
MYC, Cell Competition, and Cell Death in Cancer: The Inseparable Triad


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MYC and Cell Growth
3. MYC and Cell Death
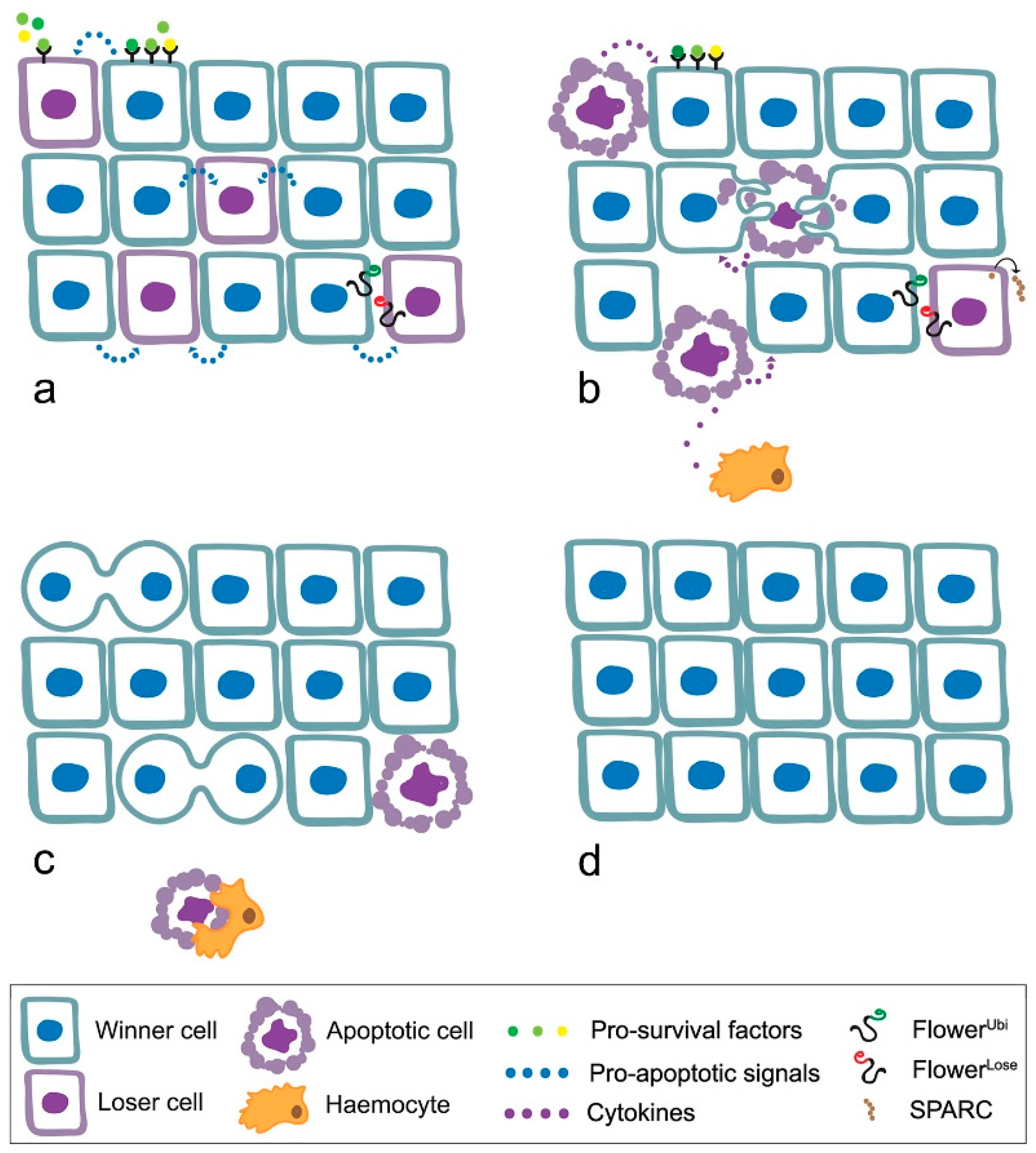
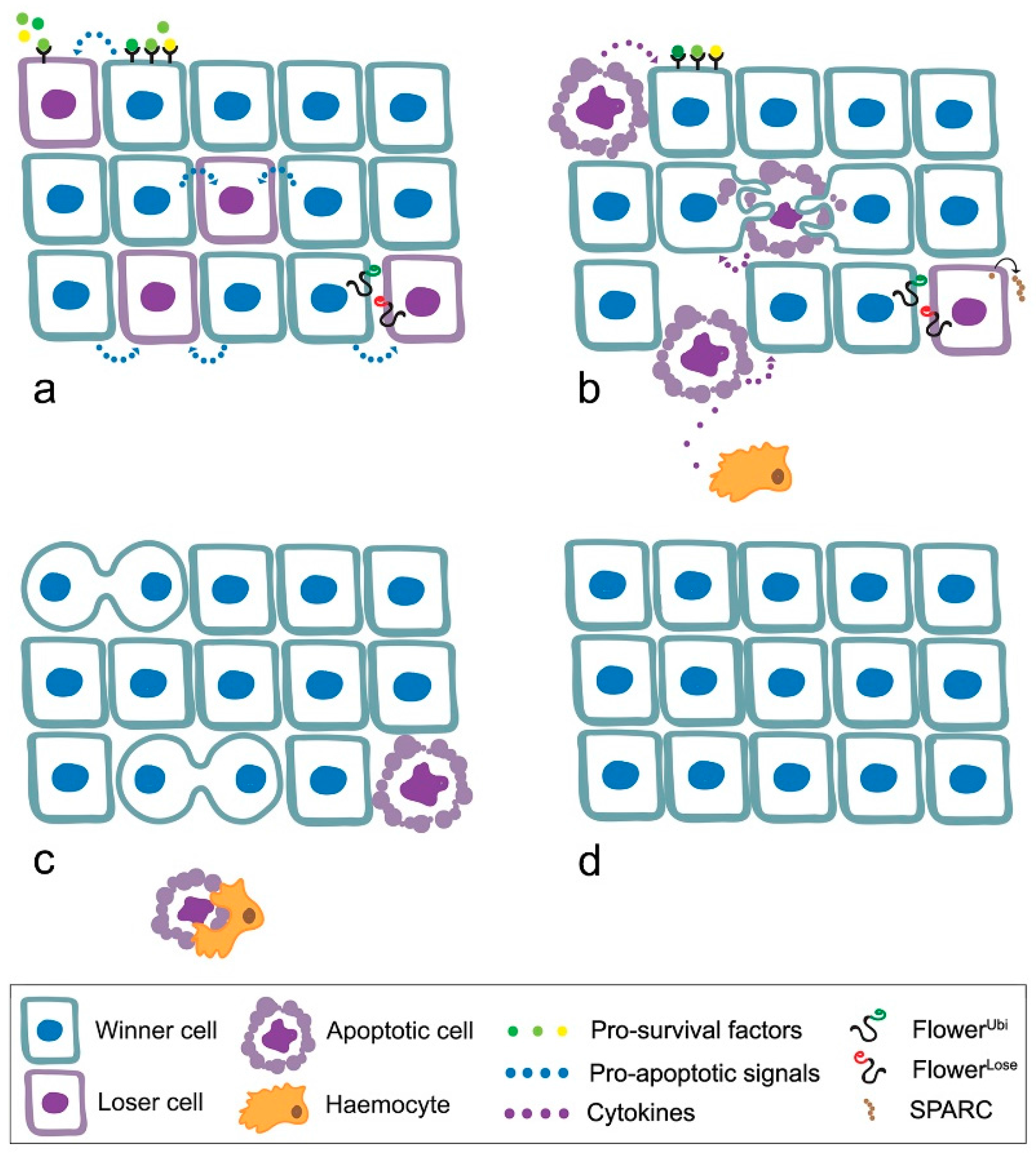
4. MYC Enters Cell Competition
- loser cells suffer from shortage of survival/growth factors such as the Drosophila TGFβ orthologue Decapentaplegic (Dpp) [52];
- the high contact tension at the interface of winner and loser cell leads to the elimination of loser cells through cell-cell intercalation [59];
- local tissue crowding can induce mechanical competition, independent of known markers of cell fitness [60];
- winner cells can acquire the ability to engulf adjacent losers [61];
- elimination of the loser cells leads to overproliferation of the winners [54].
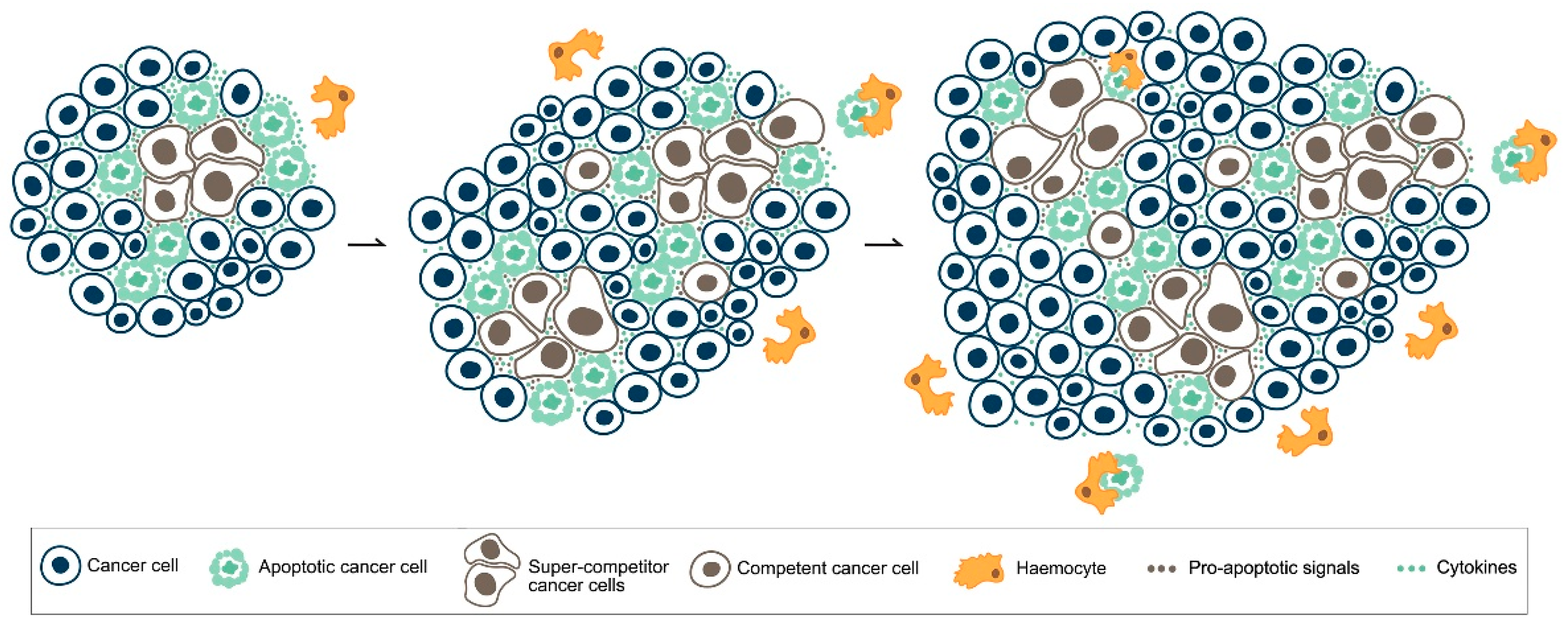
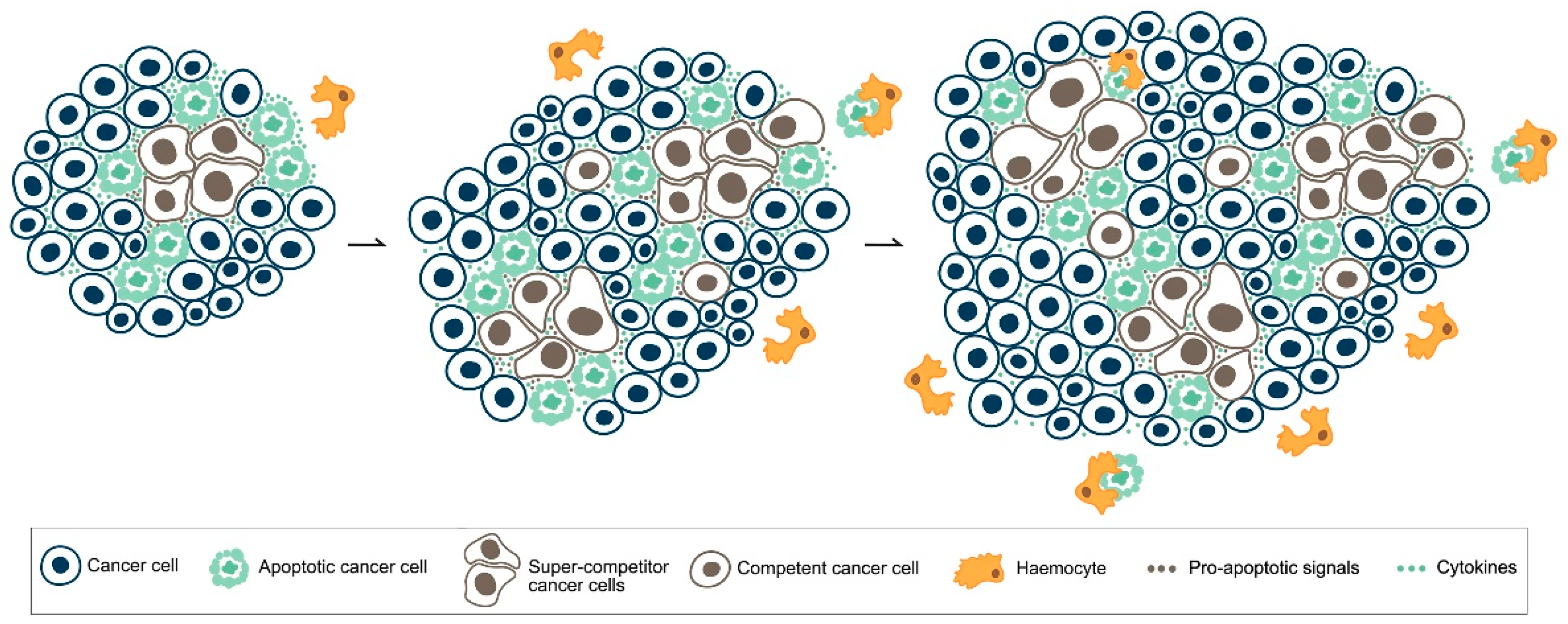
5. MYC, Cell Competition and Cancer
6. Apoptotic Cell Death in Cancer: What Side Does It Stand on?
7. Final Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dang, C.V. c-MYC target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P.; Shiio, Y.; Cheng, P.F.; Parkhurst, S.M.; Eisenman, R.N. MYC and max homologs in Ddrosophila. Science 1996, 274, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Benassayag, C.; Montero, L.; Colombie, N.; Gallant, P.; Cribbs, D.; Morello, D. Human c-MYC isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 2005, 25, 9897–9909. [Google Scholar] [CrossRef] [PubMed]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-MYC regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P. MYC/Max/Mad in invertebrates: The evolution of the max network. Curr. Top. Microbiol. Immunol. 2006, 302, 235–253. [Google Scholar] [PubMed]
- Garcia-Bellido, A.; Ripoll, P.; Morata, G. Developmental compartmentalisation of the wing disk of Drosophila. Nat. New Biol. 1973, 245, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Prober, D.A.; Edgar, B.A.; Eisenman, R.N.; Gallant, P. Drosophila MYC regulates cellular growth during development. Cell 1999, 98, 779–790. [Google Scholar] [CrossRef]
- Pierce, S.B.; Yost, C.; Britton, J.S.; Loo, L.W.; Flynn, E.M.; Edgar, B.A.; Eisenman, R.N. dDMycYC is required for larval growth and endoreplication in Drosophila. Development 2004, 131, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.S.; Li, L.; Orian, A.; Eisenman, R.N.; Edgar, B.A. MYC-dependent regulation of ribosomal rna RNA synthesis during Drosophila development. Nat. Cell Biol. 2005, 7, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Teleman, A.A.; Hietakangas, V.; Sayadian, A.C.; Cohen, S.M. Nutritional control of protein biosynthetic capacity by insulin via MYC in Drosophila. Cell Metab. 2008, 7, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Edgar, B.A.; Grewal, S.S. Nutritional control of gene expression in Drosophila larvae via tor, MYC and a novel cis-regulatory element. BMC Cell Biol. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Parisi, F.; Riccardo, S.; Daniel, M.; Saqcena, M.; Kundu, N.; Pession, A.; Grifoni, D.; Stocker, H.; Tabak, E.; Bellosta, P. Drosophila insulin and target of rapaMYCin (tor) pathways regulate gsk3 beta activity to control MYC stability and determine MYC expression in vivo. BMC Biol. 2011, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Delanoue, R.; Slaidina, M.; Leopold, P. The steroid hormone ecdysone controls systemic growth by repressing dMYC function in Drosophila fat cells. Dev. Cell 2010, 18, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Smith-Bolton, R.K.; Worley, M.I.; Kanda, H.; Hariharan, I.K. Regenerative growth in Ddrosophila imaginal discs is regulated by wingless and MYC. Dev. Cell 2009, 16, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, O.; Quinn, L.M. Controlling the master: Chromatin dynamics at the myc promoter integrate developmental signaling. Genes 2017, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Neto-Silva, R.M.; de Beco, S.; Johnston, L.A. Evidence for a growth-stabilizing regulatory feedback mechanism between MYC and yorkie, the Drosophila homolog of yap. Dev. Cell 2010, 19, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Ziosi, M.; Baena-Lopez, L.A.; Grifoni, D.; Froldi, F.; Pession, A.; Garoia, F.; Trotta, V.; Bellosta, P.; Cavicchi, S. dMYC functions downstream of yorkie to promote the supercompetitive behavior of hippo pathway mutant cells. PLoS Genet. 2010, 6, e1001140. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances MYC protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple ras-dependent phosphorylation pathways regulate MYC protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Moberg, K.H.; Mukherjee, A.; Veraksa, A.; Artavanis-Tsakonas, S.; Hariharan, I.K. The drosophila Drosophila F box protein archipelago regulates dMYC protein levels in vivo. Curr. Biol. CB 2004, 14, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-MYC degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Galletti, M.; Riccardo, S.; Parisi, F.; Lora, C.; Saqcena, M.K.; Rivas, L.; Wong, B.; Serra, A.; Serras, F.; Grifoni, D.; et al. Identification of domains responsible for ubiquitin-dependent degradation of dMYC by glycogen synthase kinase 3β and casein kinase 1 kinases. Mol. Cell. Biol. 2009, 29, 3424–3434. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct thresholds govern MYC’s biological output in vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Levens, D. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harb. Perspect. Med. 2013, 3, a014233. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Harris, A.W.; Bath, M.L.; Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between MYC and bcl-2. Nature 1990, 348, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Sorcinelli, M.D.; Beard, C.; Korsmeyer, S.J. Antiapoptotic bcl-2 is required for maintenance of a model leukemia. Cancer Cell 2004, 6, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Knezevich, S.; Ludkovski, O.; Salski, C.; Lestou, V.; Chhanabhai, M.; Lam, W.; Klasa, R.; Connors, J.M.; Dyer, M.J.; Gascoyne, R.D.; et al. Concurrent translocation of BCL2 and MYC with a single immunoglobulin locus in high-grade B-cell lymphomas. Leukemia 2005, 19, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Pfeiffer, H.K.; Mellert, H.S.; Stanek, T.J.; Sussman, R.T.; Kumari, A.; Yu, D.; Rigoutsos, I.; Thomas-Tikhonenko, A.; Seidel, H.E.; et al. Inhibition of the single downstream target bag1 BAG1 activates the latent apoptotic potential of MYC. Mol. Cell. Biol. 2011, 31, 5037–5045. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. MYC signaling via the arf tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mmdm2-p53 tumor suppressor pathway in MYC-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gu, L.; Zhang, H.; Zhou, M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle 2011, 10, 2994–3002. [Google Scholar] [CrossRef] [PubMed]
- Trudel, M.; Lanoix, J.; Barisoni, L.; Blouin, M.J.; Desforges, M.; L’Italien, C.; D’Agati, V. C-MYC-induced apoptosis in polycystic kidney disease is Bcl-2 and p53 independent. J. Exp. Med. 1997, 186, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Amanullah, A.; Liebermann, D.A.; Hoffman, B. P53-independent apoptosis associated with c-MYC-mediated block in myeloid cell differentiation. Oncogene 2000, 19, 2967–2977. [Google Scholar] [CrossRef] [PubMed]
- Boone, D.N.; Qi, Y.; Li, Z.; Hann, S.R. Egr1 mediates p53-independent c-MYC-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 632–637. [Google Scholar] [CrossRef] [PubMed]
- De la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila MYC regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef]
- Montero, L.; Muller, N.; Gallant, P. Induction of apoptosis by Drosophila MYC. Genesis 2008, 46, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Steiger, D.; Furrer, M.; Schwinkendorf, D.; Gallant, P. Max-independent functions of MYC in Drosophila melanogaster. Nat. Genet. 2008, 40, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.J.; Huh, J.R.; Muro, I.; Yu, H.; Wang, L.; Wang, S.L.; Feldman, R.M.; Clem, R.J.; Muller, H.A.; Hay, B.A. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat. Cell Biol. 2002, 4, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Silke, J.; Leevers, S.J.; Evan, G.I. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 2000, 19, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, M.H.; Nordstrom, W.; Tsang, G.; Kwan, E.; Rubin, G.M.; Abrams, J.M. Drosophila p53 binds a damage response element at the reaper locus. Cell 2000, 101, 103–113. [Google Scholar] [CrossRef]
- Zhang, C.; Casas-Tinto, S.; Li, G.; Lin, N.; Chung, M.; Moreno, E.; Moberg, K.H.; Zhou, L. An intergenic regulatory region mediates Drosophila MYC-induced apoptosis and blocks tissue hyperplasia. Oncogene 2015, 34, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cohen, S.M. The hippo pathway acts via p53 and microRNAs to control proliferation and proapoptotic gene expression during tissue growth. Biol. Open 2013, 2, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Morata, G.; Ripoll, P. Minutes: Mutants of Drosophila autonomously affecting cell division rate. Dev. Biol. 1975, 42, 211–221. [Google Scholar] [CrossRef]
- Marygold, S.J.; Roote, J.; Reuter, G.; Lambertsson, A.; Ashburner, M.; Millburn, G.H.; Harrison, P.M.; Yu, Z.; Kenmochi, N.; Kaufman, T.C.; et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 2007, 8, R216. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.S. Genetic mosaic techniques for studying Drosophila development. Development 2003, 130, 5065–5072. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P. Parameters of cell competition in the compartments of the wing disc of Drosophila. Dev. Biol. 1979, 69, 182–193. [Google Scholar] [CrossRef]
- Moreno, E.; Basler, K.; Morata, G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 2002, 416, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.R.; Saunders, T.L.; Tarle, S.A.; Glaser, T. Ribosomal protein Ll24 defect in belly spot and tail (Bst), a mouse Minute. Development 2004, 131, 3907–3920. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Basler, K. dMYC transforms cells into super-competitors. Cell 2004, 117, 117–129. [Google Scholar] [CrossRef]
- Senoo-Matsuda, N.; Johnston, L.A. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila MYC. Proc. Natl. Acad. Sci. USA 2007, 104, 18543–18548. [Google Scholar] [CrossRef] [PubMed]
- Rhiner, C.; Lopez-Gay, J.M.; Soldini, D.; Casas-Tinto, S.; Martin, F.A.; Lombardia, L.; Moreno, E. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila. Dev. Cell 2010, 18, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Portela, M.; Casas-Tinto, S.; Rhiner, C.; Lopez-Gay, J.M.; Dominguez, O.; Soldini, D.; Moreno, E. Drosophila sparc is a self-protective signal expressed by loser cells during cell competition. Dev. Cell 2010, 19, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.M.; Rhiner, C.; Lopez-Gay, J.M.; Buechel, D.; Hauert, B.; Moreno, E. Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell 2015, 160, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Levayer, R.; Hauert, B.; Moreno, E. Cell mixing induced by MYC is required for competitive tissue invasion and destruction. Nature 2015, 524, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Levayer, R.; Dupont, C.; Moreno, E. Tissue crowding induces caspase-dependent competition for space. Curr. Biol. CB 2016, 26, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Baker, N.E. Engulfment is required for cell competition. Cell 2007, 129, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Lolo, F.N.; Casas-Tinto, S.; Moreno, E. Cell competition time line: Winners kill losers, which are extruded and engulfed by hemocytes. Cell Rep. 2012, 2, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Casas-Tinto, S.; Lolo, F.N.; Moreno, E. Active JNK-dependent secretion of Drosophila Tyrosyl-tRNA synthetase by loser cells recruits haemocytes during cell competition. Nat. Commun. 2015, 6, 10022. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Antala, B.; Shrivastava, N. In silico screening of alleged miRNAs associated with cell competition: An emerging cellular event in cancer. Cell. Mol. Biol. Lett. 2015, 20, 798–815. [Google Scholar] [CrossRef] [PubMed]
- Rhiner, C.; Diaz, B.; Portela, M.; Poyatos, J.F.; Fernandez-Ruiz, I.; Lopez-Gay, J.M.; Gerlitz, O.; Moreno, E. Persistent competition among stem cells and their daughters in the Drosophila ovary germline niche. Development 2009, 136, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- De la Cova, C.; Senoo-Matsuda, N.; Ziosi, M.; Wu, D.C.; Bellosta, P.; Quinzii, C.M.; Johnston, L.A. Supercompetitor status of Drosophila MYC cells requires p53 as a fitness sensor to reprogram metabolism and promote viability. Cell Metab. 2014, 19, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Claveria, C.; Giovinazzo, G.; Sierra, R.; Torres, M. MYC-driven endogenous cell competition in the early mammalian embryo. Nature 2013, 500, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Di-Gregorio, A.; George, N.; Pozzi, S.; Sanchez, J.M.; Pernaute, B.; Rodriguez, T.A. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev. Cell 2013, 26, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Villa del Campo, C.; Claveria, C.; Sierra, R.; Torres, M. Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep. 2014, 8, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Villa Del Campo, C.; Lioux, G.; Carmona, R.; Sierra, R.; Munoz-Chapuli, R.; Claveria, C.; Torres, M. MYC overexpression enhances of epicardial contribution to the developing heart and promotes extensive expansion of the cardiomyocyte population. Sci. Rep. 2016, 6, 35366. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A. Socializing with MYC: Cell competition in development and as a model for premalignant cancer. Cold Spring Harb. Perspect. Med. 2014, 4, a014274. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Laconi, E.; Doratiotto, S.; Vineis, P. The microenvironments of multistage carcinogenesis. Semin. Cancer Biol. 2008, 18, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.E.; Li, W. Cell competition and its possible relation to cancer. Cancer Res. 2008, 68, 5505–5507. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E. Is cell competition relevant to cancer? Nat. Rev. Cancer 2008, 8, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, D.; Bellosta, P. Drosophila MYC: A master regulator of cellular performance. Biochim. Biophys. Acta 2015, 1849, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, A.; Bowling, S.; Rodriguez, T.A. Cell competition and its role in the regulation of cell fitness from development to cancer. Dev. Cell 2016, 38, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Froldi, F.; Ziosi, M.; Garoia, F.; Pession, A.; Grzeschik, N.A.; Bellosta, P.; Strand, D.; Richardson, H.E.; Pession, A.; Grifoni, D. The lethal giant larvae tumour suppressor mutation requires dMYC oncoprotein to promote clonal malignancy. BMC Biol. 2010, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Feng, Y.; Chen, X.; Li, W.; Xue, L. MYC inhibits JNK-mediated cell death in vivo. Apoptosis Int. J. Progr. Cell Death 2017, 22, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.; Perez-Garijo, A.; Calleja, M.; Morata, G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 14651–14656. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Schroeder, M.C.; Kango-Singh, M.; Tao, C.; Halder, G. Tumor suppression by cell competition through regulation of the Hippo pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.J.; Bajpai, A.; Alam, M.A.; Gupta, R.P.; Harsh, S.; Pandey, R.K.; Goel-Bhattacharya, S.; Nigam, A.; Mishra, A.; Sinha, P. Epithelial neoplasia in Drosophila entails switch to primitive cell states. Proc. Natl. Acad. Sci. USA 2013, 110, E2163–E2172. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Hong, A.W.; Guan, K.L. Disease implications of the Hippo/YAP pathway. Trends Mol. Med. 2015, 21, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Doggett, K.; Grusche, F.A.; Richardson, H.E.; Brumby, A.M. Loss of the Drosophila cell polarity regulator Sscribbled promotes epithelial tissue overgrowth and cooperation with oncogenic Ras-Raf through impaired hippo pathway signaling. BMC Dev. Biol. 2011, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Eichenlaub, T.; Cohen, S.M.; Herranz, H. Cell competition drives the formation of metastatic tumors in a Drosophila model of epithelial tumor formation. Curr. Biol. CB 2016, 26, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Arias, L.; Saavedra, V.; Morata, G. Cell competition may function either as tumour-suppressing or as tumour-stimulating factor in Drosophila. Oncogene 2013, 33, 4377–4384. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Targeting apoptosis for anticancer therapy. Semin. Cancer Biol. 2015, 31, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D.; Gorenc, T.; Steller, H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the wingless signaling pathways. Dev. Cell 2004, 7, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.A.; Perez-Garijo, A.; Morata, G. Apoptosis in Drosophila: Compensatory proliferation and undead cells. Int. J. Dev. Biol. 2009, 53, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garijo, A.; Martin, F.A.; Morata, G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 2004, 131, 5591–5598. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.R.; Guo, M.; Hay, B.A. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase dronc in a nonapoptotic role. Curr. Biol. CB 2004, 14, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Diwanji, N.; Bergmann, A. The beneficial role of extracellular reactive oxygen species in apoptosis-induced compensatory proliferation. Fly 2016, 11, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Suijkerbuijk, S.J.; Kolahgar, G.; Kucinski, I.; Piddini, E. Cell competition drives the growth of intestinal adenomas in Drosophila. Curr. Biol. CB 2016, 26, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Sollazzo, M.; de Biase, D.; Bellosta, P.; Pession, A.; Grifoni, D. Intra-tumoural apoptosis following myc-mediated cell competition shapes cancer mass and aggressiveness. Unpublished work. 2017. [Google Scholar]
- Pantziarka, P. Emergent properties of a computational model of tumour growth. PeerJ 2016, 4, e2176. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.A.; Petrova, S.; Pound, J.D.; Voss, J.J.; Melville, L.; Paterson, M.; Farnworth, S.L.; Gallimore, A.M.; Cuff, S.; Wheadon, H.; et al. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr. Biol. CB 2015, 25, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Sun, Y.; Wang, J.; Zhao, X.; Wang, X.; Hao, X. Extent, relationship and prognostic significance of apoptosis and cell proliferation in synovial sarcoma. Eur. J. Cancer Prev. 2006, 15, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Jalalinadoushan, M.; Peivareh, H.; Azizzadeh Delshad, A. Correlation between apoptosis and histological grade of transitional cell carcinoma of urinary bladder. Urol. J. 2004, 1, 177–179. [Google Scholar] [PubMed]
- Naresh, K.N.; Lakshminarayanan, K.; Pai, S.A.; Borges, A.M. Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: A hypothesis to support this paradoxical association. Cancer 2001, 91, 578–584. [Google Scholar] [CrossRef]
- Feng, X.; Yu, Y.; He, S.; Cheng, J.; Gong, Y.; Zhang, Z.; Yang, X.; Xu, B.; Liu, X.; Li, C.Y.; et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017, 385, 12–20. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Giacomo, S.; Sollazzo, M.; Paglia, S.; Grifoni, D. MYC, Cell Competition, and Cell Death in Cancer: The Inseparable Triad. Genes 2017, 8, 120. https://doi.org/10.3390/genes8040120
Di Giacomo S, Sollazzo M, Paglia S, Grifoni D. MYC, Cell Competition, and Cell Death in Cancer: The Inseparable Triad. Genes. 2017; 8(4):120. https://doi.org/10.3390/genes8040120
Chicago/Turabian StyleDi Giacomo, Simone, Manuela Sollazzo, Simona Paglia, and Daniela Grifoni. 2017. "MYC, Cell Competition, and Cell Death in Cancer: The Inseparable Triad" Genes 8, no. 4: 120. https://doi.org/10.3390/genes8040120
APA StyleDi Giacomo, S., Sollazzo, M., Paglia, S., & Grifoni, D. (2017). MYC, Cell Competition, and Cell Death in Cancer: The Inseparable Triad. Genes, 8(4), 120. https://doi.org/10.3390/genes8040120