1. Introduction
Ankylosing spondylitis (AS) is a progressive and debilitating seronegative spondiloarthritis, involving primarily the sacro-iliac joints and the spine and frequently the peripheral joints. Extra-articular tissues involvement, such as skin, tendons or eyes is often present [
1]. AS has a strong genetic background being associated with the major histocompatibility complex (MHC) class I allele HLA-B27 in more than 90% of the patients [
2].
The disease prevalence ranges the 0.02%–0.35% depending on the race, with a higher prevalence in males than in females [
1].
According to the Assessment of SpondyloArthritis International Society/European League Against Rheumatism (ASAS/EULAR) and American College of Rheumatology/ Spondylitis Association of America/ Spondyloarthritis Research and Treatment Network (ACR/SAA/SPARTAN), there is a recommendation for patients with a predominantly active axial manifestation of AS that they should be treated for at least three months with nonsteroidal anti-inflammatory drugs (NSAIDs) followed by a tumor necrosis factor (TNF)-alpha blocker in case of non response or of NSAIDs-related side effects, while patients with both axial and peripheral involvement need treatment with TNF-blockers and eventually with disease-modifying antirheumatic drugs (DMARDS) in case of intolerance or controindication to anti-TNF-alpha treatment [
3,
4].
Treatment with the TNF blockers, infliximab, etanercept, adalimumab, golimumab and certolizumab, leads to a good improvement in clinical symptoms, in C-reactive protein (CRP) levels and in magnetic resonance imaging (MRI)-detectable inflammation of the sacroiliac joints [
5].
In particular, Adalimumab (ADA) is a humanized monoclonal antibody against TNF-alpha, able to block both monomeric and trimeric soluble forms of TNF-alpha and cell-surface TNF-alpha, inducing cell apoptosis.
In this work, we have analyzed the transcriptome of Peripheral Blood Cells (PBCs) from patients with AS in order to identify possible mechanisms involved in the pathogenesis of the disease.
Previous studies on gene expression analyses in PBCs have identified gene signatures specifically associated to several diseases including Rheumatoid Arthritis (RA) and Systemic Lupus Erythematosus (SLE) [
6,
7]. Moreover, in a previous work on microarray analysis in Psoriatic Arthritis (PsA), we analyzed Differentially expressed genes (DEGs) in PBCs and in cells derived from synovial biopsies, and found that gene expression profiles of the cells obtained from the inflamed synovium are similar to those of PBCs, indicating that PBCs can be a good substitute of the target tissues to analyze disease-associated gene expression [
8].
High-throughput methodologies, including microarray, have increased our understanding of many diseases. However, while several studies on gene array analyses revealed transcriptional profiles of many single genes differentially expressed in AS compared to healthy subjects, such studies have not analyzed gene product interactions and molecular pathways possibly related to the disease onset. A careful evaluation of such networks can provide a deeper insight in the understanding of disease pathogenesis. In this study, we implemented the gene-array study with networks and modules analyses to uncover possible molecular pathways associated to AS and to a good response to ADA therapy.
This is indeed a novel approach since other studies used a conventional gene expression analysis to study, at gene level, the response to TNF inhibitors in rheumatoid arthritis [
9,
10] and to our knowledge, there are no such studies of ankylosing spondylitis.
In AS we found deregulated genes that are involved in important pathways; moreover, such genes are differently modulated in patients that show a good response to treatment compared to non-responders. Interestingly, in patients with a good response to treatment, we also observed that ADA normalized the altered expression of genes involved in AS-associated pathways.
3. Results
In order to identify gene expression profiles associated with AS, we compared the gene expression profiles of PBC samples obtained from 10 AS patients before treatment with ADA with 10 PBC samples obtained from healthy age and sex matched donors.
We found that 740 modulated transcripts satisfied the Bonferroni–corrected
p value criterion (
p ≤ 0.01) and the fold change criterion (FC ≥ |1.5|), showing robust and statistically significant variation between AS and healthy controls samples. In particular, 615 and 125 transcripts resulted to be over- and underexpressed respectively. The complete list of modulated genes can be found in
Supplementary Materials Table S2.
The Gene Ontology analysis showed that a large number of the modulated transcripts can be ascribed to biological processes that may play a role in AS, including: cell proliferation, extracellular matrix organization, angiogenesis, immune response, inflammatory response, interleukin-6 production, ossification, bone remodeling and signal transduction. Among these processes we noticed that signal transduction and immune response were the most represented biological processes. We also observed that immune response encompassed several Th-17-related genes including CD28 molecule, CD3 molecule epsilon (CD3E) and gamma (CD3G) subunits, CD4 molecule, transforming growth factor beta 1 (TGFB1), interleukin 6 receptor (IL6R), interleukin 6 signal transducer (IL6ST).
The signal transduction functional class included genes that may be ascribed to different subcategories including: inflammation mediated by chemokine and cytokine signaling pathway, Jak-Stat signaling pathway, Wnt signaling pathway, mirogen-activated protein kinase (MAPK) cascade, tumor necrosis factor-mediated signaling pathway, TLR signaling pathway, cytokine-mediated signaling pathway, Type I interferon signaling pathway, fibroblast growth factor receptor signaling pathway, T-cell receptor signaling pathway, small GTPase mediated signal transduction, transforming growth factor (TGFB) signaling, Notch signaling and phosphatidylinositol-mediated signaling.
Table 1 and
Table 2 show a detailed selection of DEGs within the above-mentioned processes. The tables also include GeneBank accession numbers and fold changes.
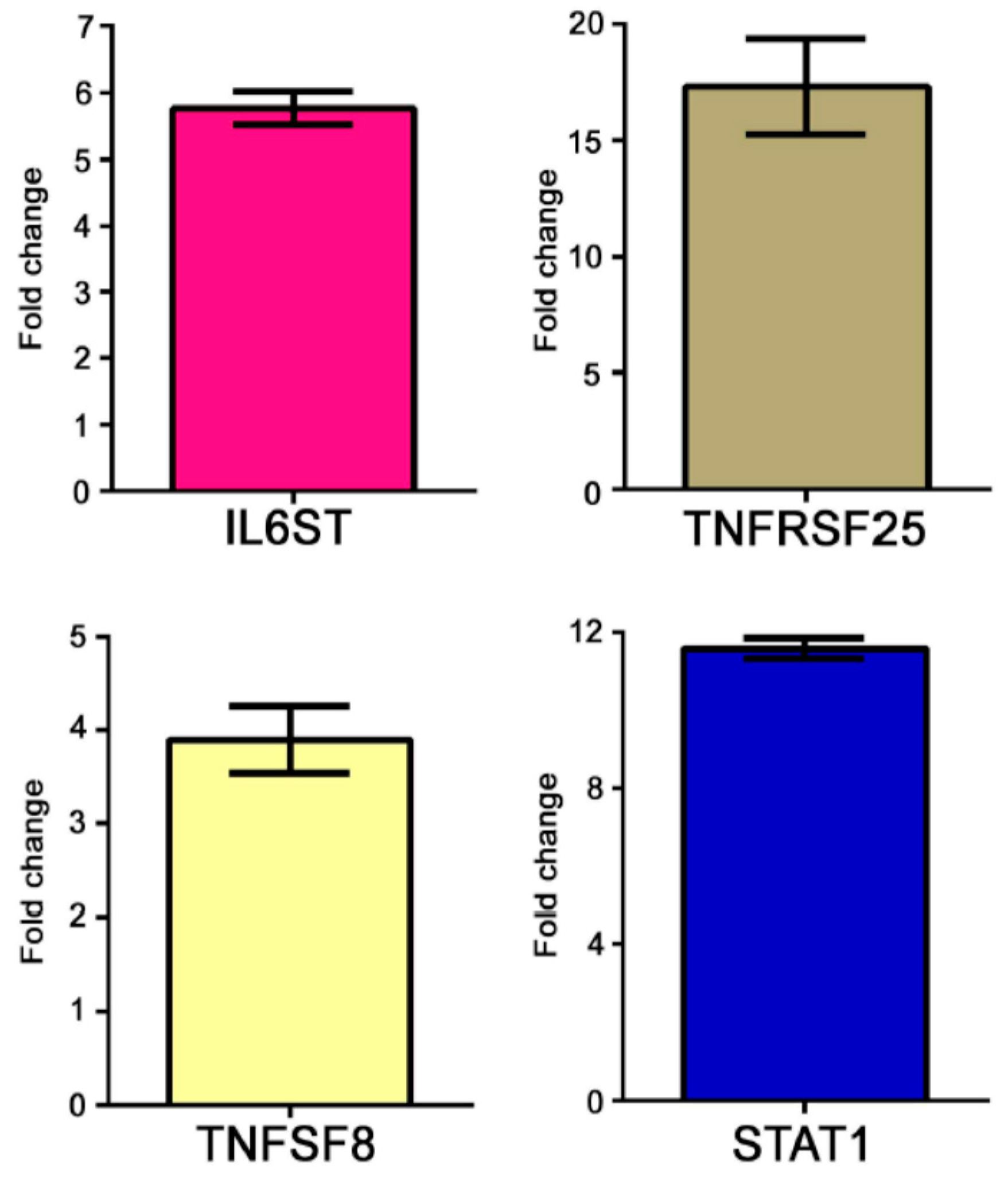
The modulation of some genes observed by gene array analysis was validated by Q-PCR (
Figure 1).
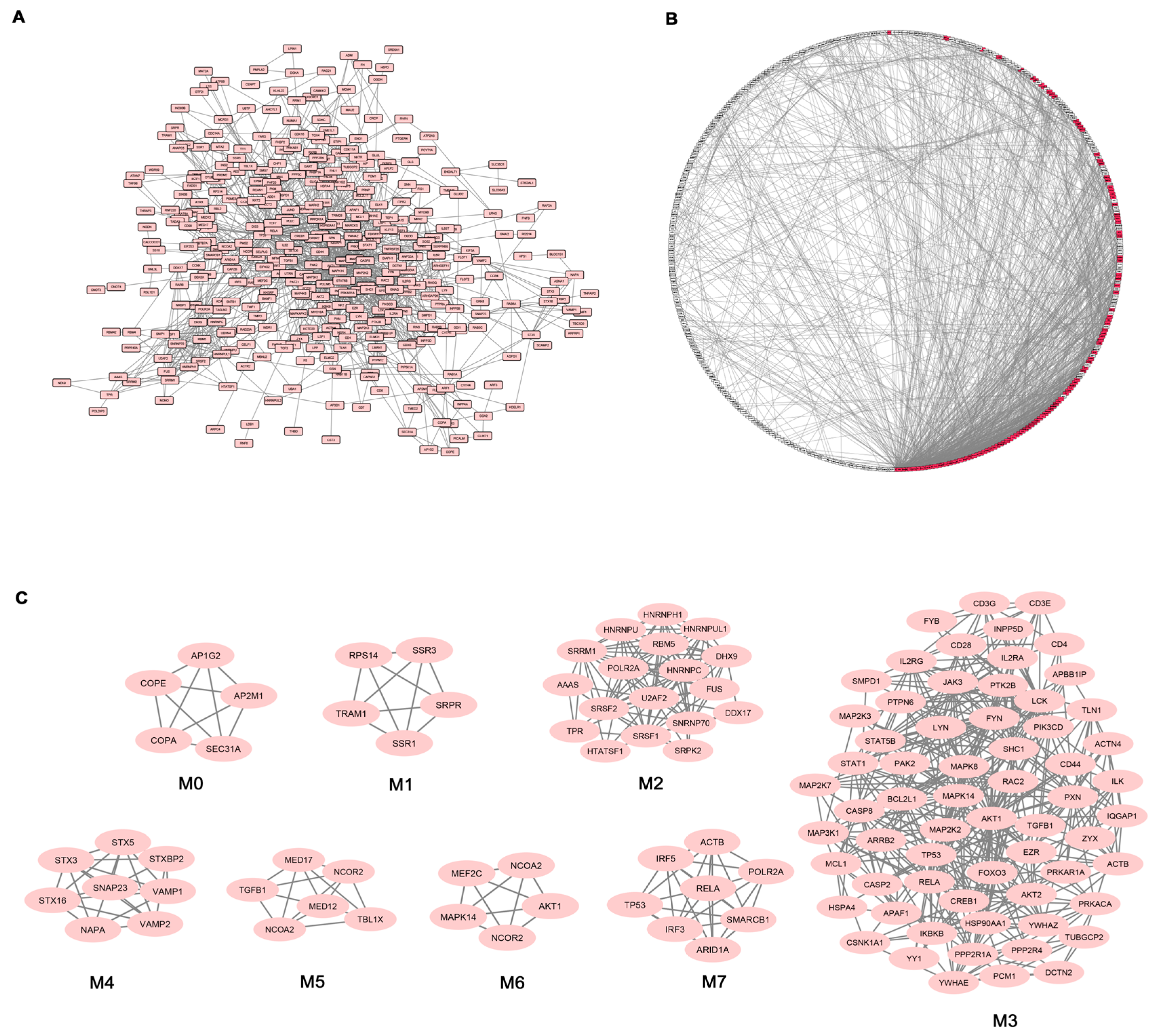
Given the high number of DEGs and the corresponding functional classes, to make our study more informative, we decided to complement the gene expression profiling of AS PBC with the study of functional interactions between the protein products of modulated genes. To this aim, a protein-protein interaction (PPI) network comprising 401 genes (nodes) and 1345 pairs of interactions (edges) was obtained (
Figure 2A). In
Figure 2B the interconnected genes are represented in a circular graph and are ordered based on their degree of connectivity, so genes with a similar degree of connectivity are placed close to each other. We then submitted the PPI network to a modular analysis to find set of densely interconnected nodes (modules) that are expected to participate in multiple biological activities in a coordinated manner. We identified eight modules (
k = 5) that are graphically represented in
Figure 2C.
Figure 2B shows that the genes belonging to the eight modules (highlighted in red) are distributed (concentrated) over an area in which the highest degrees of connectivity are present, thus confirming that they are highly connected genes.
We next performed functional enrichment analysis to identify association of genes, in each module, with different “GO terms” and pathways. All the significantly enriched categories for each module are shown in
Supplementary Materials Table S3 and included pathogenetically relevant processes such as: T-cell receptor signaling (module M3), TLRs signaling (modules M3 and M7), small GTPase signaling (module M3) Wnt signaling (module M7), p38 MAP kinase cascade (modules M3 and M6) Jak-stat signaling (module M3) fibroblast growth factor receptor signaling (module M3) inflammation mediated by chemokine and cytokine signaling pathway (modules M3 and M7) VEGF signaling (modules M3 and M6) angiogenesis (module M3).
In order to evaluate the effect of treatment on gene expression in AS patients, we analysed the transcriptional profiles obtained from the same AS patients seven days after ADA administration.
When the same Bonferroni-corrected
p value and fold change criteria were applied, 571 genes were differentially expressed in AS patients after treatment compared to healthy controls, in particular 492 and 79 transcripts resulted to be up- and down-regulated respectively (
Supplementary Materials Table S4). The protein products of 297 DEGs, among the above mentioned genes, were connected in a PPI network involving 819 pairs of interactions (
Figure 3), from which 7 modules can be extracted.
We next performed GO biological process and pathways enrichment analysis of genes included in the seven modules. Modules and the resulting enriched functional categories are listed in
Supplementary Materials Table S5.
Afterwards, we compared the lists of processes enriched in modules from the two analysis (samples before treatment versus healthy controls and samples after treatment versus healthy controls) and we found that several pathways and biological processes were enriched only in the pre-treatment associated modules (
Table 3).
In particular, we found that after treatment the enrichment in p38 MAPK (p value = 0.007), P53 pathway feedback loops 1 (p value = 0.005), PI3 kinase pathway (p value = 0.015) and Wnt signaling pathways (p value < 0.001) disappeared. In addition, we observed that all pathways that showed an enrichment only in the pre-treatment condition were associated to the pre-treatment associated modules M3, M6 and M7.
Many biological processes were also influenced by the therapy, since they were no longer enriched after treatment. These processes were associated to the pre-treatment module M3 and included: fibroblast growth factor receptor signaling (p value < 0.001), small GTPase mediated signal transduction (p value = 0.040), immune effector process (p value = 0.007), leukocyte migration (p value = 0.032), positive regulation of T-cell proliferation (p value = 0.035), response to interleukin-2 (p value = 0.001), T-cell costimulation (p value < 0.001), T-cell differentiation (p value = 0.015), T-cell receptor signaling (p value < 0.001), and phosphatidylinositol-mediated signaling pathway (p value = 0.002).
Although the patients enrolled in the study were homogeneous as far as clinical features and stage of the disease concern, we could identify two groups who showed a different response to treatment.
The first group (group 1) consisted of 6 patients who had a very good response to the treatment with disease remission and the second group (group 2) of four patients who showed a poor response to the treatment.
We then performed a gene array analysis by dividing patients in two groups, according to their response to the treatment, and comparing the expression profiles of two groups, before and after treatment, to those of the healthy control samples.
Before treatment we observed that in the first group of patients 689 genes, and in the second group 987 genes were modulated (
Supplementary Materials Tables S6 and S7). After treatment, a modulation of 846 transcripts in the first group of patients and of 1077 genes in the second group was observed compared to the control group (
Supplementary Materials Tables S8 and S9). When we compared genes modulated pre- and post-treatment in the two groups, we observed that 306 genes in the first group and 119 genes in the second group, returned to baseline expression levels after treatment, showing an expression level comparable to that of healthy controls.
To finely dissect the effect of ADA treatment on gene expression profiles in the two patients groups, we decided to functionally analyze these genes and we found that ADA had an impact on genes involved in signaling events that may be related to the pathogenesis of the disease. In particular, we compared the two lists of biological signaling in which DEGs, that were corrected by the treatment, were involved (
Table 4).
We found that some of these categories were shared by the two groups of patients, (i.e., immune response, inflammatory response, angiogenesis, MAP kinase, Jak-Stat small GTPase, T-cell receptor, Notch and TGF-beta signaling) while others were only associated to group 1 (i.e., interleukin 6 production, cytokine mediated, NIK/NF-Kappa B, Toll-like receptor, Tumor necrosis factor, Type 1 interferon, and Wnt signaling) or to group 2 (i.e., EGF receptor, Ephrin receptor, Fc-gamma receptor signaling and positive regulation of BMP and VEGF signaling).
A selection of the above-mentioned genes, which were corrected by the treatment in the two groups of patients, referred to as “genes modulated only before treatment is reported in
Table 5 and
Table 6, where transcripts are grouped according to the Gene Ontology classification criteria. These tables also show the fold changes resulting from the comparison of pre- or post-treatment condition versus healthy controls.
Then we also performed a functional enrichment analysis to detect biological processes that were significantly enriched in genes corrected by the treatment.
Some of the enriched biological classes were related to the immune response and, in particular, to the activation of B and T-cell response, as indicated by the enrichment in T-cell activation (
p value = 0.005), positive regulation of B-cell activation (
p value = 0.041) and B-cell cytokine production (
p value = 0.024) categories in the first group, and by the enrichment in T-cell activation (
p value = 0.038) and positive regulation of B-cell activation (
p value = 0.015) classes in the second group (
Table 7). Genes involved in inflammation were also well represented in the two datasets modulated only before treatment and the two classes “inflammatory response” (
p value = 0.026) and “NLRP3 inflammasome complex assembly” (
p value = 0.006) were enriched in the first and in the second group, respectively.
In addition, we also identified functional classes that were enriched only in one of the two groups of patients. These classes were: positive regulation of cytokine production (p value = 0.012) positive regulation of type I interferon production (p value = 0.017) Toll-like receptor signaling pathway (p value = 0.039) positive regulation of interleukin-2 production (p value = 0.001) cellular response to unfolded protein (p value = 0.001) in the first group and, angiogenesis (p value = 0.002) response to transforming growth factor beta (p value = 0.010) and G-protein coupled receptor signaling pathway (p value = 0.031) in the second group.
4. Discussion
In this work, we first compared the gene expression profiles of 10 AS patients before treatment with 10 healthy controls. Secondly, we analysed the gene expression before and after ADA therapy, in order to identify biological processes that may be modulated by the drug. Finally, we compared gene profiles of patients characterized by a different response to the treatment.
Our results showed that AS has a profound impact on gene expression, since a large number of genes are modulated in AS samples. Moreover, DEGs have a role in biological processes that may be pathogenetically relevant to both the disease onset and progression.
The pathological features of AS are supposed to originate from several events that may interact, including biomechanical insult, genetic background, infections, inflammation and immune response, even though the precise mechanisms of the immune activation are still a matter of discussion [
24].
We here identified a large number (40) of genes that have well-documented roles in the immune response, including for example TNFSF8/CD30L, interleukin-32 and several Th17-lymphocytes-related genes. TNFSF8 is a TNF-family member expressed on activated peripheral blood T-cells, B-cells, neutrophils, mast cells, monocytes, and macrophages and it is the ligand for CD30, a molecule that is present at high levels in AS patients compared to healthy controls [
25]. The CD30/CD30L signaling system has been implicated in the pathogenesis of several autoimmune and inflammatory diseases and it has been recently associated to the pathogenesis of rheumatoid arthritis synovitis [
26].
Interleukin-32 is a proinflammatory cytokine produced by T lymphocytes, natural killer cells, blood monocytes and fibroblast-like synoviocytes in the joints. It is present in the synovial fluid of AS patients at higher levels compared with RA or osteoarthritis patients and induces osteoblast differentiation leading to pathological bone growth [
27].
Th17 cells have been associated to several autoimmune disorders including SLE, RA and psoriasis where they play a role in the pathogenesis of the disease [
28,
29,
30]. Recent studies have demonstrated that Th17 cells are involved also in the pathogenesis of AS [
31].
It is well known that the pathogenesis of AS is characterized by three phases: inflammation, erosion and abnormal bone outgrowth [
32]. In this regard our data seem to recapitulate all these pathogenetic stages, showing deregulation of genes involved in the inflammatory process as well of transcripts that play a role in bone remodeling (i.e., bone growth and bone erosion).
Although the precise events that prime inflammation in spondyloarthritis is still unclear, recent findings provided evidence of the involvement of immune pathways, including the lately described IL-17/IL-23 pathway, NF-κB activation and antigen presentation [
33] reintroducing the issue of autoimmune versus autoinflammatory disease. To date, however, no immunological pathway has emerged as the master regulator that may finely orchestrate the disease pathogenesis.
Interestingly, we found a remarkable number of DEGs included genes acting in commonly known immunological signalings such as TNF, MAP kinase cascade, Wnt, Toll-like receptor, TGF beta, T-cell receptor, Type I interferon, and Jak-stat signaling.
TNF-alpha is one of the main actors in the inflammatory scenario during the disease progression. Interestingly, here we show up-regulation of several genes involved in the TNF-alpha signaling including TNFRSF25. This molecule is a tumor necrosis factor receptor mainly expressed on T-cells [
34] that has been associated with autoimmune diseases such as RA. Moreover, it has been reported that the absence of TNFRSF25 protects from the development of severe bone disorders in experimental antigen-induced arthritis (AIA) [
35]. Along with the upregulation of TNF-related genes we also found overexpression of genes involved in interleukin-6 production and signal transduction (including IL6R and IL6ST) together with overexpression of transcripts of the MAP kinases cascade, two well-documented down-stream events of the biologic response to TNF [
36]. In particular, it has been shown that high serum levels of IL-6 may associate with the development of AS [
36]. The triggering of mitogen-activated kinase (MAPK) signaling cascade leads to activation of downstream JUN N-terminal kinase (JNK) and p38/MAPK14 [
37], which is involved in the proinflammatory activity of interleukin 1 (IL-1) and TNF-alpha [
38]. The transcript for this kinase was 1.8 fold increased in AS samples. Interestingly, it has been shown that inhibition of p38 can restore chondrocyte proliferation in inflammatory bone diseases such as spondyloarthritis [
38].
The Wnt signaling components are critically involved in normal bone homeostasis, and in particular in new bone formation [
32]. Moreover, it has been recently suggested that Wnt signaling may take part to the process of ankylosis in spondyloarthritis [
32].
Deregulation of TLR signaling has been already described in AS [
39] and there is emerging evidence of a potential role of TLRs in AS [
40].
The up-regulation of genes involved in TGF-beta signaling is not surprising, since TGF-beta is important for bone formation and has been detected in biopsy specimens from sacroiliac joints of patient with AS where it may play a role in articular cartilage fibrosis and ossification [
41]. Moreover, this cytokine is also involved in adaptive immune responses by regulating effector and regulatory CD4
+ T-cell responses [
42]. The importance of T-cell response is highlighted in our AS samples by the up-regulation of several T-cell related genes involved in T-cell receptor signaling including CD3g molecule, gamma (CD3G) and T-cell receptor alpha constant (TRAC). In particular, the association of this signaling pathway with AS has been already suggested by Chen et al. in a previous whole-blood gene expression study [
43]. Among the modulated genes, we also found overexpression of transcripts for members of type I IFN signaling, a network typically associated with autoimmune diseases such as SLE, RA, Crohn’s disease and Sjogren syndrome [
44,
45,
46,
47,
48,
49,
50].
In this regard, the co-presence of type I IFN signaling and Th-17 related genes may be suggestive for an autoimmune origin of AS, since the co-activity of IFN and Th17 pathways is typical of autoimmunity and has been described both in human diseases and in animal models [
51,
52,
53].
Another signaling pathway relevant to autoimmunity is the Jak-stat pathway [
54] that is represented in our dataset by the upregulation of STAT1 and STAT5b. It has been described that STAT1 gain-of-function mutations are associated to autoimmune response [
54] and that this molecule is differentially expressed in AS patients by transcriptome network analysis [
55]. In addition we also found modulation of genes involved in fibroblast growth factor receptor, small GTPase and Notch signalings. It is worthwhile mentioning that the first favours the formation of new bone in AS patients [
56], the second triggers proliferation of rheumatoid synovial fibroblasts [
57] and the Notch signaling is involved in ligament ossification of hip joints in AS [
58].
In the next step of our data analysis we found that DEG products were connected in a wide PPI network from which we obtained eight functional modules, containing the most functionally interconnected genes. These modules were significantly enriched in several of the above mentioned pathologically relevant processes and pathways including: T-cell receptor signaling, TLRs signaling, small GTPase signaling, Wnt signaling, p38 MAP kinase cascade and Jak-stat signaling.
Moreover, this analysis was paralleled by the comparison of post-treatment samples to healthy controls and, in this case, the resulted PPI network included seven modules.
Interestingly, by comparing pathways and biological processes enrichments of modules derived from the pre- and the post-treatment conditions, we identified three pre-treatment-associated modules (M3, M6, and M7) towards which the drug seemed to direct its activity. Indeed several crucial processes and pathways, enriched in these modules, were no longer enriched after treatment, including p38 MAP kinase, Wnt, FGF receptor, small GTPase, T-cell receptor and PI3 kinase signaling.
To more finely dissect the impact of treatment on expression profiles of AS samples, we separately analyzed the two groups of patients that were characterized by a different response to the treatment.
The two analyses showed that a quite different number of genes were modulated before treatment in the two datasets. In particular, after comparing genes modulated before and after treatment in the two groups of patients, we found a different number of genes that returned to a normal expression level after treatment.
Focusing our analysis on these genes, we realized that globally, in both groups of patients, the treatment had an impact on many pathologically relevant biological processes, including immune response, inflammatory response, angiogenesis and on several signaling pathways (i.e., Jak-Stat signaling, MAP kinase cascade and Small GTPase signaling). However we also observed that only in the responders group (group 1), genes involved in Toll-like receptor signaling, tumor necrosis factor signaling, Type I interferon signaling and Wnt signaling returned to normal.
Moreover, when we specifically analyzed the categories enriched in genes corrected by the treatment in the two groups of patients, we observed, in both data sets, an enrichment in the inflammatory response and in the lymphocytes activation processes, but we also observed that several categories were enriched only in the group 1, including regulation of interleukin-2 production, cellular response to unfolded protein, positive regulation of type I interferon production and toll-like receptor signaling.
These results suggest that the differences emerged from the separated analysis of the two groups of patients, may be partially related to the different response to treatment. In particular, we have identified altered processes and signaling that may be more strongly associated to AS pathogenesis since they are corrected by the treatment in patients with a good response.
We suggest that these biological processes may be the target of novel therapies if our results are confirmed in further experiments and in a larger number of patients.


 ,
, 


{kind=link}
{kind=link}
{kind=link}