
c-MYC—Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease



Abstract
:1. Introduction
2. c-MYC Functions in Liver Regeneration, Health and Disease: How Important or Dispensable?
3. Deconstructing c-MYC: Transgenic Models
4. Of Mice and Men—Is c-MYC Relevant for Liver Cancer?
5. Treating Liver Cancer: From Theory to Practice
6. c-MYC: Causing Liver Stiffness
7. c-MYC and Viral Hepatitis: Playing the Same Tune
8. Drinking and Thinking of c-MYC
9. Notorious c-MYC: Involvement in Pediatric Liver Cancers
10. Study in Yellow: c-MYC in Hepatic Cholestasis
11. Good News at the End
12. Final Considerations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Whitfield, J.R.; Soucek, L. Tumor microenvironment: Becoming sick of myc. Cell. Mol. Life Sci. 2012, 69, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with myc. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. C-myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.M. Retroviruses and cancer genes. Adv. Cancer Res. 1982, 37, 1–32. [Google Scholar] [PubMed]
- Bister, K.; Jansen, H.W. Oncogenes in retroviruses and cells: Biochemistry and molecular genetics. Adv. Cancer Res. 1986, 47, 99–188. [Google Scholar] [PubMed]
- Dang, C.V. C-myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.H.; Vogt, P.K. Avian acute leukemia viruses mc29 and mh2 share specific rna sequences: Evidence for a second class of transforming genes. Proc. Natl. Acad. Sci. USA 1979, 76, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.S.; Lai, M.M.; Vogt, P.K. Genome of avian myelocytomatosis virus mc29: Analysis by heteroduplex mapping. Proc. Natl. Acad. Sci. USA 1979, 76, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Sheiness, D.; Bishop, J.M. DNA and rna from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J. Virol. 1979, 31, 514–521. [Google Scholar] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Evan, G. Myc-is this the oncogene from hell? Cancer Cell 2002, 1, 406–408. [Google Scholar] [CrossRef]
- Thompson, N.L.; Mead, J.E.; Braun, L.; Goyette, M.; Shank, P.R.; Fausto, N. Sequential protooncogene expression during rat liver regeneration. Cancer Res. 1986, 46, 3111–3117. [Google Scholar] [PubMed]
- Fausto, N.; Mead, J.E.; Braun, L.; Thompson, N.L.; Panzica, M.; Goyette, M.; Bell, G.I.; Shank, P.R. Proto-oncogene expression and growth factors during liver regeneration. Symp. Fundam. Cancer Res. 1986, 39, 69–86. [Google Scholar] [PubMed]
- Sanders, J.A.; Gruppuso, P.A. Nucleolar localization of hepatic c-myc: A potential mechanism for c-myc regulation. Biochim. Biophys. Acta 2005, 1743, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Baena, E.; Gandarillas, A.; Vallespinos, M.; Zanet, J.; Bachs, O.; Redondo, C.; Fabregat, I.; Martinez, A.C.; de Alboran, I.M. C-myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc. Natl. Acad. Sci. USA 2005, 102, 7286–7291. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.A.; Schorl, C.; Patel, A.; Sedivy, J.M.; Gruppuso, P.A. Postnatal liver growth and regeneration are independent of c-myc in a mouse model of conditional hepatic c-myc deletion. BMC Physiol. 2012, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xiang, Y.; Potter, J.; Dinavahi, R.; Dang, C.V.; Lee, L.A. Conditional deletion of c-myc does not impair liver regeneration. Cancer Res. 2006, 66, 5608–5612. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hu, H.; Braren, R.; Fong, S.Y.; Trumpp, A.; Carlson, T.R.; Wang, R.A. C-myc in the hematopoietic lineage is crucial for its angiogenic function in the mouse embryo. Development 2008, 135, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Muakkassah-Kelly, S.F.; Jans, D.A.; Lydon, N.; Bieri, F.; Waechter, F.; Bentley, P.; Staubli, W. Electroporation of cultured adult rat hepatocytes with the c-myc gene potentiates DNA synthesis in response to epidermal growth factor. Exp. Cell Res. 1988, 178, 296–306. [Google Scholar] [CrossRef]
- Factor, V.M.; Jensen, M.R.; Thorgeirsson, S.S. Coexpression of c-myc and transforming growth factor alfa in the liver promotes early replicative senescence and diminishes regenerative capacity after partial hepatectomy in transgenic mice. Hepatology 1997, 26, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Sanderson, N.D.; Nagy, P.; Marino, P.A.; Merlino, G.; Thorgeirsson, S.S. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: Interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993, 53, 1719–1723. [Google Scholar] [PubMed]
- Freimuth, J.; Gassler, N.; Moro, N.; Gunther, R.W.; Trautwein, C.; Liedtke, C.; Krombach, G.A. Application of magnetic resonance imaging in transgenic and chemical mouse models of hepatocellular carcinoma. Mol. Cancer 2010, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S.; Santoni-Rugiu, E. Transgenic mouse models in carcinogenesis: Interaction of c-myc with transforming growth factor alpha and hepatocyte growth factor in hepatocarcinogenesis. Br. J. Clin. Pharmacol. 1996, 42, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Sanderson, N.D.; Ton, P.; Thorgeirsson, S.S. Molecular analyses of liver tumors in c-myc transgenic mice and c-myc and tgf-alpha double transgenic mice. Cancer Lett. 1996, 106, 43–49. [Google Scholar] [CrossRef]
- Conner, E.A.; Lemmer, E.R.; Sanchez, A.; Factor, V.M.; Thorgeirsson, S.S. E2f1 blocks and c-myc accelerates hepatic ploidy in transgenic mouse models. Biochem. Biophys. Res. Commun. 2003, 302, 114–120. [Google Scholar] [CrossRef]
- Klocke, R.; Bartels, T.; Jennings, G.; Brand, K.; Halter, R.; Strauss, M.; Paul, D. Lack of p53 accelerates hepatocarcinogenesis in transgenic mice constitutively overexpressing c-myc in the liver. FASEB J. 2001, 15, 1404–1406. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.; Komatsubara, K.; Bellovin, D.I.; Kurobe, M.; Sylvester, K.; Felsher, D.W. Hepatotoxin-induced changes in the adult murine liver promote myc-induced tumorigenesis. PLoS ONE 2008, 3, e2493. [Google Scholar] [CrossRef] [PubMed]
- Terradillos, O.; Billet, O.; Renard, C.A.; Levy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis b virus x gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Li, Q.; Dang, C.V.; Lee, L.A. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-myc in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 11198–11202. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Myc on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, E.P.; Quaife, C.J.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L. Oncogene-induced liver neoplasia in transgenic mice. Oncogene 1989, 4, 715–724. [Google Scholar] [PubMed]
- Hermeking, H.; Eick, D. Mediation of c-myc-induced apoptosis by p53. Science 1994, 265, 2091–2093. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.S.; Wiman, K.G. Myc and e2f1 induce p53 through p14arf-independent mechanisms in human fibroblasts. Oncogene 2003, 22, 4993–5005. [Google Scholar] [CrossRef] [PubMed]
- Heindryckx, F.; Colle, I.; Van Vlierberghe, H. Experimental mouse models for hepatocellular carcinoma research. Int. J. Exp. Pathol. 2009, 90, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Tonjes, R.R.; Lohler, J.; O’Sullivan, J.F.; Kay, G.F.; Schmidt, G.H.; Dalemans, W.; Pavirani, A.; Paul, D. Autocrine mitogen igegf cooperates with c-myc or with the hcs locus during hepatocarcinogenesis in transgenic mice. Oncogene 1995, 10, 765–768. [Google Scholar] [PubMed]
- Ladu, S.; Calvisi, D.F.; Conner, E.A.; Farina, M.; Factor, V.M.; Thorgeirsson, S.S. E2f1 inhibits c-myc-driven apoptosis via pik3ca/akt/mtor and cox-2 in a mouse model of human liver cancer. Gastroenterology 2008, 135, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Van Calcar, S.; Qu, C.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A global transcriptional regulatory role for c-myc in burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.T.; Stadler, B.M.; Kowalik, T.F. Mir-17 and mir-20a temper an e2f1-induced g1 checkpoint to regulate cell cycle progression. Oncogene 2009, 28, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Zeller, K.I.; Zhao, X.; Lee, C.W.; Chiu, K.P.; Yao, F.; Yustein, J.T.; Ooi, H.S.; Orlov, Y.L.; Shahab, A.; Yong, H.C.; et al. Global mapping of c-myc binding sites and target gene networks in human b cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17834–17839. [Google Scholar] [CrossRef] [PubMed]
- Santoni-Rugiu, E.; Duro, D.; Farkas, T.; Mathiasen, I.S.; Jaattela, M.; Bartek, J.; Lukas, J. E2f activity is essential for survival of myc-overexpressing human cancer cells. Oncogene 2002, 21, 6498–6509. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; Sears, R.; Huang, E.; Rempel, R.; Nuckolls, F.; Park, C.H.; Giangrande, P.; Wu, L.; Saavedra, H.I.; Field, S.J.; et al. Myc requires distinct e2f activities to induce s phase and apoptosis. Mol. Cell 2001, 8, 105–113. [Google Scholar] [CrossRef]
- Akita, H.; Marquardt, J.U.; Durkin, M.E.; Kitade, M.; Seo, D.; Conner, E.A.; Andersen, J.B.; Factor, V.M.; Thorgeirsson, S.S. Myc activates stem-like cell potential in hepatocarcinoma by a p53-dependent mechanism. Cancer Res. 2014, 74, 5903–5913. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Casey, S.C.; Felsher, D.W. Inactivation of myc reverses tumorigenesis. J. Intern. Med. 2014, 276, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. Myc oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.Y.; Lai, P.L.; Hsu, H.C. Amplification of the c-myc gene in human hepatocellular carcinoma: Biologic significance. J. Formos. Med. Assoc. 1993, 92, 866–870. [Google Scholar] [PubMed]
- Schlaeger, C.; Longerich, T.; Schiller, C.; Bewerunge, P.; Mehrabi, A.; Toedt, G.; Kleeff, J.; Ehemann, V.; Eils, R.; Lichter, P.; et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 2008, 47, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Poon, T.C.; Wong, N.; Lai, P.B.; Rattray, M.; Johnson, P.J.; Sung, J.J. A tumor progression model for hepatocellular carcinoma: Bioinformatic analysis of genomic data. Gastroenterology 2006, 131, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Kawate, S.; Fukusato, T.; Ohwada, S.; Watanuki, A.; Morishita, Y. Amplification of c-myc in hepatocellular carcinoma: Correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology 1999, 57, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Liu, C.R.; Lee, C.N.; Chan, T.S.; Liu, H.E. Targeting c-myc as a novel approach for hepatocellular carcinoma. World J. Hepatol. 2010, 2, 16–20. [Google Scholar] [PubMed]
- Wang, Y.; Wu, M.C.; Sham, J.S.; Zhang, W.; Wu, W.Q.; Guan, X.Y. Prognostic significance of c-myc and aib1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer 2002, 95, 2346–2352. [Google Scholar] [CrossRef] [PubMed]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, H.G.; Lee, Y.H.; Sears, N.C.; Coulouarn, C.; Conner, E.A.; Factor, V.M.; Roskams, T.; Thorgeirsson, S.S. Central role of c-myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, P.E.; Wink, J.; Alers, J.C.; JN, I.J.; Schalm, S.W.; de Man, R.A.; van Dekken, H. Molecular cytogenetic evaluation of virus-associated and non-viral hepatocellular carcinoma: Analysis of 26 carcinomas and 12 concurrent dysplasias. J. Pathol. 2000, 192, 207–215. [Google Scholar] [CrossRef]
- Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Frau, M.; Asara, G.; Serra, S.; Muntoni, M.D.; Seddaiu, M.A.; Daino, L.; Feo, F.; et al. Down-regulation of c-myc and cyclin d1 genes by antisense oligodeoxy nucleotides inhibits the expression of e2f1 and in vitro growth of hepg2 and morris 5123 liver cancer cells. Carcinogenesis 2004, 25, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Jiang, C.; Cai, Y.; Kim, J.H.; Tanaka, N.; Ward, J.M.; Shah, Y.M.; Gonzalez, F.J. Role of myc in hepatocellular proliferation and hepatocarcinogenesis. J. Hepatol. 2014, 60, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Transient excess of myc activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3940–3944. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.; Deb-Basu, D.; Cherry, A.; Turner, S.; Ford, J.; Felsher, D.W. Defective double-strand DNA break repair and chromosomal translocations by myc overexpression. Proc. Natl. Acad. Sci. USA 2003, 100, 9974–9979. [Google Scholar] [CrossRef] [PubMed]
- Neiman, P.E.; Kimmel, R.; Icreverzi, A.; Elsaesser, K.; Bowers, S.J.; Burnside, J.; Delrow, J. Genomic instability during myc-induced lymphomagenesis in the bursa of fabricius. Oncogene 2006, 25, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Prochownik, E.V. C-myc: Linking transformation and genomic instability. Curr. Mol. Med. 2008, 8, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Atkuri, K.R.; Deb-Basu, D.; Adler, A.S.; Chang, H.Y.; Herzenberg, L.A.; Felsher, D.W. Myc can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 2006, 66, 6598–6605. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. Myc inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Gentles, A.J.; Elchuri, S.; Sahoo, D.; Soen, Y.; Sharpe, O.; Perez, O.D.; Chang, M.; Mitchel, D.; Robinson, W.H.; et al. Genomic and proteomic analysis reveals a threshold level of myc required for tumor maintenance. Cancer Res. 2008, 68, 5132–5142. [Google Scholar] [CrossRef] [PubMed]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A myc-aurora kinase a protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, C.M.; Gunther, E.J.; Boxer, R.B.; Hartman, J.L.; Sintasath, L.; Moody, S.E.; Cox, J.D.; Ha, S.I.; Belka, G.K.; Golant, A.; et al. C-myc induces mammary tumorigenesis by means of a preferred pathway involving spontaneous kras2 mutations. Nat. Med. 2001, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-myc protein requires dimerization with max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Amati, B.; Dalton, S.; Brooks, M.W.; Littlewood, T.D.; Evan, G.I.; Land, H. Transcriptional activation by the human c-myc oncoprotein in yeast requires interaction with max. Nature 1992, 359, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, A.V.; Hu, C.D.; Kerppola, T.K. Visualization of myc/max/mad family dimers and the competition for dimerization in living cells. Mol. Cell. Biol. 2004, 24, 4294–4308. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Lee, W.M.; Chen, L.L.; Dang, C.V. Max: Functional domains and interaction with c-myc. Genes Dev. 1992, 6, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Myc and max proteins possess distinct transcriptional activities. Nature 1992, 359, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Hopewell, R.; Ziff, E.B. The nerve growth factor-responsive pc12 cell line does not express the myc dimerization partner max. Mol. Cell. Biol. 1995, 15, 3470–3478. [Google Scholar] [CrossRef] [PubMed]
- Steiger, D.; Furrer, M.; Schwinkendorf, D.; Gallant, P. Max-independent functions of myc in drosophila melanogaster. Nat. Genet. 2008, 40, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Liu, J.D.; Chow, J.M.; Liu, C.R.; Liu, H.E. Small-molecule c-myc inhibitor, 10058-f4, inhibits proliferation, downregulates human telomerase reverse transcriptase and enhances chemosensitivity in human hepatocellular carcinoma cells. Anticancer Drugs 2007, 18, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Parise, R.A.; Joseph, E.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the myc-max disruptor, 10058-f4 [z,e]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother. Pharmacol. 2009, 63, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight myc-max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Hurley, L.H. Targeting myc expression through g-quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microrna repression by myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-myc-regulated micrornas modulate e2f1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Slack, F.J. Micrornas: Small molecules with big roles—c. Elegans to human cancer. Biol. Cell 2008, 100, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microrna delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Ji, J.; Li, X.; Ding, N.; Wu, H.; Liu, Y.; Wang, X.W.; Calvisi, D.F.; Song, G.; Chen, X. Distinct anti-oncogenic effect of various micrornas in different mouse models of liver cancer. Oncotarget 2015, 6, 6977–6988. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L.; Bernstein, B.E. Signaling network model of chromatin. Cell 2002, 111, 771–778. [Google Scholar] [CrossRef]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The brd4 extraterminal domain confers transcription activation independent of ptefb by recruiting multiple proteins, including nsd3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. Bet bromodomain inhibition as a therapeutic strategy to target c-myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Molecular mechanisms of hepatic fibrosis and principles of therapy. J. Gastroenterol. 1997, 32, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, Y.A.; Hu, W.; Cubero, F.J.; Haas, U.; Freimuth, J.; Tacke, F.; Trautwein, C.; Liedtke, C. Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Aiba, N.; Nambu, S.; Inoue, K.; Sasaki, H. Hypomethylation of the c-myc oncogene in liver cirrhosis and chronic hepatitis. Gastroenterol. Jpn. 1989, 24, 270–276. [Google Scholar] [PubMed]
- Chung, S.I.; Moon, H.; Kim, D.Y.; Cho, K.J.; Ju, H.L.; Kim do, Y.; Ahn, S.H.; Han, K.H.; Ro, S.W. Development of a transgenic mouse model of hepatocellular carcinoma with a liver fibrosis background. BMC Gastroenterol. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Himeno, Y.; Fukuda, Y.; Hatanaka, M.; Imura, H. Expression of oncogenes in human liver disease. Liver 1988, 8, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Balsano, C.; Avantaggiati, M.L.; Natoli, G.; De Marzio, E.; Will, H.; Perricaudet, M.; Levrero, M. Full-length and truncated versions of the hepatitis b virus (hbv) x protein (px) transactivate the cmyc protooncogene at the transcriptional level. Biochem. Biophys. Res. Commun. 1991, 176, 985–992. [Google Scholar] [CrossRef]
- Lakhtakia, R.; Kumar, V.; Reddi, H.; Mathur, M.; Dattagupta, S.; Panda, S.K. Hepatocellular carcinoma in a hepatitis b ‘x’ transgenic mouse model: A sequential pathological evaluation. J. Gastroenterol. Hepatol. 2003, 18, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, W.; Ko, C.; Ryu, W.S. Hepatitis b virus x protein enhances myc stability by inhibiting scf (skp2) ubiquitin e3 ligase-mediated myc ubiquitination and contributes to oncogenesis. Oncogene 2016, 35, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, N.; Tsunedomi, R.; Tamesa, T.; Okada, T.; Sakamoto, K.; Hamaguchi, T.; Yamada-Okabe, H.; Miyamoto, T.; Uchimura, S.; Hamamoto, Y.; et al. Involvement of c-myc-regulated genes in hepatocellular carcinoma related to genotype-c hepatitis b virus. J. Cancer Res. Clin. Oncol. 2006, 132, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Huppi, K.; Pitt, J.J.; Wahlberg, B.M.; Caplen, N.J. The 8q24 gene desert: An oasis of non-coding transcriptional activity. Front. Genet. 2012, 3, 69. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yang, Y.; Zhang, L.; Tang, G.; Wang, Y.; Xue, G.; Zhou, W.; Sun, S. Characterization of the genotype and integration patterns of hepatitis b virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015, 61, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.; Kumar, V. Specific inhibition of gene expression and transactivation functions of hepatitis b virus x protein and c-myc by small interfering rnas. FEBS Lett. 2004, 560, 210–214. [Google Scholar] [CrossRef]
- Hung, L.; Kumar, V. Antisense regulation of expression and transactivation functions of the tumorigenic hbx and c-myc genes. Biochem. Biophys. Res. Commun. 2006, 344, 293–299. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma and hepatitis c in the united states. Hepatology 2002, 36, S74–S83. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis c virus-induced activation of beta-catenin promotes c-myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Borro, P. Alcohol and gastrointestinal oncology. World J. Gastrointest. Oncol. 2010, 2, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Borro, P.; Ancarani, O.; Sumberaz, A. Human carcinogenesis and alcohol in hepato-gastroenterology. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 512–518. [Google Scholar] [PubMed]
- Testino, G.; Leone, S.; Borro, P. Alcohol and hepatocellular carcinoma: A review and a point of view. World J. Gastroenterol. 2014, 20, 15943–15954. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127, S87–S96. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; Bassendine, M.F. Genetic predisposition to alcoholic liver disease. Gut 1992, 33, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Devor, E.J.; Reich, T.; Cloninger, C.R. Genetics of alcoholism and related end-organ damage. Semin. Liver Dis. 1988, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, D.W. Genetic influences in alcoholism. Adv. Intern. Med. 1987, 32, 283–297. [Google Scholar] [PubMed]
- Nevzorova, Y.A.; Cubero, F.J.; Hu, W.; Hao, F.; Haas, U.; Ramadori, P.; Gassler, N.; Hoss, M.; Strnad, P.; Zimmermann, H.W.; et al. Enhanced expression of c-myc in hepatocytes promotes initiation and progression of alcoholic liver disease. J. Hepatol. 2016, 64, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Buendia, M.A.; Bourre, L.; Cairo, S. Myc target mirs and liver cancer: Small molecules to get myc sick. Gastroenterology 2012, 142, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Comerford, S.A.; Hinnant, E.A.; Chen, Y.; Bansal, H.; Klapproth, S.; Rakheja, D.; Finegold, M.J.; Lopez-Terrada, D.; O’Donnell, K.A.; Tomlinson, G.E.; et al. Hepatoblastoma modeling in mice places nrf2 within a cancer field established by mutant beta-catenin. JCI Insight 2016, 1, e88549. [Google Scholar] [CrossRef] [PubMed]
- Cairo, S.; Armengol, C.; De Reynies, A.; Wei, Y.; Thomas, E.; Renard, C.A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of wnt/beta-catenin and myc signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Cairo, S.; Wang, Y.; de Reynies, A.; Duroure, K.; Dahan, J.; Redon, M.J.; Fabre, M.; McClelland, M.; Wang, X.W.; Croce, C.M.; et al. Stem cell-like micro-rna signature driven by myc in aggressive liver cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 20471–20476. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, T.W.; Peng, J.; Tang, X.; Ko, K.S.; Xia, M.; Aller, M.A. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology 2011, 141, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, T.W.; Ko, K.S.; Xia, M.; Lu, S.C. Switch from mnt-max to myc-max induces p53 and cyclin d1 expression and apoptosis during cholestasis in mouse and human hepatocytes. Hepatology 2009, 49, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Roberts, L.R. Myc, max, and mnt: Molecular mechanisms of enhancement of cholangiocarcinogenesis by cholestasis. Gastroenterology 2011, 141, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, T.; Wang, J.; Li, T.W.; Fan, W.; Peng, H.; Krishnan, A.; Gores, G.J.; Mato, J.M.; Lu, S.C. Deregulated methionine adenosyltransferase alpha1, c-myc, and maf proteins together promote cholangiocarcinoma growth in mice and humans(double dagger). Hepatology 2016, 64, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Fernandez-Barrena, M.G.; Avila, M.A. New molecular interactions of c-myc in cholangiocarcinoma may open new therapeutic opportunities. Hepatology 2016, 64, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Radziuk, J.; Pye, S. Hepatic glucose uptake, gluconeogenesis and the regulation of glycogen synthesis. Diabetes Metab. Res. Rev. 2001, 17, 250–272. [Google Scholar] [CrossRef] [PubMed]
- Vaulont, S.; Vasseur-Cognet, M.; Kahn, A. Glucose regulation of gene transcription. J. Biol. Chem. 2000, 275, 31555–31558. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; Pujol, A.; Gregori, X.; Riu, E.; Visa, J.; Bosch, F. Evidence from transgenic mice that myc regulates hepatic glycolysis. FASEB J. 1995, 9, 1067–1078. [Google Scholar] [PubMed]
- Riu, E.; Bosch, F.; Valera, A. Prevention of diabetic alterations in transgenic mice overexpressing myc in the liver. Proc. Natl. Acad. Sci. USA 1996, 93, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Riu, E.; Ferre, T.; Mas, A.; Hidalgo, A.; Franckhauser, S.; Bosch, F. Overexpression of c-myc in diabetic mice restores altered expression of the transcription factor genes that regulate liver metabolism. Biochem. J. 2002, 368, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Riu, E.; Ferre, T.; Hidalgo, A.; Mas, A.; Franckhauser, S.; Otaegui, P.; Bosch, F. Overexpression of c-myc in the liver prevents obesity and insulin resistance. FASEB J. 2003, 17, 1715–1717. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of cdk4 as a target of c-myc. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; van Steensel, B.; Delrow, J.; Bussemaker, H.J.; Li, L.; Sawado, T.; Williams, E.; Loo, L.W.; Cowley, S.M.; Yost, C.; et al. Genomic binding by the drosophila myc, max, mad/mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
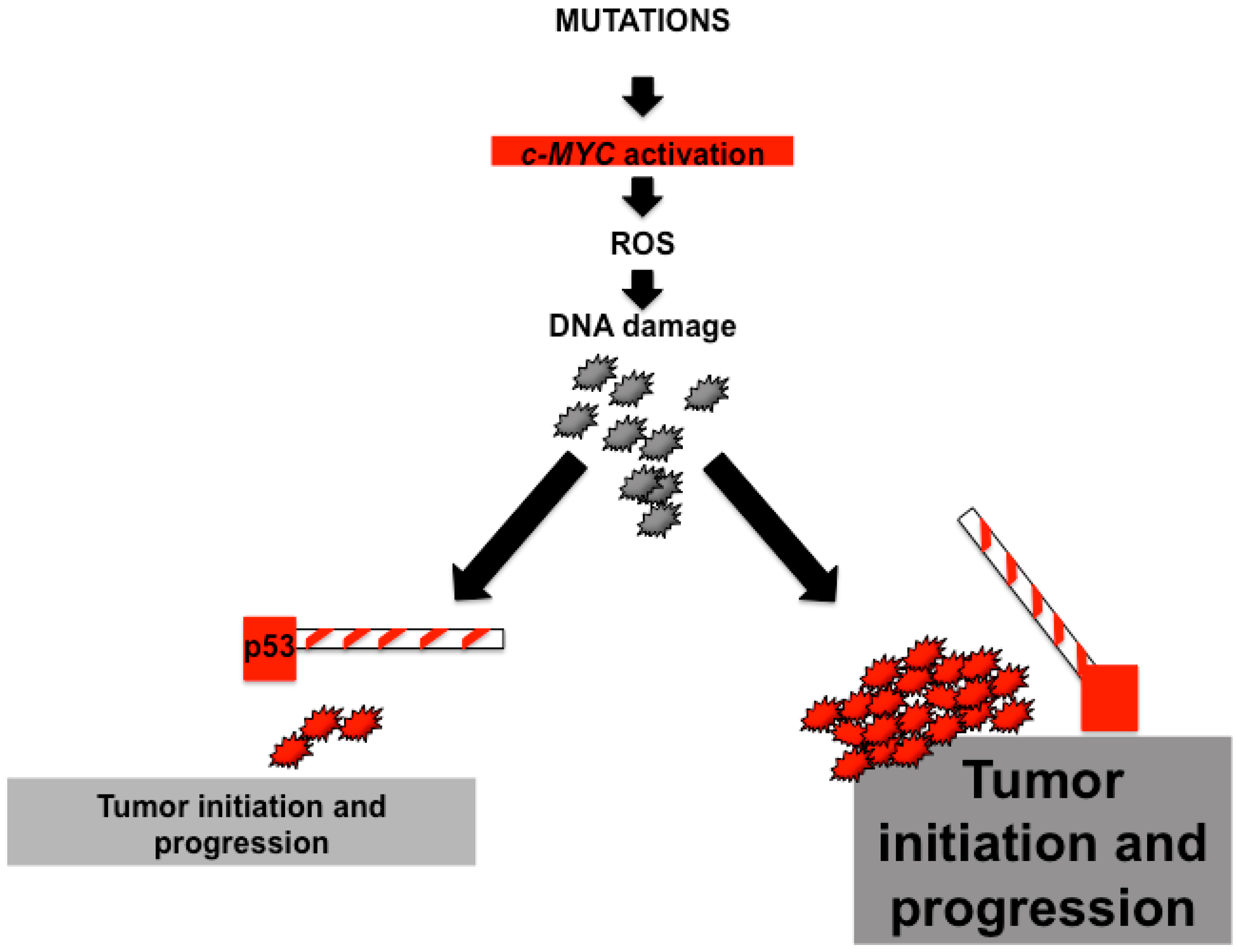

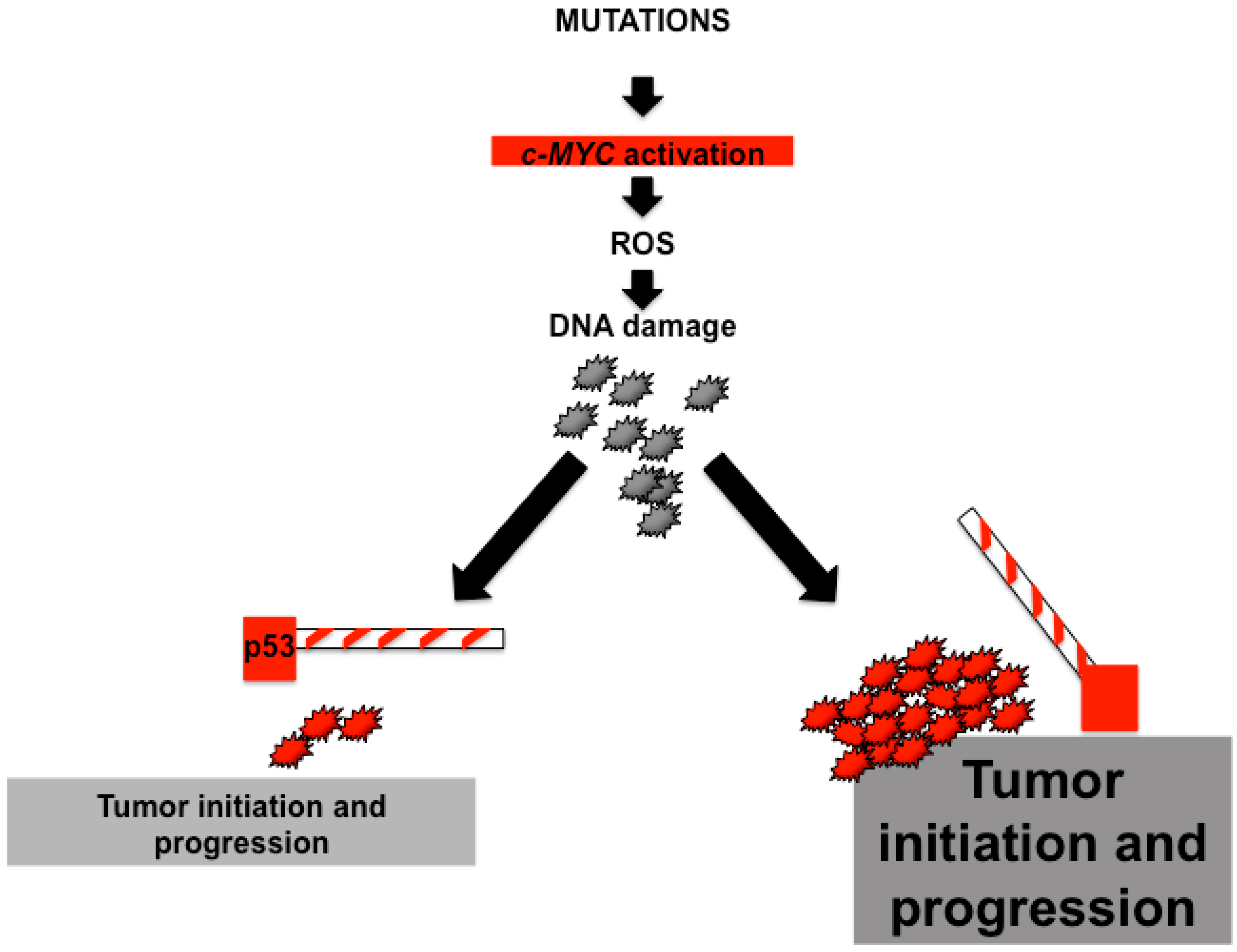{kind=link}
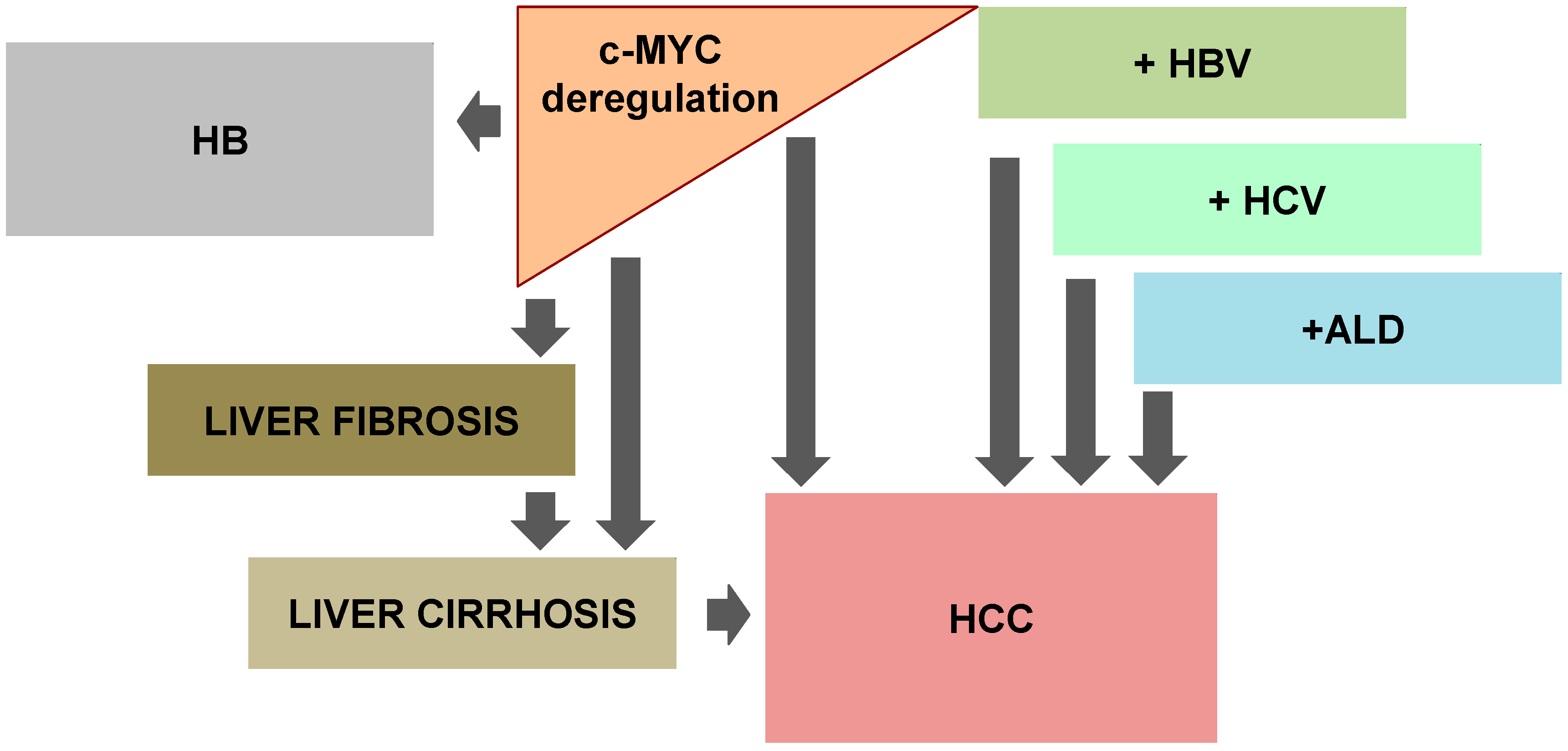
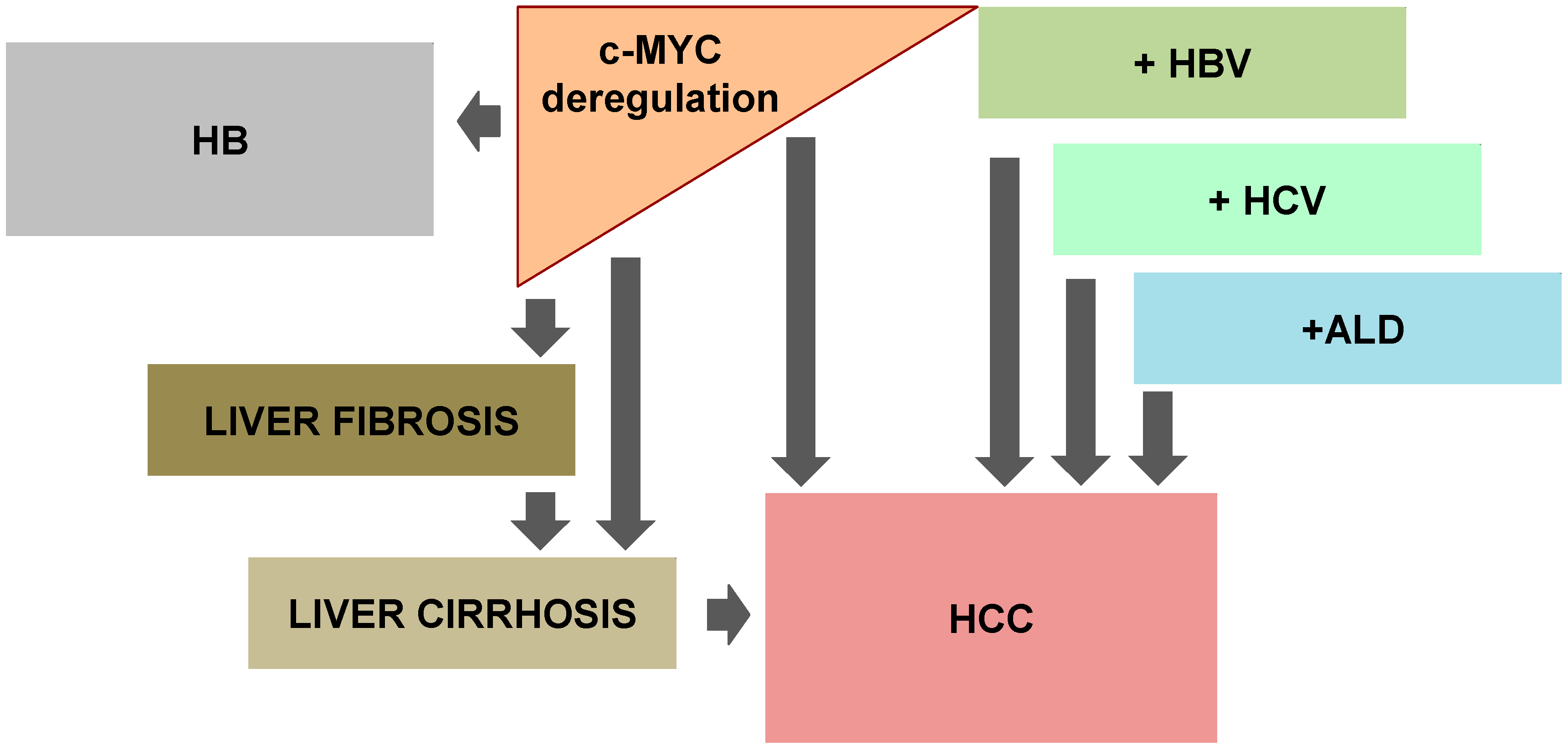{kind=link}
| Transgenic Mice | Phenotype | Time to Tumor Development | References |
|---|---|---|---|
| alb-Cre+/c-MYCtg (hepatocyte-specific) | HCC development albeit with a long latency period | 45 weeks of age—less than 40%; 65 weeks—80% of mice have a minimum of one liver tumor | [22,23,24] |
| alb-Cre+/c-MYCtg/TGFαtg (hepatocyte-specific) | Dramatic acceleration of HCC neoplasia | 17 weeks—20% with tumor 40 weeks—100% with tumor | [23,25,26] |
| alb-Cre+/c-MYCtg/HGFαtg (hepatocyte-specific) | Delayed appearance of preneoplastic lesions and prevention of malignancy | 12 months of age—15% of mice display dysplastic cells; 16 months—67% affected by mild hepatic dysplasia | [25] |
| alb-Cre+/c-MYC/tgE2F1tg (hepatocyte-specific) | Aggressive tumor phenotype | 26–35 weeks—100% of mice have tumors | [27] |
| alb-Cre+/c-MYCtg/p53−/− | Accelerated increase in size and malignancy of HCCs | 250 days—100% moribund with tumor burden | [28] |
| alb-Cre+/c-MYCtg/IgEGFtg/p53−/− | Accelerated HCC growth | 58 days—100% moribund with tumor burden | [28] |
| c-MYCtg tetracycline-regulated, liver specific) + DDC | Accelerated HCC | 31 days—100% moribund with tumor burden | [29] |
| c-MYCtg (tetracycline-regulated, liver-specific) + CCl4 | Accelerated HCC | 40 days—100% moribund with tumor burden | [29] |
| WHV/c-MYC/HBx | Accelerate tumor development | 60 weeks—100% mice developed large tumors | [30] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, K.; Cubero, F.J.; Nevzorova, Y.A. c-MYC—Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes 2017, 8, 123. https://doi.org/10.3390/genes8040123
Zheng K, Cubero FJ, Nevzorova YA. c-MYC—Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes. 2017; 8(4):123. https://doi.org/10.3390/genes8040123
Chicago/Turabian StyleZheng, Kang, Francisco Javier Cubero, and Yulia A. Nevzorova. 2017. "c-MYC—Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease" Genes 8, no. 4: 123. https://doi.org/10.3390/genes8040123
APA StyleZheng, K., Cubero, F. J., & Nevzorova, Y. A. (2017). c-MYC—Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes, 8(4), 123. https://doi.org/10.3390/genes8040123