
A Transcriptomic Comparison of Two Bambara Groundnut Landraces under Dehydration Stress

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Site Descriptions and Experimental Design
2.3. RNA Extraction
2.4. cRNA and Genomic DNA Affymetrix Labelling and Hybridisation
2.5. Probe Selection and Identification of Differentially Expressed Genes
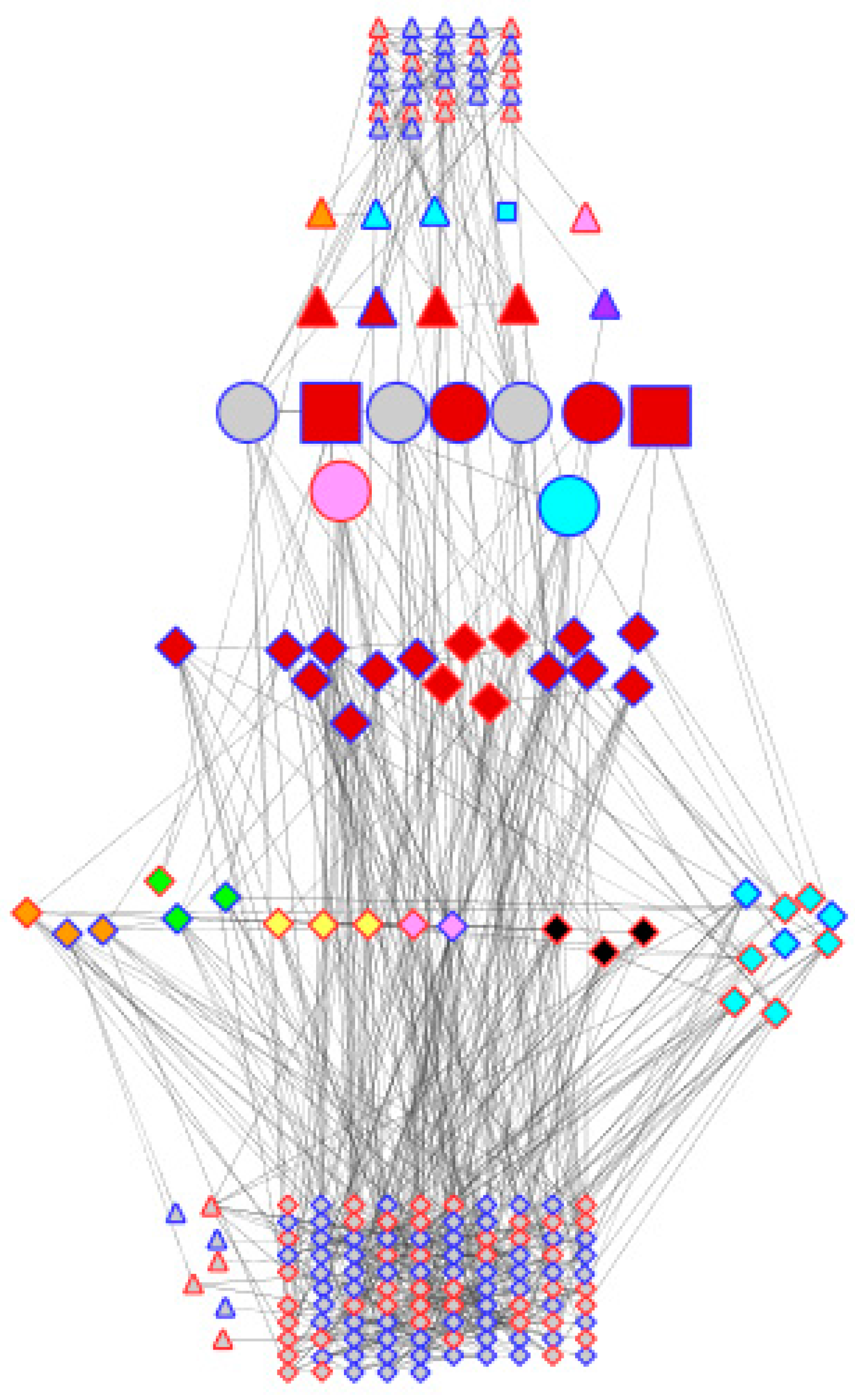
2.6. Construction of the Co-Expression Network
2.7. Expression Validation of Differentially Expressed Genes Using Real-Time qPCR
3. Results
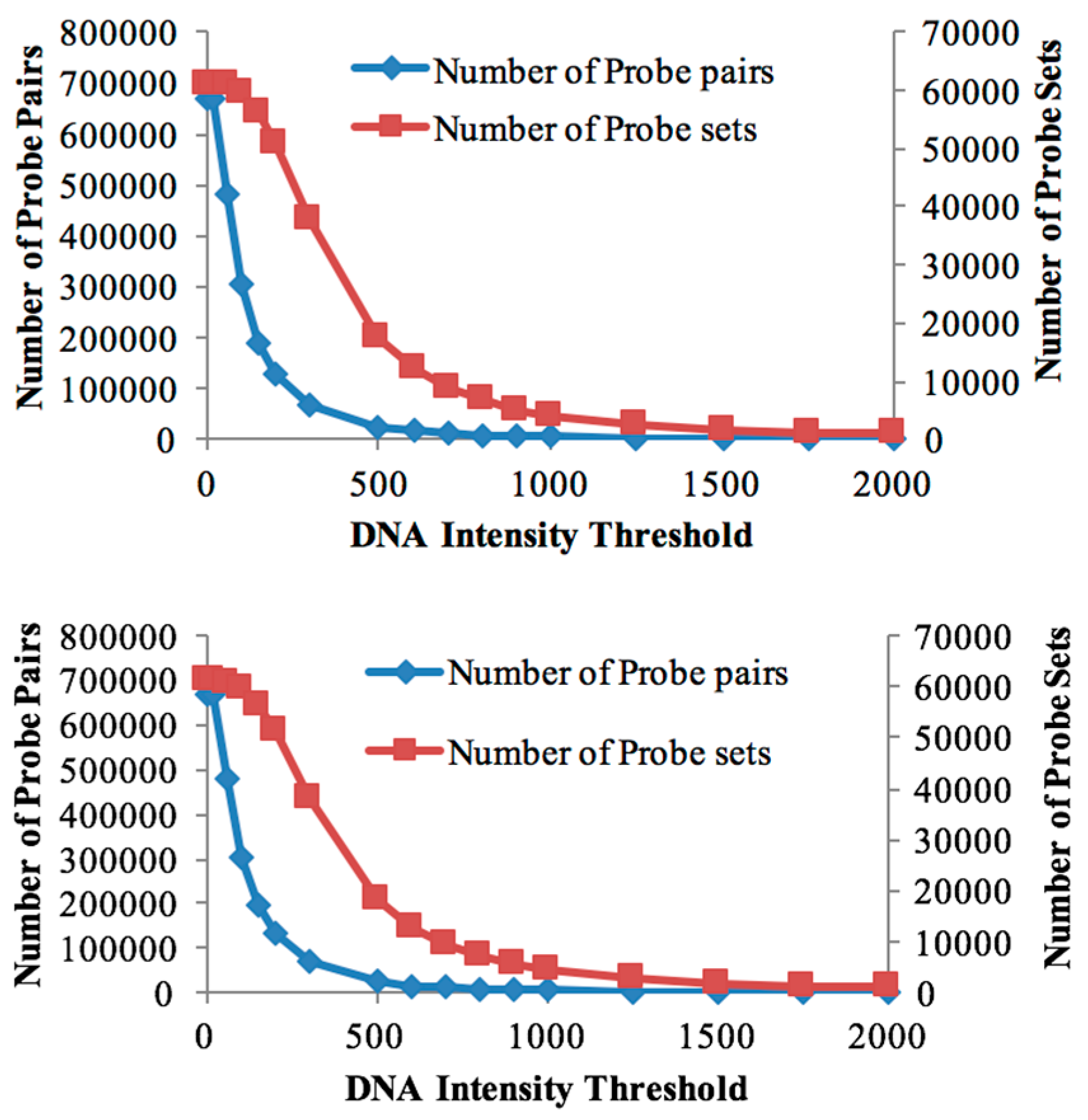
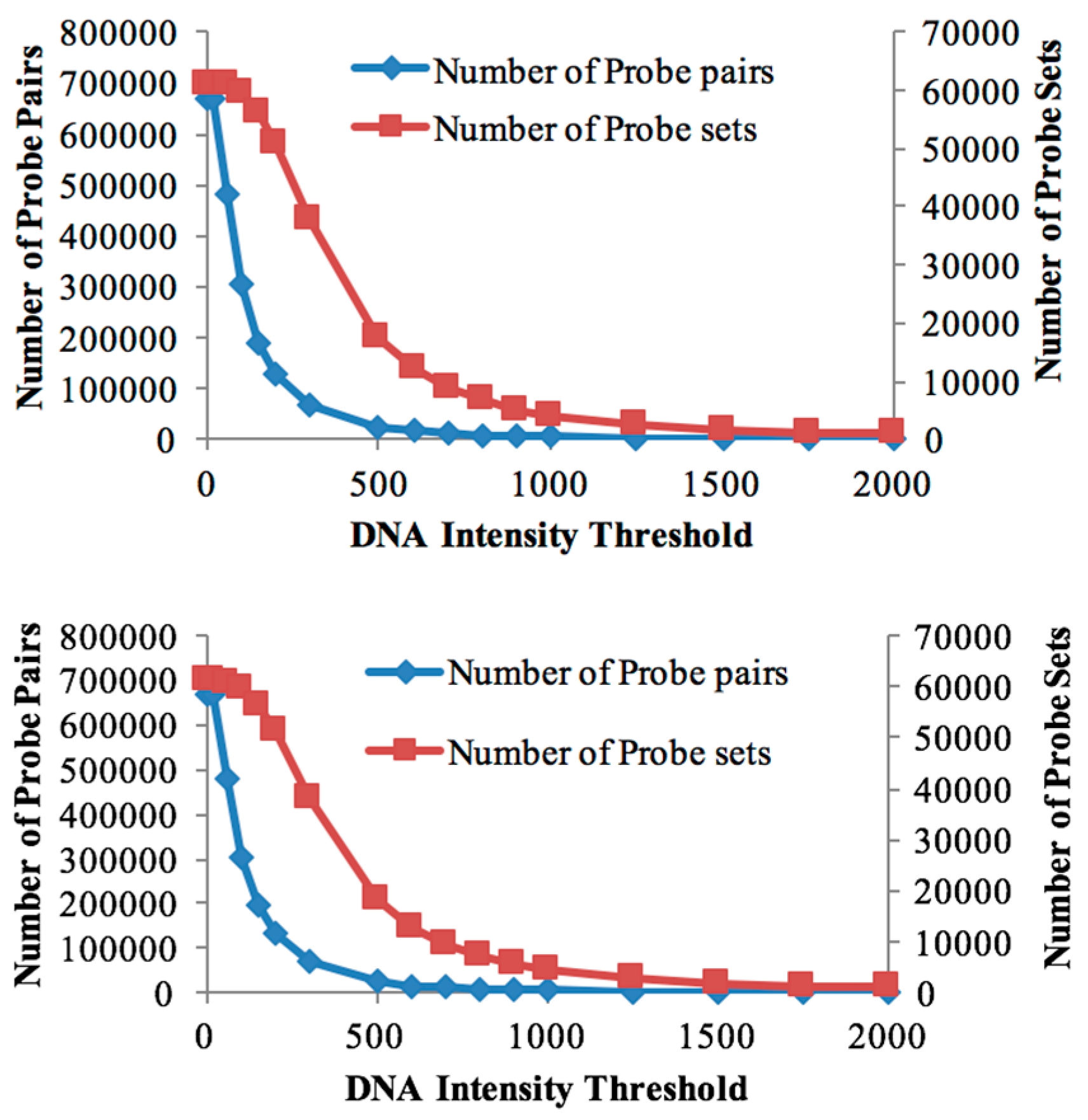
3.1. Probe Selection Based on gDNA
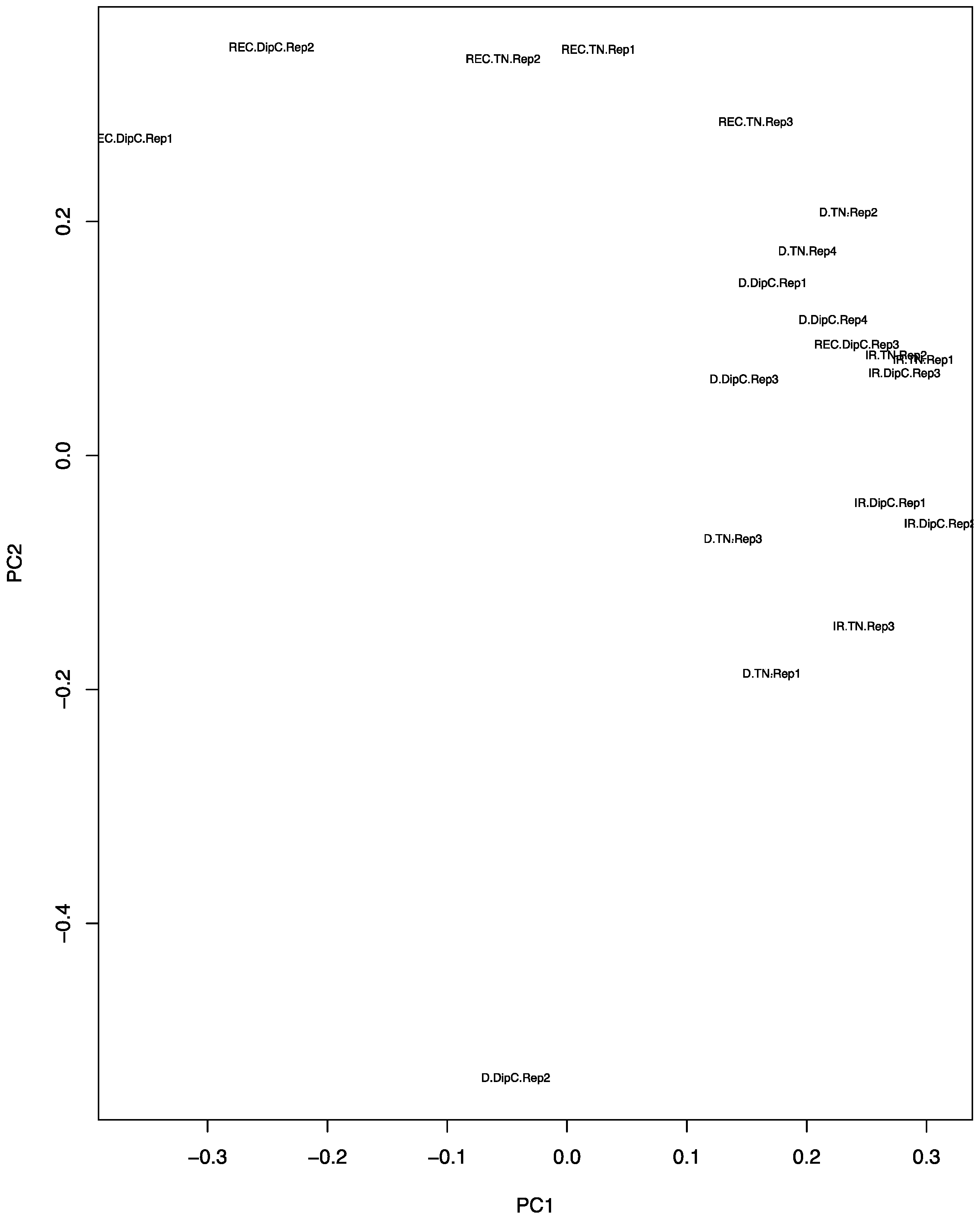
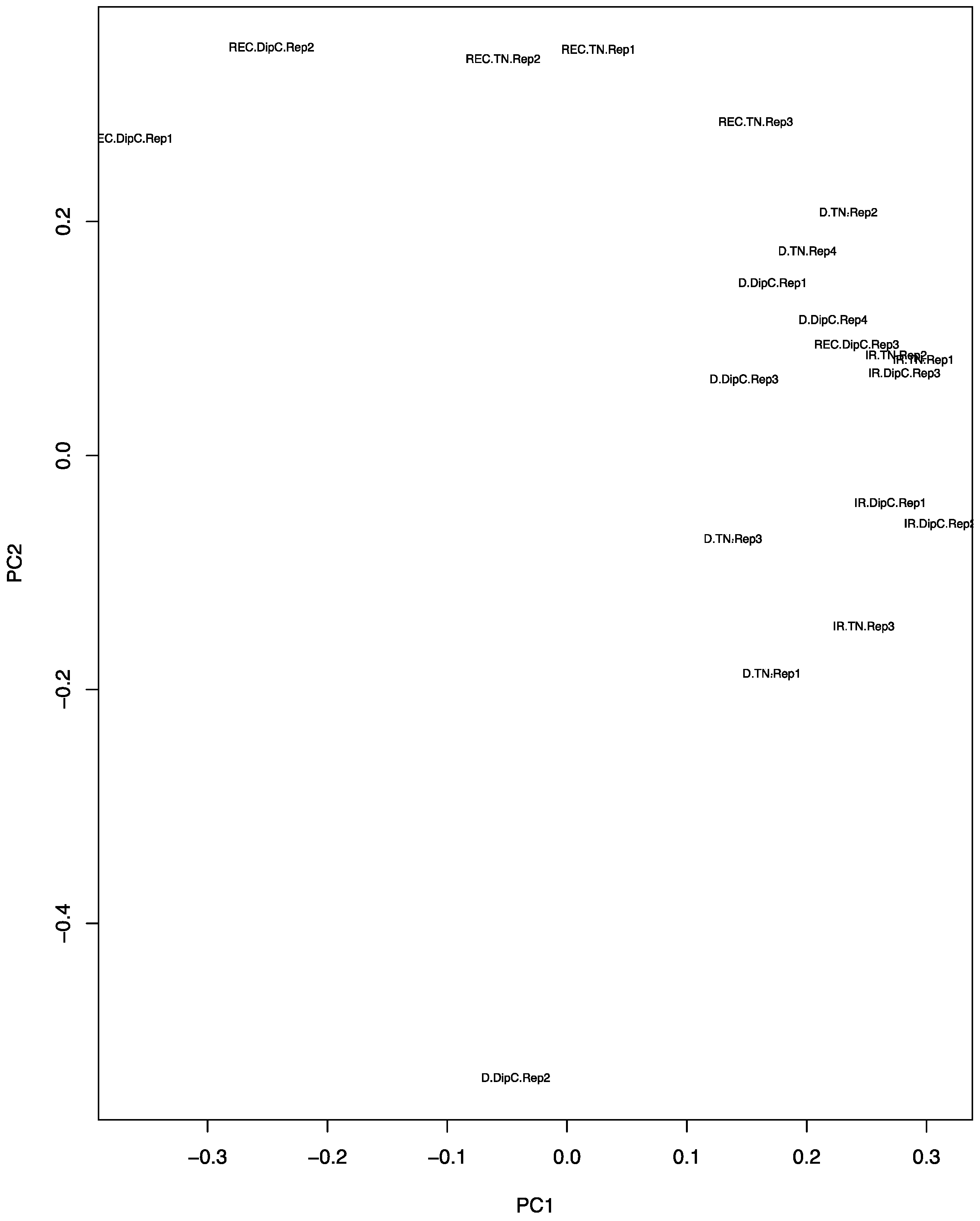
3.2. Principal Component Analysis
3.3. Gene Expression Under Water-Sufficient Conditions
3.4. Identification of Differentially Expressed Genes
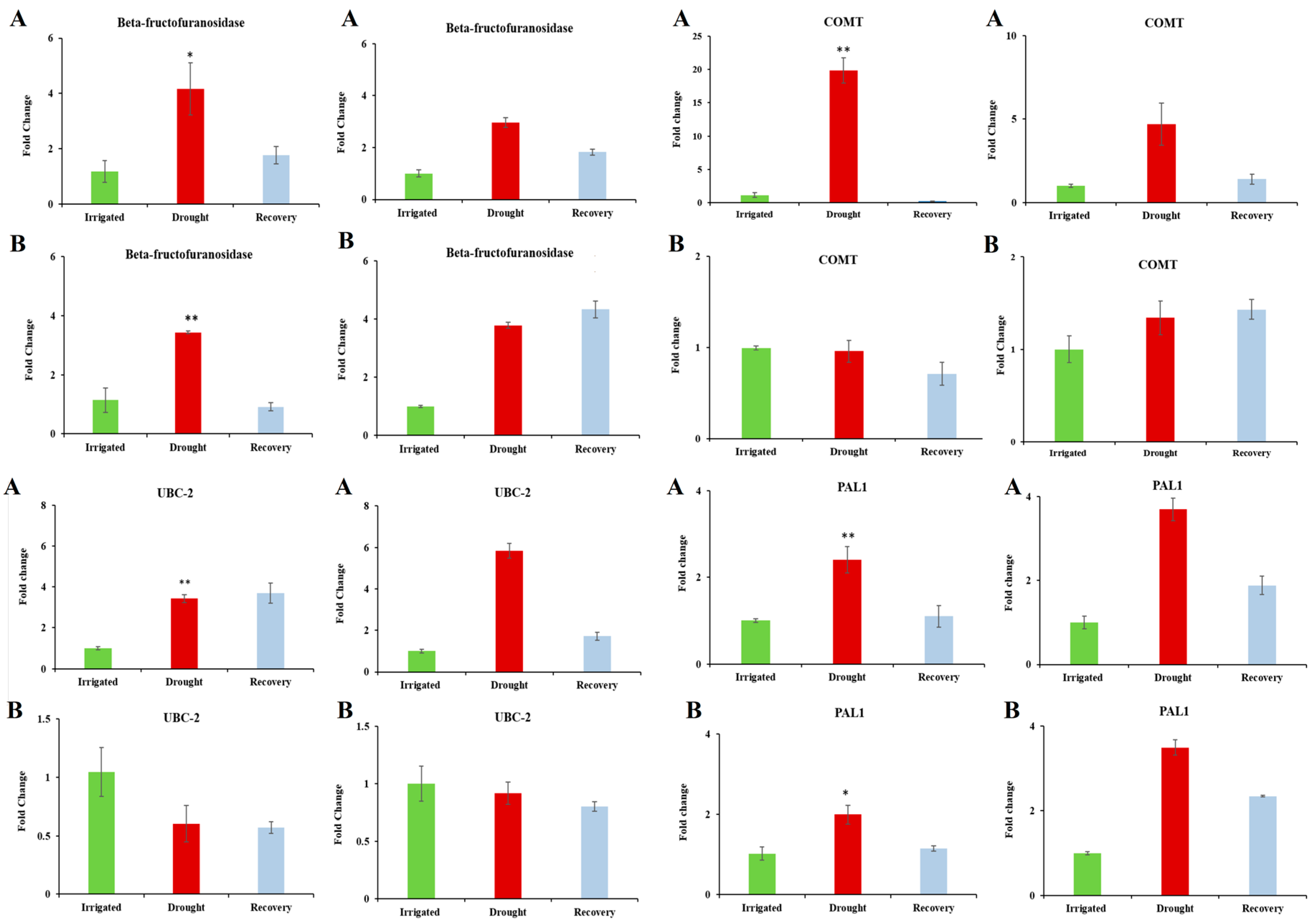
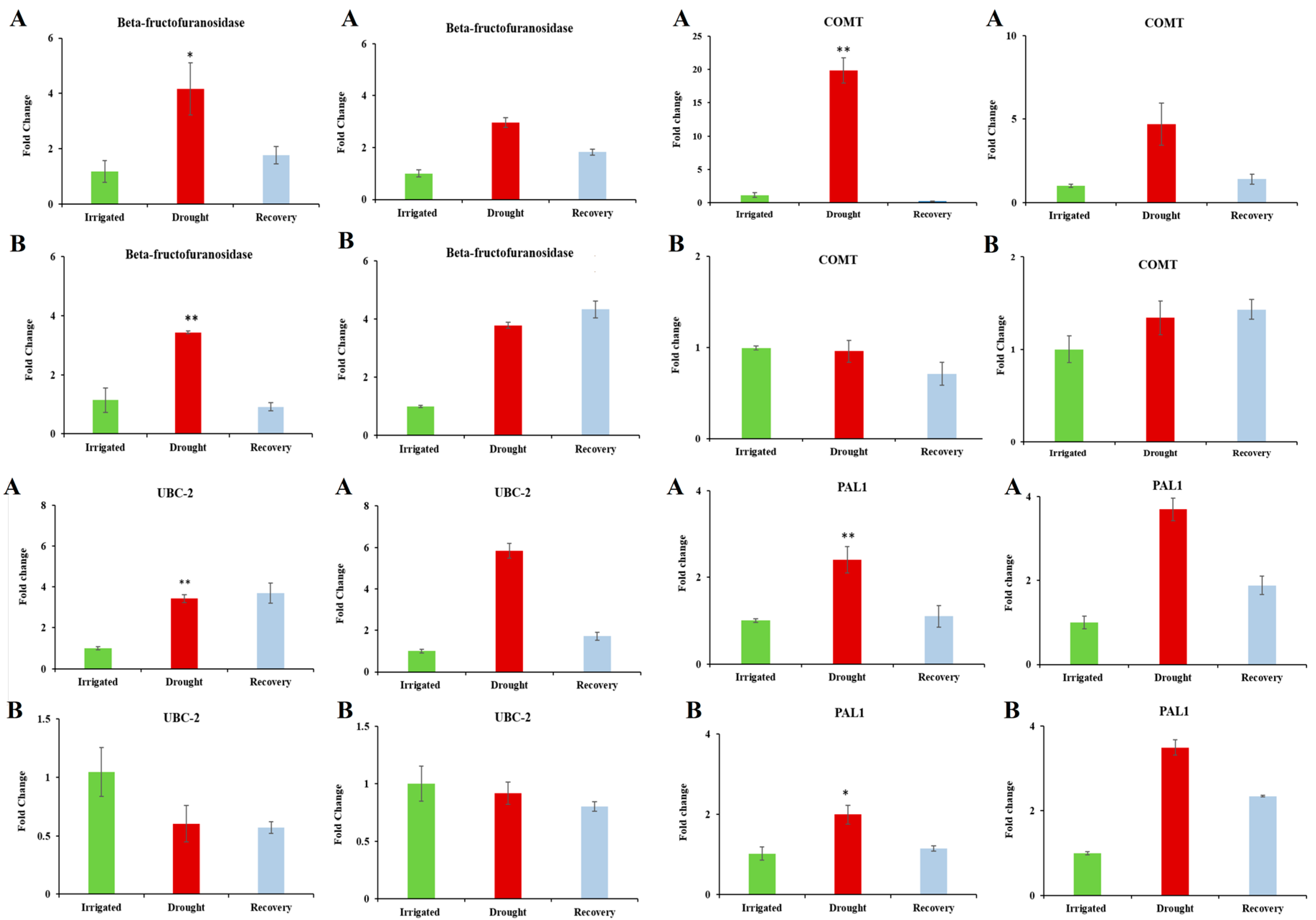
3.5. Confirmation of Candidate Dehydration-Associated Genes by Real-Time qRT-PCR
3.6. Transcription Factors Associated with Dehydration Stress
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ocampo, E.; Robles, R. Drought tolerance in mungbean. I. Osmotic adjustment in drought stressed mungbean. Philipp. J. Crop Sci. 2000, 25, 1–5. [Google Scholar]
- Subbarao, G.V.; Chauhan, Y.S.; Johansen, C. Patterns of osmotic adjustment in pigeonpea—Its importance as a mechanism of drought resistance. Eur. J. Agron. 2000, 12, 239–249. [Google Scholar] [CrossRef]
- Kooyers, N.J. The evolution of drought escape and avoidance in natural herbaceous populations. Plant Sci. 2015, 234, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Harb, A.; Krishnan, A.; Ambavaram, M.M.R.; Pereira, A. Molecular and physiological analysis of drought stress in Arabidopsis reveals early responses leading to acclimation in plant growth. Plant Physiol. 2010, 154, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Al Shareef, I.; Sparkes, D.; Azam-Ali, S. Temperature and drought stress effects on growth and development of bambara groundnut (Vigna subterranea L.). Exp. Agric. 2013, 50, 72–89. [Google Scholar] [CrossRef]
- Eslami, S.V.; Gill, G.S.; McDonald, G. Effect of water stress during seed development on morphometric characteristics and dormancy of wild radish (Raphanus raphanistrum L.) seeds. Int. J. Plant Prod. 2012, 4, 159–168. [Google Scholar]
- Gaur, M.P.; Krishnamurthy, L.; Kashiwagi, J. Improving drought-avoidance root traits in chickpea (Cicer arietinum); current status of research at ICRISAT. Plant Prod. Sci. 2008, 11, 3–11. [Google Scholar] [CrossRef]
- Gupta, N.K.; Gupta, S.; Kumar, A. Effect of water stress on physiological attributes and their relationship with growth and yield of wheat cultivars at different stages. J. Agron. Crop Sci. 2001, 186, 55–62. [Google Scholar] [CrossRef]
- Liu, F.; Jensen, C.R.; Andersen, M.N. Hydraulic and chemical signals in the control of leaf expansion and stomatal conductance in soybean exposed to drought stress. Funct. Plant Biol. 2003, 30, 65–73. [Google Scholar] [CrossRef]
- Ludlow, M. Strategies of response to water stress. In Structural and Functional Responses to Environmental Stresses: Water Shortage; Kreeeb, K.H., Richter, H., Hinckley, T.M., Eds.; SPB Academic publishing: The Hague, The Netherlands, 1989; pp. 269–282. [Google Scholar]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Mwale, S.S.; Azam-Ali, S.N.; Massawe, F.J. Growth and development of bambara groundnut (Vigna subterranea) in response to soil moisture. Eur. J. Agron. 2007, 26, 345–353. [Google Scholar] [CrossRef]
- Umezawa, T.; Fujita, M.; Fujita, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Engineering drought tolerance in plants: Discovering and tailoring genes to unlock the future. Curr. Opin. Biotechnol. 2006, 17, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants. Front Plant Sci. 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Poets, A.M.; Fang, Z.; Clegg, M.T.; Morrell, P.L. Barley landraces are characterized by geographically heterogeneous genomic origins. Genome Biol. 2015, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.R.; Upadhyaya, H.D.; Ramu, P.; Deshpande, S.; Hash, C.T.; Bonnette, J.; Juenger, T.E.; Hyma, K.; Acharya, C.; Mitchell, S.E.; et al. Genome-environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 2015, 1, e1400218. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhu, X.; Yu, J.; Wang, L.; Zhang, Y.; Li, D.; Zhou, R.; Zhang, X. Identification of sesame genomic variations from genome comparison of landrace and variety. Front. Plant Sci. 2016, 7, 1169. [Google Scholar] [CrossRef] [PubMed]
- Valliyodan, B.; Nguyen, H.T. Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr. Opin. Plant Biol. 2006, 9, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Dodig, D.; Zorić, M.; Kandić, V.; Perović, D.; Šurlan-Momirović, G. Comparison of responses to drought stress of 100 wheat accessions and landraces to identify opportunities for improving wheat drought resistance. Plant Breed. 2012, 131, 369–379. [Google Scholar] [CrossRef]
- Collinson, S.T.; Clawson, E.J.; Azam-Ali, S.N.; Black, C.R. Effects of soil moisture deficits on the water relations of bambara groundnut (Vigna subterranea L. Verdc.). J. Exp. Bot. 1997, 48, 877–884. [Google Scholar] [CrossRef]
- Linnemann, A.; Azam-ALI, S. Bambara groundnut (Vigna subterraneanea); Williams, J.T., Ed.; Chapman and Hall: London, UK, 1993. [Google Scholar]
- Mazahib, A.M.; Nuha, M.O.; Salawa, I.S.; Babiker, E.E. Some nutritional attributes of bambara groundnut as influenced by domestic processing. Int. Food Res. J. 2013, 20, 1165–1171. [Google Scholar]
- Heller, J. Bambara Groundnut: Vigna subterranea (l.) Verdc. Promoting the Conservation and Use of Under-Utilized and Neglected Crops; IPGRI: Harare, Zimbabwe, 1997. [Google Scholar]
- Mabhaudhi, T.; Modi, A.T.; Beletse, Y.G. Growth, phenological and yield responses of a bambara groundnut (Vigna subterranea L. Verdc) landrace to imposed water stress: II. Rain shelter conditions. Afr. Crop Sci. J. 2013, 39, 191–198. [Google Scholar] [CrossRef]
- Collinson, S.T.; Azam-Ali, S.N.; Chavula, K.M.; Hodson, D.A. Growth, development and yield of bambara groundnut (Vigna subterranea) in response to soil moisture. J. Agric. Sci. 1996, 126, 307. [Google Scholar] [CrossRef]
- Nayamudeza, P. Crop water use and the root systems of bambara groundnut (Vigna subterranea (L.) verdc.) and groundnut (Arachis hypogaea (L.)) in response to irrigation and drought. Master’s Thesis, The University of Nottingham, Nottingham, UK, 1989. [Google Scholar]
- Berchie, J.N. Evaluation of five bambara groundnut (Vigna subterranea (L.) verdc.) landraces to heat and drought stress at Tono-Navrongo, Upper East Region of Ghana. Afr. J. Agric. Res. 2012, 7, 250–256. [Google Scholar] [CrossRef]
- Vurayai, R.; Emongor, V.; Moseki, B.; Emongor, V.; Moseki, B. Physiological responses of bambara groundnut (Vigna subterranea L. Verdc) to short periods of water stress during different developmental stages. Asian J. Agric. Sci. 2011, 3, 37–43. [Google Scholar]
- Chai, H.H. Developing new approaches for transcriptomics and genomics—Using major resources developed in model species for research in crop species. Ph.D. Thesis, The University of Nottingham, Nottingham, UK, 2014. [Google Scholar]
- Chai, H.H.; Massawe, F.; Mayes, S. Effects of mild drought stress on the morpho-physiological characteristics of a bambara groundnut segregating population. Euphytica 2015, 208, 225–236. [Google Scholar] [CrossRef]
- Buckley, B.A. Comparative environmental genomics in non-model species: Using heterologous hybridization to DNA-based microarrays. J. Exp. Biol. 2007, 210, 1602–1606. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.W.; Graham, N.S.; Vanholme, B.; Swennen, R.; May, S.T.; Keulemans, J. Heterologous oligonucleotide microarrays for transcriptomics in a non-model species; a proof-of-concept study of drought stress in musa. BMC Genomics 2009, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Pariset, L.; Chillemi, G.; Bongiorni, S.; Spica, V.R.; Valentini, A. Microarrays and high-throughput transcriptomic analysis in species with incomplete availability of genomic sequences. New Biotechnol. 2009, 25, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.P.; Broadley, M.R.; Craigon, D.J.; Higgins, J.; Emmerson, Z.F.; Townsend, H.J.; White, P.J.; May, S.T. Using genomic DNA-based probe-selection to improve the sensitivity of high-density oligonucleotide arrays when applied to heterologous species. Plant Methods 2005, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.S.; May, S.T.; Daniel, Z.C.; Emmerson, Z.F.; Brameld, J.M.; Parr, T. Use of the affymetrix human genechip array and genomic DNA hybridisation probe selection to study ovine transcriptomes. Animal 2011, 5, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Scharf, E.; Kondo, T. Genespring tm: Tools for analyzing data microarray expression. Genome Inform. 2001, 12, 227–229. [Google Scholar]
- Maere, S.; Heymans, K.; Kuiper, M. Bingo: A cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wei, H.; Zhao, P.X. DeGNServer: Deciphering genome-scale gene networks through high performance reverse engineering analysis. Biomed. Res. Int. 2013, 2013, 856325. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Fan, C.; Li, H.; Zhang, Q.; Fu, Y.-F. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol. Biol. 2009, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nijhawan, A.; Tyagi, A.K.; Khurana, J.P. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time pcr. Biochem. Biophys. Res. Commun. 2006, 345, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-blast: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Bonthala, V.S.; Mayes, K.; Moreton, J.; Blythe, M.; Wright, V.; May, S.T.; Massawe, F.; Mayes, S.; Twycross, J. Identification of gene modules associated with low temperatures response in bambara groundnut by network-based analysis. PLoS ONE 2016, 11, e0148771. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Meng, F.R.; Zhang, C.Y.; Zhang, N.; Sun, M.S.; Ren, J.P.; Niu, H.B.; Wang, X.; Yin, J. Comparative analysis of water stress-responsive transcriptomes in drought-susceptible and -tolerant wheat (Triticum aestivum L.). J. Plant Biol. 2012, 55, 349–360. [Google Scholar] [CrossRef]
- Majlath, I.; Darko, E.; Palla, B.; Nagy, Z.; Janda, T.; Szalai, G. Reduced light and moderate water deficiency sustain nitrogen assimilation and sucrose degradation at low temperature in durum wheat. J. Plant Physiol. 2016, 191, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Vaittinen, M.; Kaminska, D.; Kakela, P.; Eskelinen, M.; Kolehmainen, M.; Pihlajamaki, J.; Uusitupa, M.; Pulkkinen, L. Downregulation of cpped1 expression improves glucose metabolism in vitro in adipocytes. Diabetes 2013, 62, 3747–3750. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.-i.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JMJC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, N.; Virlouvet, L.; Riethoven, J.-J.; Fromm, M.; Avramova, Z. Four distinct types of dehydration stress memory genes in Arabidopsis thaliana. BMC Plant Biol. 2013, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Michniewicz, M.; Bergmann, D.C.; Wang, Z.Y. Brassinosteroid regulates stomatal development by GSK3-mediated inhibition of a MAPK pathway. Nature 2012, 482, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Brutnell, T.P.; Sawers, R.J.H.; Mant, A.; Langdale, J.A. Bundle sheath defective2, a novel protein required for post-translational regulation of the RBCL gene of maize. Plant Cell 1999, 11, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, A. Effects of drought on the activity of phenylalanine ammonia lyase in the leaves and roots of maize inbreds. Aust. J. Basic Appl. Sci. 2011, 5, 952–956. [Google Scholar]
- Gerhardt, L.B.d.A.; Magioli, C.; Perez, A.B.U.C.M.; Margis, R.; Sachetto-Martins, G.; Margis-Pinheiro, M. AtCHITIV gene expression is stimulated under abiotic stresses and is spatially and temporally regulated during embryo development. Genetics Mol. Biol. 2004, 27, 118–123. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, X.; Liu, Y.; Roth, C.; Copeland, C.; McFarlane, H.E.; Huang, S.; Lipka, V.; Wiermer, M.; Li, X. Mitochondrial atPAM16 is required for plant survival and the negative regulation of plant immunity. Nat. Commun. 2013, 4, 2558. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C. Jasmonates: An update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, Y.; Liu, Z.; Kong, D.; Duan, M.; Luo, L. Genome-wide identification and analysis of drought-responsive micrornas in Oryza sativa. J. Exp. Bot. 2010, 61, 4157–4168. [Google Scholar] [CrossRef] [PubMed]
- Scherle, P.; Behrens, T.; Staudt, L.M. Ly-gdi, a gdp-dissociation inhibitor of the rhoa GTP-binding protein, is expressed preferentially in lymphocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 7568–7572. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Bush, D.R. Lht1, a lysine- and histidine-specific amino acid transporter in arabidopsis. Plant Physiol. 1997, 115, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, W.C.; Xu, Y.Q.; Li, G.J.; Liao, Y.; Fu, F.L. Differential expression of candidate genes for lignin biosynthesis under drought stress in maize leaves. J. Appl. Genetics 2009, 50, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Oliver, D.J. H-protein of the glycine decarboxylase multienzyme complex: Complementary DNA encoding the protein from Arabidopsis thaliana. Plant Physiol. 1992, 98, 1518–1519. [Google Scholar] [CrossRef] [PubMed]
- Ledger, S.; Strayer, C.; Ashton, F.; Kay, S.A.; Putterill, J. Analysis of the function of two circadian-regulated constans-like genes. Plant J. 2001, 26, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Eamens, A.L.; Wook Kim, K.; Waterhouse, P.M. Drb2, drb3 and drb5 function in a non-canonical microRNA pathway in Arabidopsis thaliana. Plant Signal. Behav. 2012, 7, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Ades, S.E.; Grigorova, I.L.; Gross, C.A. Regulation of the alternative sigma factor σ(e) during initiation, adaptation, and shutoff of the extracytoplasmic heat shock response in Escherichia coli. J. Bacteriol. 2003, 185, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Kato, H.; Ogawa, T.; Kurachi, A.; Nakagawa, Y.; Kobayashi, H. Sigma factor phosphorylation in the photosynthetic control of photosystem stoichiometry. Proc. Natl. Acad. Sci. USA 2010, 107, 10760–10764. [Google Scholar] [CrossRef] [PubMed]
- Periappuram, C.; Steinhauer, L.; Barton, D.L.; Taylor, D.C.; Chatson, B.; Zou, J. The plastidic phosphoglucomutase from arabidopsis. A reversible enzyme reaction with an important role in metabolic control. Plant Physiol. 2000, 122, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef] [PubMed]
- Wedel, N.; Soll, J.; Paap, B.K. Cp12 provides a new mode of light regulation of Calvin cycle activity in higher plants. Proc. Natl. Acad. Sci. USA 1997, 94, 10479–10484. [Google Scholar] [CrossRef] [PubMed]
- Lorick, K.L.; Jensen, J.P.; Fang, S.; Ong, A.M.; Hatakeyama, S.; Weissman, A.M. Ring fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. USA 1999, 96, 11364–11369. [Google Scholar] [CrossRef] [PubMed]
- Lindemose, S.; Shea, C.; Jensen, K.M.; Skriver, K. Structure, function and networks of transcription factors involved in abiotic stress responses. Int. J. Mol. Sci. 2013, 14, 5842–5878. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Yamamoto, H.; Shikanai, T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. BBA Bioenerg. 2011, 1807, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, J.; Adrian, J.; Gissot, L.; Coupland, G.; Yu, D.; Turck, F. Elevated levels of MYB30 in the phloem accelerate flowering in arabidopsis through the regulation of flowering locus t. PLoS ONE 2014, 9, e89799. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Aalen, R.B.; Audenaert, D.; Beeckman, T.; Broadley, M.R.; Butenko, M.A.; Cano-Delgado, A.I.; de Vries, S.; Dresselhaus, T.; Felix, G.; et al. Tackling drought stress: Receptor-like kinases present new approaches. Plant Cell 2012, 24, 2262–2278. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Rabara, R.C.; Reese, R.N.; Miller, M.A.; Rohila, J.S.; Subramanian, S.; Shen, Q.J.; Morandi, D.; Bucking, H.; Shulaev, V.; et al. A toolbox of genes, proteins, metabolites and promoters for improving drought tolerance in soybean includes the metabolite coumestrol and stomatal development genes. BMC Genomics 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.J.; Guang, Y. Inositol 1,4,5-trisphosphate 3-kinases: Functions and regulations. Cell Res 2005, 15, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Tanaka, M.; Seki, M.; Ham le, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Differential gene expression in soybean leaf tissues at late developmental stages under drought stress revealed by genome-wide transcriptome analysis. PLoS ONE 2012, 7, e49522. [Google Scholar] [CrossRef] [PubMed]
- Grundy, J.; Stoker, C.; Carré, I.A. Circadian regulation of abiotic stress tolerance in plants. Front. Plant Sci. 2015, 6, 648. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shen, J.; Xu, Y.; Li, X.; Xiao, J.; Xiong, L. Ghd2, a constans-like gene, confers drought sensitivity through regulation of senescence in rice. J. Exp. Bot. 2016, 67, 5785–5798. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Kwon, Y.; Kim, J.H.; Noh, H.; Hong, S.-W.; Lee, H. A dual role for myb60 in stomatal regulation and root growth of Arabidopsis thaliana under drought stress. Plant Mol. Biol. 2011, 77, 91–103. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Threshold Value | Number of Probe Sets (Soybean Chip Hyb. to DipC gDNA) | Number of Probe Sets (Soybean Chip Hyb. to TN gDNA) | Number of Probe Pairs (Soybean Chip Hyb. to DipC gDNA) | Number of Probe Pairs (Soybean Chip Hyb. to TN gDNA) | Number of DEGs in DipC | Number of DEGs in TN |
|---|---|---|---|---|---|---|
| 20 | 61,072 | 61,072 | 670,388 | 670,388 | 6165 | 6165 |
| 60 | 60,877 | 60,895 | 479,538 | 482,352 | 6927 | 6814 |
| 100 | 59,782 | 59,835 | 302,834 | 304,708 | 7183 | 7159 |
| 150 | 56,266 | 56,511 | 190,570 | 193,522 | 7036 | 7159 |
| 200 | 51,071 | 51,319 | 129,806 | 132,521 | 6638 | 6731 |
| 300 | 37,813 | 38,000 | 66,907 | 68,106 | 5275 | 5345 |
| 500 | 17,469 | 18,176 | 23,464 | 24,693 | 2784 | 2911 |
| 600 | 12,258 | 12,930 | 15,701 | 16,650 | 2089 | 2170 |
| 700 | 8896 | 9566 | 11,193 | 12,061 | 1574 | 1673 |
| 800 | 6687 | 7208 | 8415 | 9070 | 1195 | 1291 |
| 900 | 5140 | 5657 | 6559 | 7140 | 958 | 1057 |
| 1000 | 4085 | 4482 | 5304 | 5733 | 802 | 877 |
| Water-Limited versus Water-Sufficient | Water-Limited versus Recovery | |||||
|---|---|---|---|---|---|---|
| Up-Regulated under Dehydration | Down-Regulated under Recovery | Down-Regulated under Dehydration | Up-Regulated under Recovery | Up-Regulated | Down-Regulated | |
| DipC | 80 | 68 | 109 | 94 | 340 | 146 |
| Tiga Nicuru | 28 | 22 | 53 | 42 | 294 | 97 |
| Gene Name | FDR | Fold Change | Gene Description | References |
|---|---|---|---|---|
| UP-Regulated Genes in DipC | ||||
| PAL1 (Phenylalanine ammonia-lyase 1) | 0.018 | 3.901 | Key enzyme involved in the biosynthesis of isoprenoid antioxidative and polyphenol compounds such as lignin and is involved in defense mechanism. | [53] |
| ATEP3/AtchitIV | 0.001 | 3.845 | Encodes an EP3 chitinase that is stimulated under abiotic stress. | [54] |
| TXR1(Thaxtomin A resistant 1)/ATPAM16 | 6.87 × 10-5 | 3.718 | TXR1 is a component of a dispensable transport mechanism. Involved in negative regulation of defense responses by reducing reactive oxygen species (ROS). | [55] |
| Acetyl-CoA C-acyltransferase, putative / 3-ketoacyl-CoA thiolase | 0.001 | 3.554 | Functions in Jasmonic acid synthesis which plays a role in plant response to mechanical and abiotic stress. | [56] |
| UBC-2 (ubiquitin-conjugating enzyme 2) | 0.004 | 3.407 | Ubiquitination plays a part in increasing rate of the protein breakdown. Arabidopsis plants overexpressing UBC-2 were more tolerant to dehydration stress compared to the control plants. | [57] |
| Rho GDP dissociation inhibitor 2 | 0.001 | 3.348 | Involves in the regulation of Rho protein and small GTPase mediated signal transduction. | [58] |
| Histidine amino acid transporter (LHT1) | 0.001 | 3.256 | Amino acid transmembrane transporter involved in apoplastic transport of amino acids in leaves. | [59] |
| COMT (3-Caffeic acid o methyltransferase) | 0.006 | 3.234 | Involved in lignin biosynthesis. High activation of lignifying enzymes was found in dehydration-stressed white clover (Trifolium repens L.), which lead to reduced forage growth. | [60] |
| Glycine decarboxylase complex H | 0.005 | 3.113 | Functions in photo respiratory carbon recovery. Carbon dioxide is found to be low in plants subjected to dehydration stress due to the closing of stomata in order to prevent water loss. | [61] |
| Up-Regulated Genes in TN | ||||
| Clp amino terminal domain-containing protein, putative | 0.035 | 3.778 | Protein and ATP binding. | |
| CONSTANS-LIKE 1 | 0.025 | 3.294 | Transcription factor regulating flower development and response to light stimulus. | [62] |
| DRB3 (DSRNA-BINDING PROTEIN 3) | 0.020 | 2.984 | Assists in miRNA-targeted RNA degradation. | [63] |
| SIGE (SIGMA FACTOR E) | 0.032 | 2.808 | Responds to effects of abiotic stresses. Phosphorylation of major sigma factor SIG1 in Arabidopsis thaliana inhibits the transcription of the psaA gene, which encodes photosystem-I (PS-I). This disturbs photosynthetic activity. | [64,65] |
| Reticulon family protein | 0.029 | 2.772 | Playing a role in promoting membrane curvature. | |
| Cytochrome c oxidase family protein | 0.025 | 2.727 | Essential for the assembly of functional cytochrome oxidase protein. | |
| DNA-binding S1FA family protein | 0.049 | 2.717 | Binds to the negative promoter element S1F. | |
| DNA photolyase | 0.032 | 2.667 | DNA repair enzyme. | |
| Zinc knuckle (CCHC-type) family protein | 0.040 | 2.567 | Zinc ion binding | |
| Monosaccaride transporter | 0.025 | 2.547 | Plays a role in long-distance sugar partitioning or sub-cellular sugar distribution. | |
| Nodulin MtN3 family protein | 0.025 | 2.376 | Key role in the establishment of symbiosis. | |
| Serine acetyltransferase, N-terminal | 0.040 | 2.302 | Catalyzes the formation of a cysteine precursor. |
| Gene name | FDR | Fold Change | Gene Description | References |
|---|---|---|---|---|
| Down-Regulated Genes in DipC | ||||
| Dihydroxyacetone kinase | 0.003 | 6.489 | Glycerone kinase activity | |
| Phosphoglucomutase, putative/glucose phosphomutase, putative | 0.007 | 6.471 | Involved in controlling photosynthetic carbon flow and plays essential role starch synthesis. Down regulation of photosynthesis-related gene will lead to significant reduction in plant growth. | [66] |
| Auxin-induced protein 22D AUXX-IAA | 0.003 | 4.627 | Involved in stress defense response. Many AUXX-IAA genes were found to be down-regulated in Sorghum bicolor under drought conditions. | [67] |
| CP12-1, putative | 0.014 | 4.390 | Involved in calvin cycle, therefore linked to photosynthesis. Most drastic down-regulated genes which were photosynthesis-related was observed in barley (Hordeum vulgare L.) | [68] |
| PHS2 (ALPHA-GLUCAN PHOSPHORYLASE 2). | 0.014 | 4.375 | Encodes a cytosolic alpha-glucan phosphorylase. | |
| APRR5 (PSEUDO-RESPONSE REGULATOR 5), Pseudo ARR-B family | 0.001 | 4.145 | Linked to cytokinin-mediated regulation | |
| Thiamine biosynthesis family protein | 0.002 | 4.132 | Catalyses the activation of small proteins, such as ubiquitin or ubiquitin-like proteins. | |
| Zinc finger (C3HC4-type RING finger) | 0.007 | 3.611 | Mediate ubiquitin-conjugating enzyme (UBC-2) dependent ubiquitation. | [69] |
| WRKY40 | 0.033 | 3.104 | Regulator of ABA signalling. It inhibits the expression of ABA-responsive genes ABF4, AB14, AB15, DREB1A, MYB2 and RAB18. | [70] |
| Down-Regulated Genes in TN | ||||
| AGL83 (AGAMOUS-LIKE 83) | 0.025 | 4.374 | DNA-binding transcription factor | |
| CRR23 (chlororespiratory reduction 23) | 0.025 | 3.625 | A subunit of the chloroplast NAD(P)H dehydrogenase complex, involved in PS-I cyclic electron transport. Located on the thylakoid membrane. Mutant has impaired NAD(P)H dehydrogenase activity. Part of dehydration repressing photosynthesis. | [71] |
| MYB30 (MYB DOMAIN PROTEIN 30) | 0.032 | 3.250 | Acts as a positive regulator of hypersensitive cell death and salicylic acid synthesis. Involved in the regulation of abscisic acid (ABA) signalling. | [72] |
| Photosystem II family protein, putative | 0.029 | 3.158 | Linked to photosynthesis. Down-regulation of photosynthesis-related genes during dehydration stress was observed in maize (Zea mays), which in turn leads to significant reduction in plant growth. | [73] |
| Phosphoesterase | 0.047 | 3.136 | Hydrolase activity, acting on ester bonds. | |
| Zing-finger (C3HC4-type) | 0.045 | 2.947 | Mediate ubiquitin-conjugating enzyme (UBC-2) dependent ubiquitation. | [69] |
| NHX2 (Sodium proton exchanger 2) | 0.040 | 2.742 | Involved in antiporter activity. Also involved in potassium ion homoeostasis and regulation of stomatal closure. Involved in the accumulation of K+ that drives the rapid stomatal opening. Down-regulation of genes related to stomatal regulation has been observed in soybean, which appears to be a part of dehydration response, leading to a reduction in the amount of stomata in leaves. | [74] |
| Inositol 1,3,4-trisphosphate 5/6-kinase | 0.035 | 2.090 | Part of IP3 signal transduction pathway. | [75] |
| Gene Name | FDR | Fold Change | Gene Description | References |
|---|---|---|---|---|
| Up-Regulated Genes | ||||
| Beta-fructofuranosidase | 8.90 × 10−-4 | 3.193 | Catalyses the hydrolysis of sucrose. A rise in monosaccharide content caused by the Beta-fructofuranosidase can compensate for the decline in photosynthetic carbon assimilation indicated by the decrease in net photosynthesis. | [46,47] |
| Down-Regulated Genes | ||||
| MEE59 (maternal effect embryo arrest 59) | 8.94 × 10−-4 | 8.580 | Embryo development ending in seed dormancy. | |
| Calcineurin-like phosphoesterase family protein (CPPED1). | 6.72 × 10−-4 | 5.857 | Plays inhibitory role in glucose uptake. Down-regulation of CPPED1 improves glucose metabolism. | [48] |
| Putative lysine-specific demethylase JMJD5 Jumonji/Zinc-finger-class domain containing protein | 0.003 | 4.971 | Plays role in a histone demethylation mechanism that is conserved from yeast to human. Down-regulation may lead to an increase in methylated histones and hence general down-regulation of transcription. | [49] |
| MYB-like transcription factor | 0.024 | 4.103 | Arabidopsis homolog is known to regulate stomatal opening, flower development, and plays role in circadian rhythm. | [50] |
| F-box family protein (FBL14) | 0.001 | 3.744 | Functions in signal transduction and regulation of cell cycle. | |
| BRH1 (BRASSINOSTEROID-RESPONSIVE RING-H2) | 0.007 | 2.899 | BRH1 is known to influence stomatal density. | [51] |
| Bundle-sheath defective protein 2 family/bsd2 family | 0.003 | 2.441 | Protein required for post-translational regulation of Rubisco large subunit (rbcL). | [52] |
| Mitochondrial substrate carrier family protein | 0.030 | 2.435 | Involved in energy transfer. |
| DipC | TN | ||||||
|---|---|---|---|---|---|---|---|
| Probe-Set | Name | V°Whole | V°Drought | Probe-Set | Name | V°Whole | V°Drought |
| Gma.16733.1.S1_at | WRKY40 | 68 | 17 | GmaAffx.45249.1.S1_at | CONSTANS-like 1 | 16 | 3 |
| Gma.6670.1.S1_at | PRR7 | 49 | 7 | GmaAffx.84566.2.S1_at | MYB60 | 8 | 3 |
| GmaAffx.33796.3.S1_at | Zinc-finger like C2H2 | 45 | 7 | GmaAffx.86517.1.S1_at | AGL83 | 6 | 1 |
| GmaAffx.92679.1.S1_s_at | ATAUX2-11 | 41 | 9 | Gma.1576.1.S1_at | Zinc-finger C3HC4 | 5 | 1 |
| GmaAffx.35309.1.S1_s_at | GRF2 | 35 | 6 | ||||
| GmaAffx.65059.1.S1_at | bHLH | 32 | 7 | ||||
| GmaAffx.90399.1.S1_at | C3HC4 Zinc-finger | 31 | 9 | ||||
| Gma.15774.1.S1_at | Zinc-finger C3HC4 | 26 | 3 | ||||
| GmaAffx.53180.1.S1_at | PRR7 | 25 | 9 | ||||
| GmaAffx.80492.1.S1_at | PRR5 | 9 | 2 | ||||
| GmaAffx.73009.2.S1_at | WRKY51 | 7 | 5 | ||||
| Common TFs | |||||||
| GmaAffx.60283.1.S1_at | BRH1 | 42 | 6 | ||||
| GmaAffx.9286.1.S1_s_at | MYB | 27 | 4 | ||||
| Gma.17248.1.A1_at | JMJD5 | 26 | 3 | ||||
| GmaAffx.10162.1.S1_at | MEE59 | 13 | 3 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, F.; Chai, H.H.; Ajmera, I.; Hodgman, C.; Mayes, S.; Lu, C. A Transcriptomic Comparison of Two Bambara Groundnut Landraces under Dehydration Stress. Genes 2017, 8, 121. https://doi.org/10.3390/genes8040121
Khan F, Chai HH, Ajmera I, Hodgman C, Mayes S, Lu C. A Transcriptomic Comparison of Two Bambara Groundnut Landraces under Dehydration Stress. Genes. 2017; 8(4):121. https://doi.org/10.3390/genes8040121
Chicago/Turabian StyleKhan, Faraz, Hui Hui Chai, Ishan Ajmera, Charlie Hodgman, Sean Mayes, and Chungui Lu. 2017. "A Transcriptomic Comparison of Two Bambara Groundnut Landraces under Dehydration Stress" Genes 8, no. 4: 121. https://doi.org/10.3390/genes8040121
APA StyleKhan, F., Chai, H. H., Ajmera, I., Hodgman, C., Mayes, S., & Lu, C. (2017). A Transcriptomic Comparison of Two Bambara Groundnut Landraces under Dehydration Stress. Genes, 8(4), 121. https://doi.org/10.3390/genes8040121