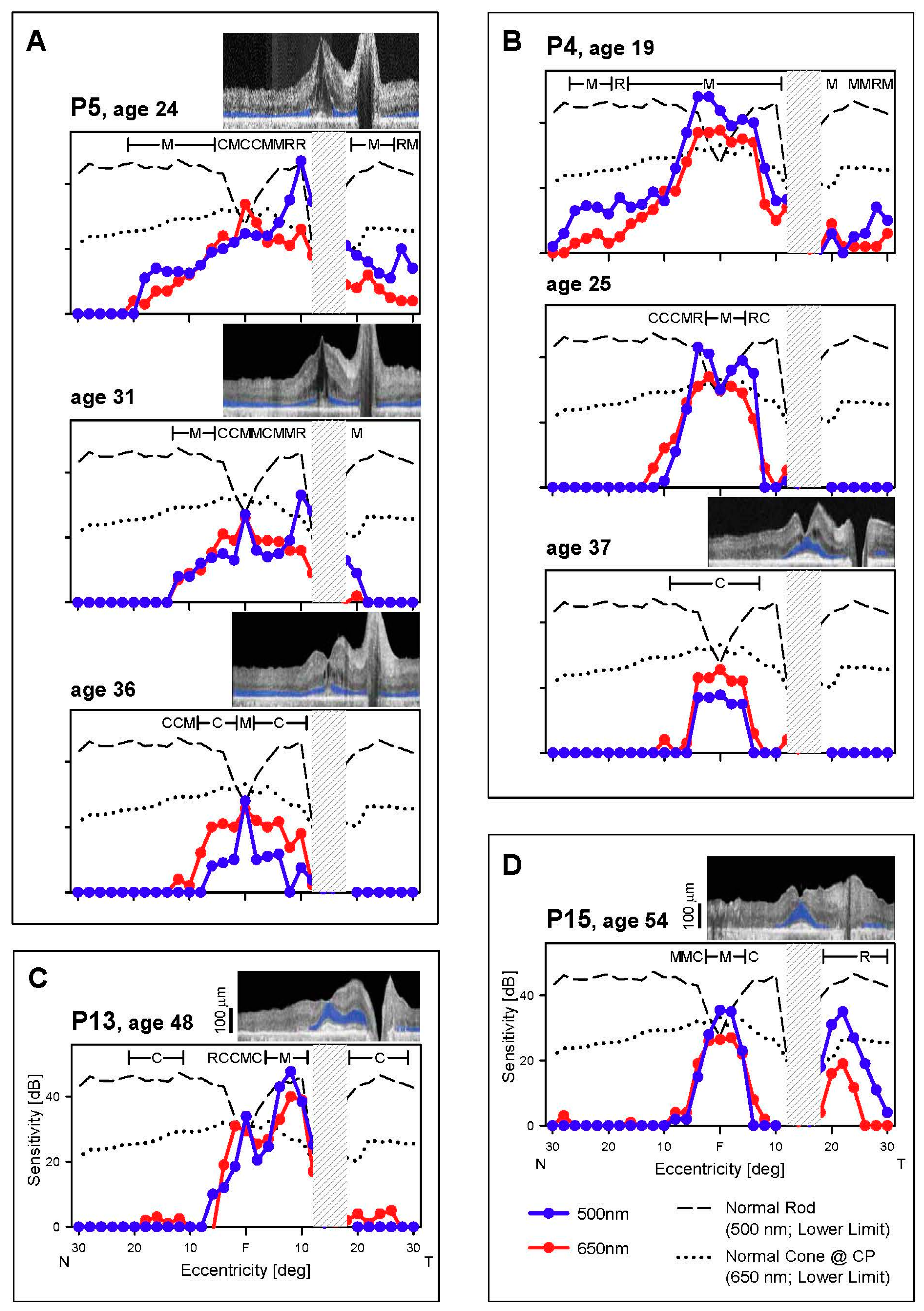
3.2. Cone and Rod Function Determined Perimetrically in EYS
3.2.1. Peripheral Cones and Rods
3.2.2. Central Cones and Rods
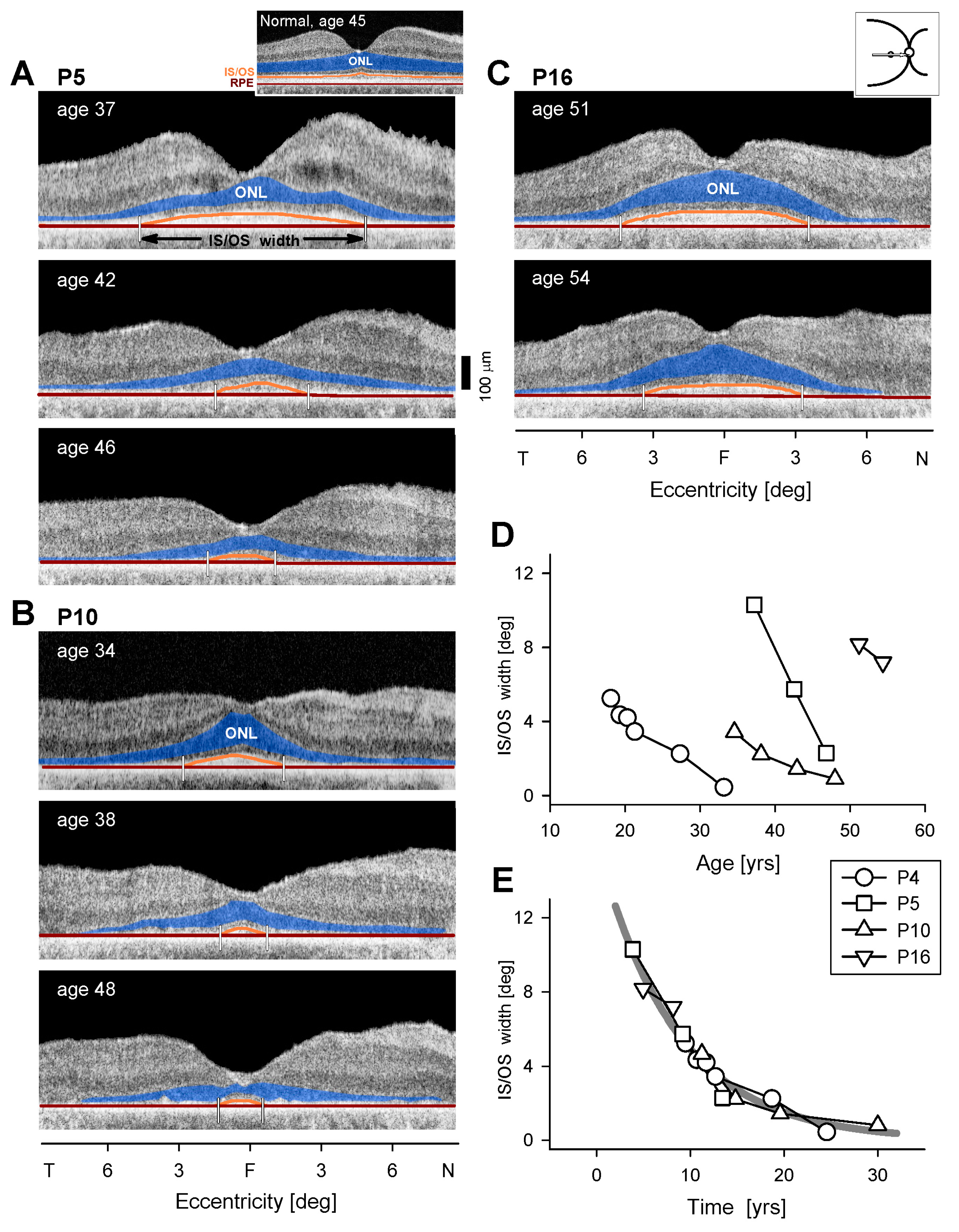
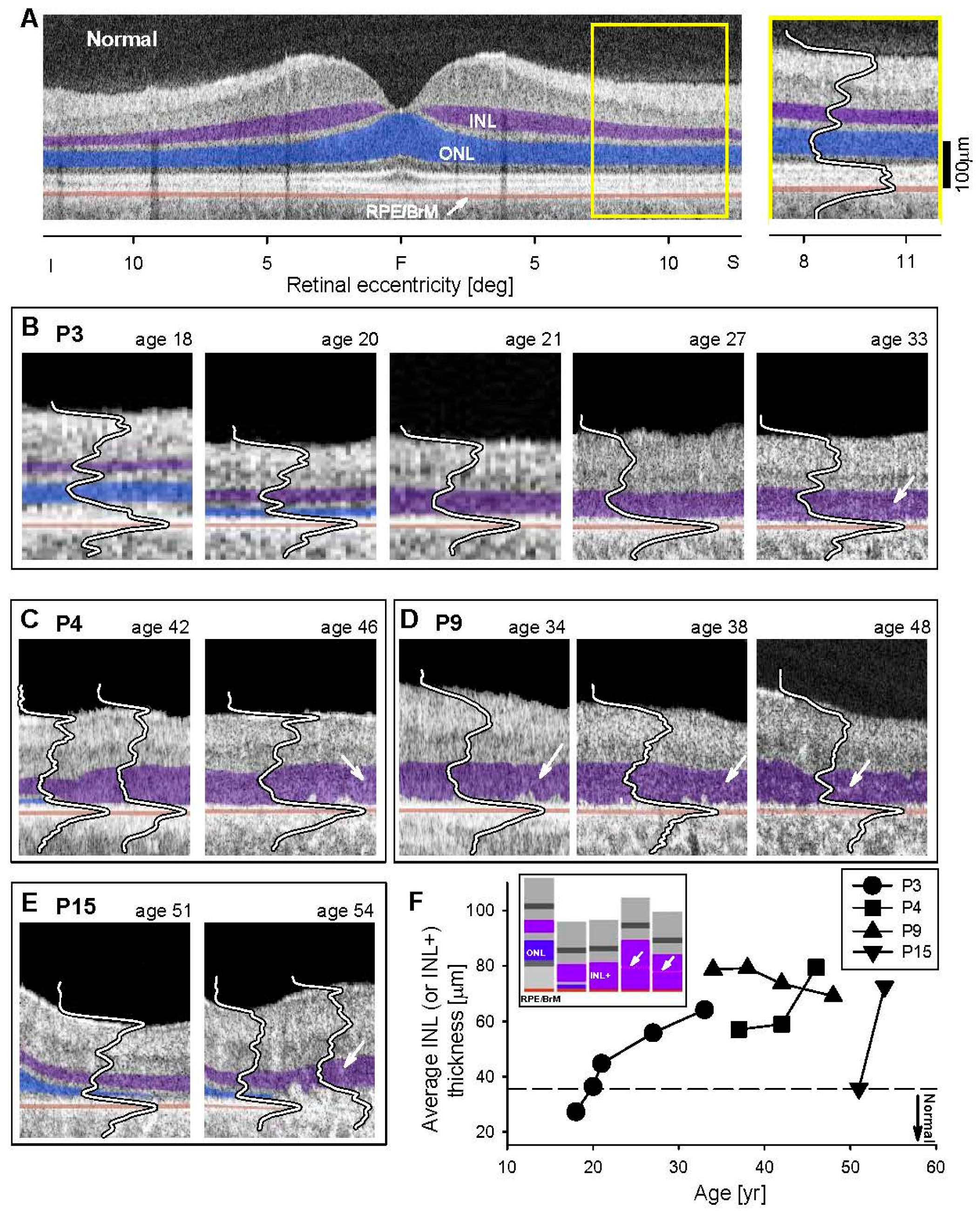
3.3. Retinal Laminar Architecture by OCT
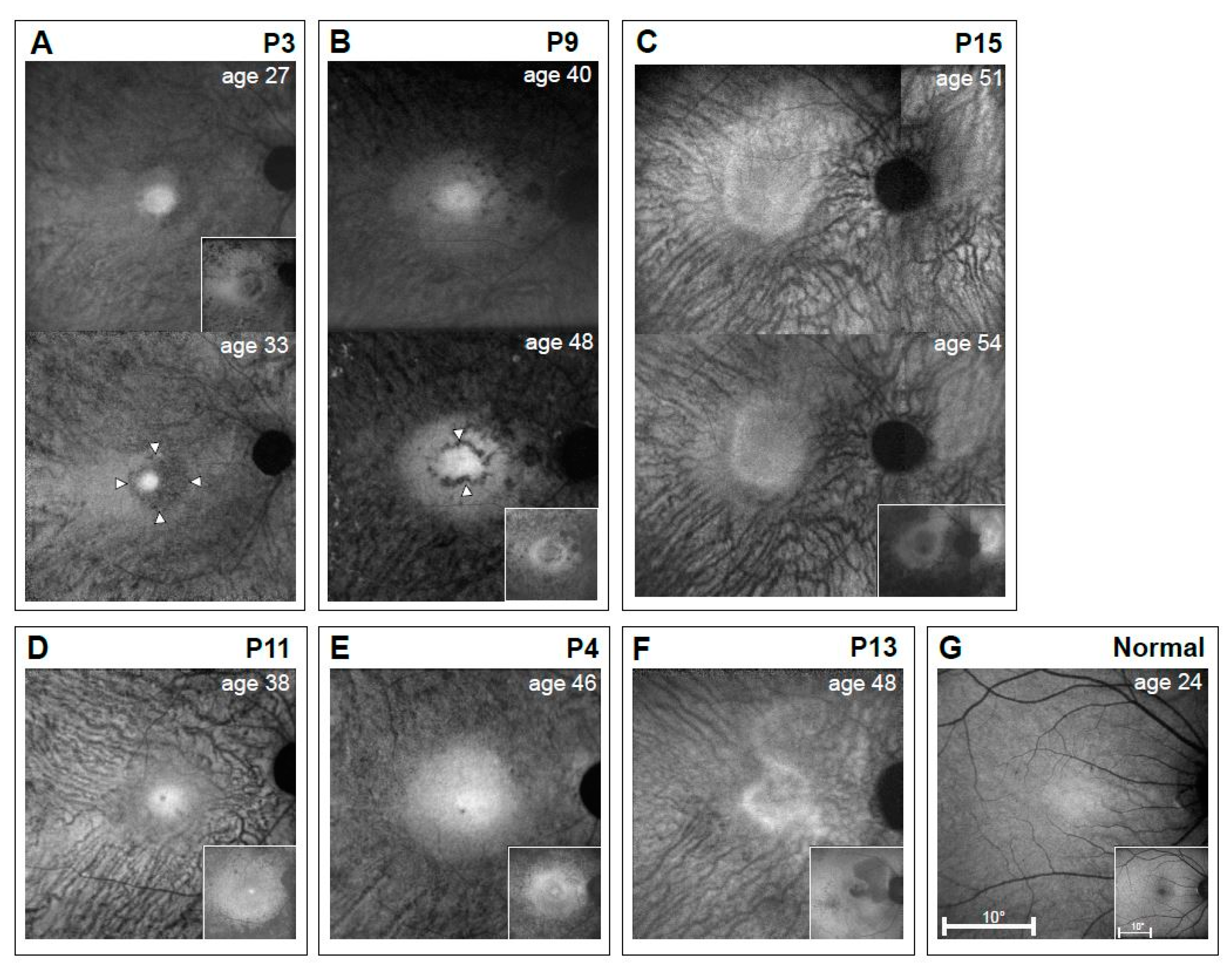
3.4. RPE Disease in EYS
4. Discussion
4.1. Comparing EYS-RP Progression to That in Other Ciliopathies
4.2. Inner Retinal Abnormalities in EYS
4.3. Mutation Spectrum in this EYS Cohort Versus Others
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abd El-Aziz, M.M.; Barragan, I.; O’Driscoll, C.A.; Goodstadt, L.; Prigmore, E.; Borrego, S.; Mena, M.; Pieras, J.I.; El-Ashry, M.F.; Safieh, L.A.; et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 2008, 40, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Sahel, J.A.; Mohand-Saïd, S.; Lancelot, M.E.; Antonio, A.; Moskova-Doumanova, V.; Nandrot, E.F.; Doumanov, J.; Barragan, I.; Antinolo, G.; et al. EYS is a major gene for rod-cone dystrophies in France. Hum. Mutat. 2010, 31, E1406–E1435. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.; Littink, K.W.; Klevering, B.J.; van den Born, L.I.; Koenekoop, R.K.; Zonneveld, M.N.; Blokland, E.A.; Strom, T.M.; Hoyng, C.B.; den Hollander, A.I.; et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008, 83, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Bandah-Rozenfeld, D.; Littink, K.W.; Ben-Yosef, T.; Strom, T.M.; Chowers, I.; den Hollander, A.I.; van den Born, L.I.; Zonneveld, M.N.; Merin, S.; Banin, E.; et al. Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; van den Born, L.I.; Koenekoop, R.K.; Collin, R.W.; Zonneveld, M.N.; Blokland, E.A.; Khan, H.; Theelen, T.; Hoyng, C.B.; Cremers, F.P.; et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology 2010, 117, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, Y.; Li, J.; Natale, B.N.; Cao, S.; Wang, D.; Amack, J.D.; Hu, H. Eyes shut homolog is required for maintaining the ciliary pocket and survival of photoreceptors in zebrafish. Biol. Open 2016, 5, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hu, X.; Liu, F.; Soares, D.C.; Liu, X.; Yu, S.; Gao, M.; Han, S.; Qin, Y.; Li, C.; et al. Ablation of EYS in zebrafish causes mislocalisation of outer segment proteins, F-actin disruption and cone-rod dystrophy. Sci. Rep. 2017, 5, 46098. [Google Scholar] [CrossRef] [PubMed]
- Bonilha, V.L.; Rayborn, M.E.; Bell, B.A.; Marino, M.J.; Pauer, G.J.; Beight, C.D.; Chiang, J.; Traboulsi, E.I.; Hollyfield, J.G.; Hagstrom, S.A. Histopathological comparison of eyes from patients with autosomal recessive retinitis pigmentosa caused by novel EYS mutations. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 253, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Weleber, R.G.; Tobler, W.R. Computerized quantitative analysis of kinetic visual fields. Am. J. Ophthalmol. 1986, 101, 461–468. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Yagasaki, K.; Feuer, W.J.; Roman, A.J. Interocular asymmetry of visual function in heterozygotes of X-linked retinitis pigmentosa. Exp. Eye Res. 1989, 48, 679–691. [Google Scholar] [CrossRef]
- Heon, E.; Alabduljalil, T.; McGuigan, D.B., III; Cideciyan, A.V.; Li, S.; Chen, S.; Jacobson, S.G. Visual function and central retinal structure in choroideremia. Investig. Ophthalmol. Vis. Sci. 2016, 57, OCT377–OCT387. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Voigt, W.J.; Parel, J.M.; Apáthy, P.P.; Nghiem-Phu, L.; Myers, S.W.; Patella, V.M. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology 1986, 93, 1604–1611. [Google Scholar] [CrossRef]
- Roman, A.J.; Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V.; Chico, J.D.; Windsor, E.A.; Gardner, L.M.; Ying, G.S.; Smilko, E.E.; Maguire, M.G.; et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: Dark-adapted thresholds as outcome measures. Exp. Eye Res. 2005, 80, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cideciyan, A.V.; Papastergiou, G.I.; Banin, E.; Semple-Rowland, S.L.; Milam, A.H.; Jacobson, S.G. Relation of optical coherence tomography to microanatomy in normal and rd chickens. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2405–2416. [Google Scholar]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Pianta, M.J.; Sumaroka, A.; Schwartz, S.B.; Smilko, E.E.; Milam, A.H.; Sheffield, V.C.; Stone, E.M. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 2003, 12, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Aleman, T.S.; Cideciyan, A.V.; Sumaroka, A.; Schwartz, S.B.; Windsor, E.A.; Traboulsi, E.I.; Heon, E.; Pittler, S.J.; Milam, A.H.; et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proc. Natl. Acad. Sci. USA 2005, 102, 6177–6182. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Aleman, T.S.; Boye, S.L.; Schwartz, S.B.; Kaushal, S.; Roman, A.J.; Pang, J.J.; Sumaroka, A.; Windsor, E.A.; Wilson, J.M.; et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA 2008, 105, 15112–15117. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Hufnagel, R.B.; Carroll, J.; Sumaroka, A.; Luo, X.; Schwartz, S.B.; Dubra, A.; Land, M.; Michaelides, M.; Gardner, J.C.; et al. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum. Gene Ther. 2013, 24, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Birch, D.G.; Locke, K.G.; Wen, Y.; Locke, K.I.; Hoffman, D.R.; Hood, D.C. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with X-linked retinitis pigmentosa. JAMA Ophthalmol. 2013, 131, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.X.; Locke, K.G.; Ramachandran, R.; Birch, D.G.; Hood, D.C. A comparison of progressive loss of the ellipsoid zone (EZ) band in autosomal dominant and X-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7417–7422. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Cai, X.C.; Lee, D.; Epstein, B.C.; Locke, K.G.; Birch, D.G.; Hood, D.C. Reliability of a manual procedure for marking the EZ endpoint location in patients with retinitis pigmentosa. Transl. Vis. Sci. Technol. 2016, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Collins, R.A.; Leavitt, B.R.; Andrews, D.F.; Hayden, M.R.; Lumsden, C.J.; McInnes, R.R. A one-hit model of cell death in inherited neuronal degenerations. Nature 2000, 406, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Ogino, K.; Gotoh, N.; Morooka, S.; Hasegawa, T.; Hata, M.; Yoshimura, N. Inner segment ellipsoid band length is a prognostic factor in retinitis pigmentosa associated with EYS mutations: 5-year observation of retinal structure. Eye 2016, 12, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Roman, M.I.; Sumaroka, A.; Schwartz, S.B.; Stone, E.M.; Jacobson, S.G. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2007, 24, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Jacobson, S.G. Autofluorescence imaging with near-infrared excitation:normalization by reflectance to reduce signal from choroidal fluorophores. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3393–3406. [Google Scholar] [CrossRef] [PubMed]
- Golovleva, I.; Jonsson, F.; Burstedt, M. Heterogeneity and complexity of EYS mutations in autosomal recessive retinitis pigmentosa in northern Sweden. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3136. [Google Scholar]
- O’Sullivan, J.; Mullaney, B.G.; Sanjeev, S.; Dickerson, J.E.; Hall, G.; O’Grady, A.; Webster, A.; Ramsden, S.C.; Black, G.C. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J. Med. Genet. 2012, 49, 322–326. [Google Scholar] [CrossRef] [PubMed]
- González-del Pozo, M.; Borrego, S.; Barragán, I.; Pieras, J.I.; Santoyo, J.; Matamala, N.; Naranjo, B.; Dopazo, J.; Antiñolo, G. Mutation screening of multiple genes in Spanish patients with autosomal recessive retinitis pigmentosa by targeted resequencing. PLoS ONE 2011, 6, e27894. [Google Scholar] [CrossRef] [PubMed]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Zelinger, L.; Bandah-Rozenfeld, D.; Shevach, E.; Harel, A.; Storm, T.; Sagi, M.; Eli, E.; Merin, S.; Banin, E.; et al. Identification of mutations causing inherited retinal degenerations in the Israeli and Palestinian populations using homozygosity mapping. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carro, R.; Corton, M.; Sánchez-Navarro, I.; Zurita, O.; Sanchez-Bolivar, N.; Sánchez-Alcudia, R.; Lelieveld, S.H.; Aller, E.; Lopez-Martinez, M.A.; López-Molina, I.; et al. Panel-based NGS reveals novel pathogenic mutations in autosomal recessive retinitis pigmentosa. Sci. Rep. 2016, 6, 19531. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, M.M.; O’Driscoll, C.A.; Kaye, R.S.; Barragan, I.; El-Ashry, M.F.; Borrego, S.; Antiñolo, G.; Pang, C.P.; Webster, A.R.; Bhattacharya, S.S. Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 20010, 51, 4266–4272. [Google Scholar]
- Srinivasan, V.J.; Monson, B.K.; Wojtkowski, M.; Bilonick, R.A.; Gorczynska, I.; Chen, R.; Duker, J.S.; Schuman, J.S.; Fujimoto, J.G. Characterization of outer retinal morphology with high-speed, ultrahigh-resolution optical coherence tomography. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Curcio, C.A. Anatomical correlates to the bands seen in the outer retina by optical coherence tomography: Literature review and model. Retina 2011, 31, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Gibbs, D.; Sumaroka, A.; Roman, A.J.; Aleman, T.S.; Schwartz, S.B.; Olivares, M.B.; Russell, R.C.; Steinberg, J.D.; et al. Retinal disease course in Usher syndrome 1B due to MYO7A mutations. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7924–7936. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Tsybovsky, Y.; Schwartz, S.B.; Windsor, E.A.; Roman, A.J.; Sumaroka, A.; Steinberg, J.D.; Jacobson, S.G.; et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum. Mol. Genet. 2009, 18, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Herrera, W.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Banin, E.; Ben-Yosef, T.; Gardner, L.M.; Sumaroka, A.; Windsor, E.A.; Schwartz, S.B.; et al. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Cideciyan, A.V.; Sumaroka, A.; Windsor, E.A.; Herrera, W.; White, D.A.; Kaushal, S.; Naidu, A.; Roman, A.J.; Schwartz, S.B.; et al. Retinal laminar architecture in human retinitis pigmentosa caused by Rhodopsin gene mutations. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Aleman, T.S.; Jacobson, S.G.; Khanna, H.; Sumaroka, A.; Aguirre, G.K.; Schwartz, S.B.; Windsor, E.A.; He, S.; Chang, B.; et al. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: Implications for therapy of Leber congenital amaurosis. Hum. Mutat. 2007, 28, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Huang, W.C.; Sumaroka, A.; Roman, A.J.; Schwartz, S.B.; Luo, X.; Sheplock, R.; Dauber, J.M.; Swider, M.; et al. TULP1 mutations causing early-onset retinal degeneration: Preserved but insensitive macular cones. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5354–5364. [Google Scholar] [CrossRef] [PubMed]
- Suto, K.; Hosono, K.; Takahashi, M.; Hirami, Y.; Arai, Y.; Nagase, Y.; Ueno, S.; Terasaki, H.; Minoshima, S.; Kondo, M.; et al. Clinical phenotype in ten unrelated Japanese patients with mutations in the EYS gene. Ophthalmic Genet. 2014, 35, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Luo, X.; Héon, E.; Lam, B.L.; Weleber, R.G.; Halder, J.A.; Affatigato, L.M.; Goldberg, J.B.; Sumaroka, A.; Schwartz, S.B.; et al. Autosomal recessive retinitis pigmentosa caused by mutations in the MAK gene. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9665–9673. [Google Scholar] [CrossRef] [PubMed]
- Van Huet, R.A.; Siemiatkowska, A.M.; Özgül, R.K.; Yücel, D.; Hoyng, C.B.; Banin, E.; Blumenfeld, A.; Rotenstreich, Y.; Riemslag, F.C.; den Hollander, A.I.; et al. Retinitis pigmentosa caused by mutations in the ciliary MAK gene is relatively mild and is not associated with apparent extra-ocular features. Acta Opthamol. 2015, 93, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Sumaroka, A.; Matsui, R.; Cideciyan, A.V.; McGuigan, D.B., III; Sheplock, R.; Schwartz, S.B.; Jacobson, S.G. Outer retinal changes including the ellipsoid zone band in Usher syndrome 1B due to MYO7A mutations. Investig. Ophthalmol. Vis. Sci. 2016, 57, OCT253–OTC261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jacobson, S.G.; Cideciyan, A.V.; Sumaroka, A.; Aleman, T.S.; Schwartz, S.B.; Windsor, E.A.; Roman, A.J.; Stone, E.M.; MacDonald, I.M. Remodeling of the human retina in choroideremia: Rab escort protein 1 (REP-1) mutations. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4113–4120. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Cideciyan, A.V.; Sumaroka, A.; Schwartz, S.B.; Roman, A.J.; Windsor, E.A.; Steinberg, J.D.; Branham, K.; Othman, M.; Swaroop, A.; et al. Inner retinal abnormalities in X-linked retinitis pigmentosa with RPGR mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4759–4765. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Sumaroka, A.; Roman, A.J.; Swider, M.; Schwartz, S.B.; Banin, E.; Stone, E.M. Human retinal disease from AIPL1 gene mutations: Foveal cone loss with minimal macular photoreceptors and rod function remaining. Investig. Ophthalmol. Vis. Sci. 2011, 52, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Sheplock, R.; Schwartz, S.B.; Stone, E.M.; Jacobson, S.G. Inner and outer retinal changes in retinal degenerations associated with ABCA4 mutations. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Matsui, R.; Sumaroka, A.; Cideciyan, A.V. Retinal structure measurements as inclusion criteria for stem cell-based therapies of retinal degenerations. Investig. Ophthalmol. Vis. Sci. 2016, 57, ORSFn1–ORSFn9. [Google Scholar] [CrossRef] [PubMed]
- Milam, A.H.; Li, Z.Y.; Fariss, R.N. Histopathology of the human retina in retinitis pigmentosa. Prog. Retin. Eye Res. 1998, 17, 175–205. [Google Scholar] [PubMed]
- Marc, R.E.; Jones, B.W.; Watt, C.B.; Strettoi, E. Neural remodeling in retinal degeneration. Prog. Retin. Eye Res. 2003, 22, 607–655. [Google Scholar] [CrossRef]
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Marmor, M.; Marc, R.E. Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 2016, 150, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Strettoi, E.; Pignatelli, V. Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2000, 97, 11020–11025. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.E.; Fletcher, E.L.; Meffin, H.; Burkitt, A.N.; Grayden, D.B.; Greferath, U. Viability of the inner retina in a novel mouse model of retinitis pigmentosa. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2010, 2010, 553–556. [Google Scholar] [PubMed]
- Barragán, I.; Borrego, S.; Pieras, J.I.; González-del Pozo, M.; Santoyo, J.; Ayuso, C.; Baiget, M.; Millan, J.M.; Mena, M.; Abd El-Aziz, M.M.; et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum. Mutat. 2010, 31, E1772–E1800. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Ishigami, C.; Takahashi, M.; Park, DH.; Hirami, Y.; Nakanishi, H.; Ueno, S.; Yokoi, T.; Hikoya, A.; Fujita, T.; et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS ONE 2012, 7, e31036. [Google Scholar] [CrossRef] [PubMed]
- Zelhof, A.C.; Hardy, R.W.; Becker, A.; Zuker, C.S. Transforming the architecture of compound eyes. Nature 2006, 443, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Alfano, G.; Kruczek, P.M.; Shah, A.Z.; Kramarz, B.; Jeffery, G.; Zelhof, A.C.; Bhattacharya, S.S. EYS is a protein associated with the ciliary axoneme in rods and cones. PLoS ONE 2016, 11, e0166397. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, X.; Sheng, X.; Gao, X.; Zhang, X.; Li, Z.; Li, H.; Liu, Y.; Rong, W.; Zhao, K.; et al. Targeted next-generation sequencing reveals novel EYS mutations in Chinese families with autosomal recessive retinitis pigmentosa. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Iwanami, M.; Oshikawa, M.; Nishida, T.; Nakadomari, S.; Kato, S. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1033–1040. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Allele 1 | Allele 2 | ||||
|---|---|---|---|---|---|---|
| Nucleotide Change | Amino Acid Change or Predicted Effect | First Reports of Mutation | Nucleotide Change | Amino Acid Change or Predicted Effect | First Reports of Mutation | |
| P1 | c.1155T>A | p.(Cys385Ter) | This Study | c.8648_8655delCATGCAGA | p.(Thr2883LysfsTer4) | [26] |
| P2 | c.2137+1G>A | p.? | This Study | c.2137+1G>A | p.? | This Study |
| P3 | c.490C>T | p.(Arg164Ter) | [27] | c.2260-1437_2847-6134del | p.(Ser754IlefsTer4) | This Study |
| P4 | c.4829_4832delCATT | p.(Ser1610PhefsTer7) | This Study | c.5928-2A>G | p.? | [28] |
| P5 a | c.7919G>A | p.(Trp2640Ter) | [1] | c.8411_8412insTT | p.(Thr2805Ter) | This Study |
| P6 | c.32dupT | p.(Met12AspfsTer14) | [29] | c.32dupT | p.(Met12AspfsTer14) | [29] |
| P7 | c.9286_9295delGTAAATATCG | p.(Val3096LysfsTer28) | [30] | c.2259+10539_2993-12013del | p.(Ser754AlafsTer6) | [1] b |
| P8 | c.490C>T | p.(Arg164Ter) | [27] | c.2826_2827delAT | p.(Val944GlyfsTer9) | [31] |
| P9 | c.2889T>A | p.(Cys963Ter) | This Study | c.2889T>A | p.(Cys963Ter) | This Study |
| P10 | c.(1766+1_17671)_(2023+1_2024-1)del | p.(Cys590TyrfsTer4) | [1] b | c.(1766+1_17671)_(2023+1_2024-1)del | p.(Cys590TyrfsTer4) | [1] b |
| P11 a | c.2259+1G>A | p.? | [8] | c.8338_8342delins c | p.(Gly2780_Ser2781 delinsTyrLysLeuTer) | This Study |
| P12 | c.6528C>A | p.(Tyr2176Ter) | This Study | c.6528C>A | p.(Tyr2176Ter) | This Study |
| P13 | c.8408dupA | p.(Asn2803LysfsTer9) | [29] | c.5834delA | p.(Lys1945SerfsTer42) | [2] |
| P14 | c.6416G>A | p.(Cys2139Tyr) | [32] | c.6416G>A | p.(Cys2139Tyr) | [32] |
| P15 | c.9286_9295delGTAAATATCG | p.(Val3096LysfsTer28) | [30] | c.9286_9295delGTAAATATCG | p.(Val3096LysfsTer28) | [30] |
| Patient/Gender | Ethnicity | Age at First and Most Recent Exam | Follow-Up Duration (Years) a | Best Corrected Visual Acuity | Refractive Error b | ||
|---|---|---|---|---|---|---|---|
| RE | LE | RE | LE | ||||
| P1/F | Russian/Ukrainian | 12 | 1 | 20/20 | 20/20 | −3.25 | −4.00 |
| 13 | 20/20 | 20/20 | −3.50 | −3.75 | |||
| P2/F | Polish/Russian | 15 | 10 | 20/20 | 20/20 | +0.25 | +0.25 |
| 25 | 20/20 | 20/20 | +0.25 | +0.25 | |||
| P3/F | Iranian/Irish, German | 18 | 15 | 20/25 | 20/25 | −2.25 | −3.00 |
| 33 | 20/80 | 20/500 | −3.75 | −3.75 | |||
| P4/M | Hispanic | 19 | 27 | 20/20 | 20/20 | −0.50 | plano |
| 46 | 20/50 | 20/50 | plano | +0.50 | |||
| P5/F | Italian | 24 | 15 | 20/30 | 20/25 | −4.25 | −4.50 |
| 39 | 20/100 | 20/100 | −2.75 | −4.00 | |||
| P6/M | East Indian/Iranian | 24 | 1 | 20/20 | 20/20 | n/a | n/a |
| 25 | 20/20 | 20/20 | n/a | n/a | |||
| P7/F | Polish/Russian | 32 | 14 | 20/50 | 20/70 | n/a | n/a |
| 46 | 20/50 | 20/80 | +2.75 | +3.75 | |||
| P8/M | Iranian | 33 | - | 20/30 | 20/50 | n/a | n/a |
| P9/F | Irish | 34 | 14 | 20/20 | 20/25 | −2.75 | −2.50 |
| 48 | 20/70 | 20/100 | −1.25 | −1.50 | |||
| P10/M | Middle Eastern c | 36 | 3 | 20/30 | 20/50 | n/a | n/a |
| 39 | 20/50 | 20/50 | n/a | n/a | |||
| P11/M | Indian d | 38 | - | 20/50 | 20/60 | −7.00 | −4.00 |
| P12/M | Italian | 40 | 6 | 20/30 | 20/20 | n/a | n/a |
| 46 | 20/25 | 20/25 | −1.50 | −2.00 | |||
| P13/F | African American | 48 | - | 20/32 | 20/25 | −6.50 | −5.75 |
| P14/M | Chinese | 47 | - | HM | HM | n/a | n/a |
| P15/M | Ashkenazi Jewish | 51 | 3 | 20/30 | 20/30 | −1.50 | −0.50 |
| 54 | 20/40 | 20/30 | −1.00 | −1.25 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGuigan, D.B.; Heon, E.; Cideciyan, A.V.; Ratnapriya, R.; Lu, M.; Sumaroka, A.; Roman, A.J.; Batmanabane, V.; Garafalo, A.V.; Stone, E.M.; et al. EYS Mutations Causing Autosomal Recessive Retinitis Pigmentosa: Changes of Retinal Structure and Function with Disease Progression. Genes 2017, 8, 178. https://doi.org/10.3390/genes8070178
McGuigan DB, Heon E, Cideciyan AV, Ratnapriya R, Lu M, Sumaroka A, Roman AJ, Batmanabane V, Garafalo AV, Stone EM, et al. EYS Mutations Causing Autosomal Recessive Retinitis Pigmentosa: Changes of Retinal Structure and Function with Disease Progression. Genes. 2017; 8(7):178. https://doi.org/10.3390/genes8070178
Chicago/Turabian StyleMcGuigan, David B., Elise Heon, Artur V. Cideciyan, Rinki Ratnapriya, Monica Lu, Alexander Sumaroka, Alejandro J. Roman, Vaishnavi Batmanabane, Alexandra V. Garafalo, Edwin M. Stone, and et al. 2017. "EYS Mutations Causing Autosomal Recessive Retinitis Pigmentosa: Changes of Retinal Structure and Function with Disease Progression" Genes 8, no. 7: 178. https://doi.org/10.3390/genes8070178
APA StyleMcGuigan, D. B., Heon, E., Cideciyan, A. V., Ratnapriya, R., Lu, M., Sumaroka, A., Roman, A. J., Batmanabane, V., Garafalo, A. V., Stone, E. M., Swaroop, A., & Jacobson, S. G. (2017). EYS Mutations Causing Autosomal Recessive Retinitis Pigmentosa: Changes of Retinal Structure and Function with Disease Progression. Genes, 8(7), 178. https://doi.org/10.3390/genes8070178