
The MYCN Protein in Health and Disease


Abstract
:1. Introduction
2. Cellular Regulation by MYCN
2.1. MYCN and the Cell Cycle
2.2. MYCN in Apoptosis and Cell Death
2.3. MYCN and Metabolism
3. MYCN and Embryonic Tumors
3.1. MYCN, Embryonic Development and Pluripotency
3.2. Neuroblastoma
3.3. Wilms’ Tumor
3.4. Retinoblastoma
3.5. Medulloblastoma
3.6. Rhabdomyosarcoma
4. Other MYCN-Related Diseases
4.1. Feingold Syndrome
4.2. Prostate Cancer
4.3. Basal Cell Carcinoma
4.4. Leukemia
4.5. Lung Cancer
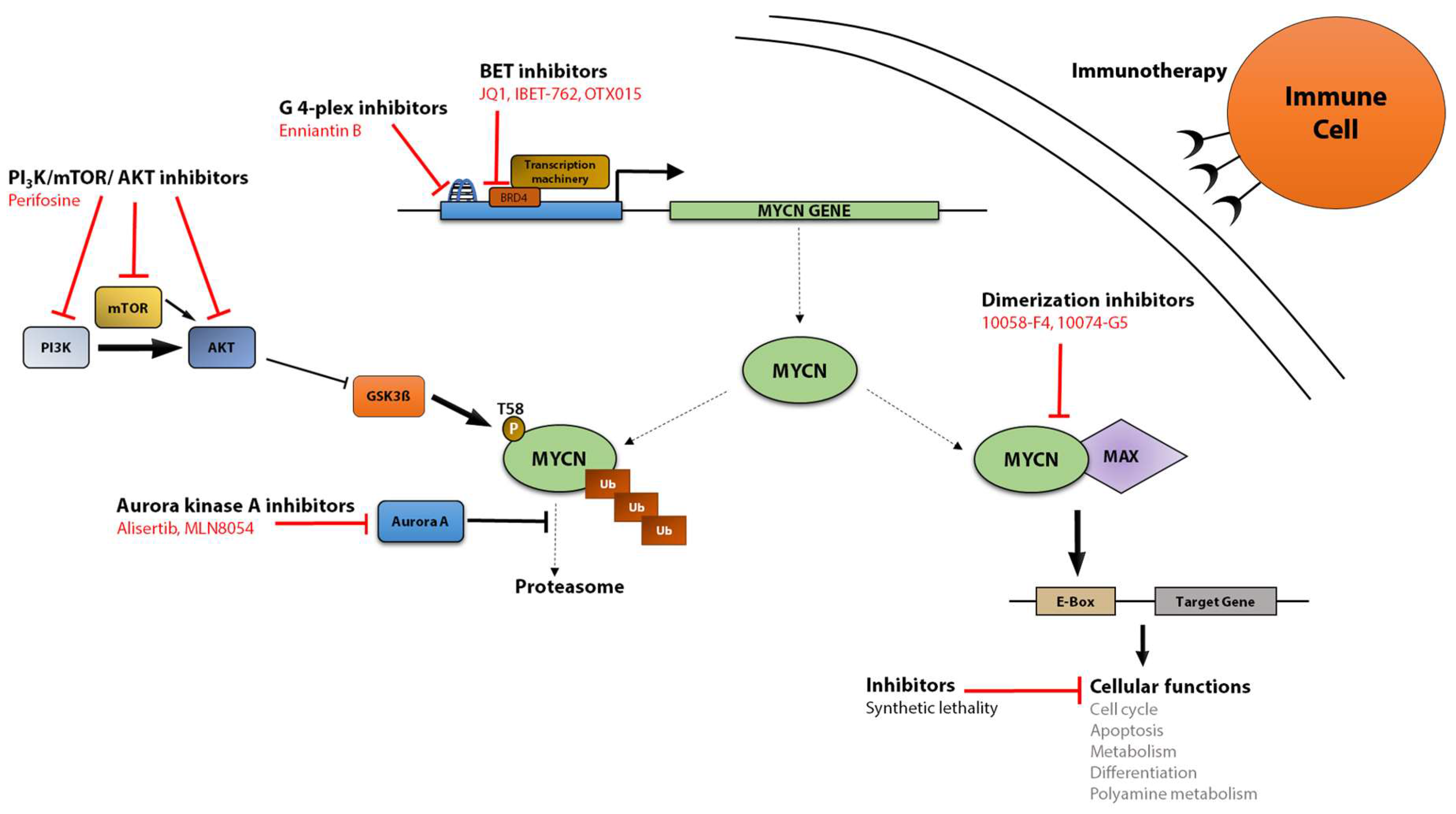
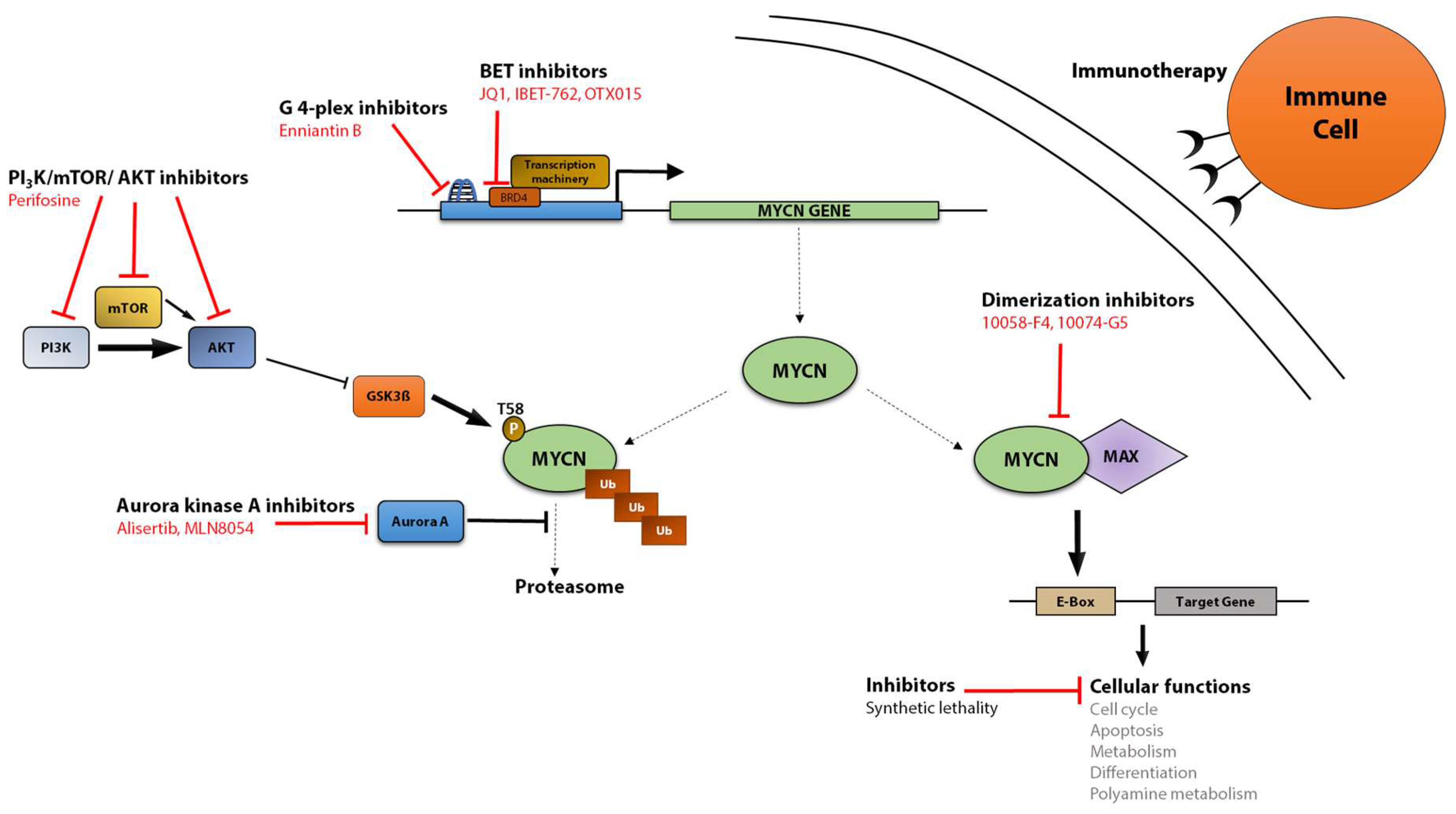
5. MYCN as a Therapeutic Target
5.1. Targeting MYCN Transcription
5.2. Targeting MYCN Protein Stability
5.3. Targeting MYCN Dimerization and Transcriptional Activity
5.4. MYCN-Based Immunotherapy
5.5. Indirect Targeting of MYCN
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mathsyaraja, H.; Eisenman, R.N. Parsing Myc paralogs in oncogenesis. Cancer Cell 2016, 29, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.R.; Ayer, D.E. Interactions between Myc and MondoA transcription factors in metabolism and tumourigenesis. Br. J. Cancer 2015, 113, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, S.; Valli, E.; Erriquez, D.; Perini, G. MYCN-mediated transcriptional repression in neuroblastoma: The other side of the coin. Front. Oncol. 2013, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.; Klarqvist, M.D.; Westermark, U.K.; Oliynyk, G.; Dzieran, J.; Kock, A.; Savatier Banares, C.; Hertwig, F.; Johnsen, J.I.; Fischer, M.; et al. Regulation of nuclear hormone receptors by MYCN-driven mirnas impacts neural differentiation and survival in neuroblastoma patients. Cell Rep. 2016, 16, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Okubo, T.; Knoepfler, P.S.; Eisenman, R.N.; Hogan, B.L. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development 2005, 132, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Varmus, H.E.; Bishop, J.M.; Grzeschik, K.H.; Naylor, S.L.; Sakaguchi, A.Y.; Brodeur, G.; Trent, J. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature 1984, 308, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Zimmerman, K.; Blank, R.D.; Alt, F.W.; D’Eustachio, P. Chromosomal location of N-myc and l-myc genes in the mouse. Oncogene Res. 1989, 4, 47–54. [Google Scholar] [PubMed]
- Zelinski, T.; Verville, G.; White, L.; Hamerton, J.L.; McAlpine, P.J.; Lewis, M. Confirmation of the assignment of MYCL to chromosome 1 in humans and its position relative to RH, UMPK and PGM1. Genomics 1988, 2, 154–156. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Crews, S.; Barth, R.; Hood, L.; Prehn, J.; Calame, K. Mouse c-myc oncogene is located on chromosome 15 and translocated to chromosome 12 in plasmacytomas. Science 1982, 218, 1319–1321. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Salwen, H.; Quasney, M.W.; Ikegaki, N.; Cowan, J.M.; Herst, C.V.; Kennett, R.H.; Rosen, S.T.; DiGiuseppe, J.A.; Brodeur, G.M. Prolonged N-myc protein half-life in a neuroblastoma cell line lacking N-myc amplification. Oncogene 1990, 5, 1821–1827. [Google Scholar] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, S.K.; Finn, G.; Hahn, W.C.; Rowitch, D.H.; Kenney, A.M. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev. Cell 2005, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schuttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Bonvini, P.; Nguyen, P.; Trepel, J.; Neckers, L.M. In vivo degradation of N-myc in neuroblastoma cells is mediated by the 26s proteasome. Oncogene 1998, 16, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Chesler, L.; Schlieve, C.; Goldenberg, D.D.; Kenney, A.; Kim, G.; McMillan, A.; Matthay, K.K.; Rowitch, D.; Weiss, W.A. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res 2006, 66, 8139–8146. [Google Scholar] [CrossRef] [PubMed]
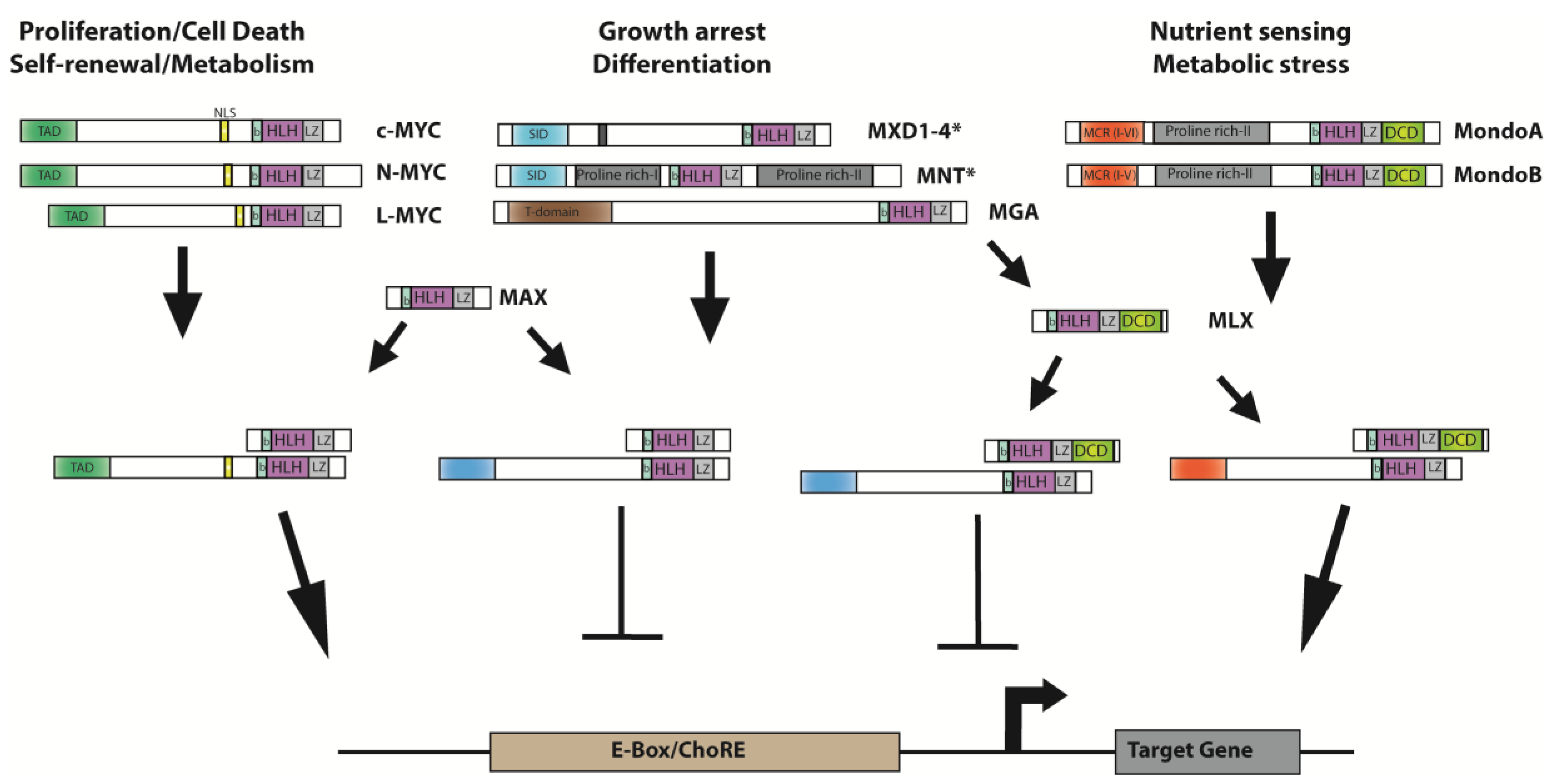
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its Interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Barisone, G.A.; Ngo, T.; Tran, M.; Cortes, D.; Shahi, M.H.; Nguyen, T.V.; Perez-Lanza, D.; Matayasuwan, W.; Diaz, E. Role of MXD3 in proliferation of DAOY human medulloblastoma cells. PLoS ONE 2012, 7, e38508. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated MYC requires MondoA/Mlx for metabolic reprogramming and tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Meroni, G.; Cairo, S.; Merla, G.; Messali, S.; Brent, R.; Ballabio, A.; Reymond, A. Mlx, a new max-like bHLHZip family member: The center stage of a novel transcription factors regulatory pathway? Oncogene 2000, 19, 3266–3277. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Dynlacht, B.D. New insights into cyclins, CDKs, and cell cycle control. Semin. Cell Dev. Biol. 2005, 16, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Peirce, S.K.; Findley, H.W. High level MycN expression in non-MYCN amplified neuroblastoma is induced by the combination treatment nutlin-3 and doxorubicin and enhances chemosensitivity. Oncol. Rep. 2009, 22, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Dittrich, O.; Kiermaier, A.; Dohmann, K.; Menkel, A.; Eilers, M.; Luscher, B. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001, 15, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H.; Dissmeyer, N.; Schnittger, A. Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 2013, 23, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.; Tan, F.; Cassano, H.; Lee, J.; Lee, K.C.; Thiele, C.J. Use of RNA interference to elucidate the effect of MYCN on cell cycle in neuroblastoma. Pediatr. Blood Cancer 2008, 50, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Cage, T.A.; Chanthery, Y.; Chesler, L.; Grimmer, M.; Knight, Z.; Shokat, K.; Weiss, W.A.; Gustafson, W.C. Downregulation of MYCN through PI3K inhibition in mouse models of pediatric neural cancer. Front. Oncol. 2015, 5, 111. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra251. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Stegmuller, J.; Guardavaccaro, D.; Liu, G.; Carro, M.S.; Rothschild, G.; de la Torre-Ubieta, L.; Pagano, M.; Bonni, A.; Iavarone, A. Degradation of id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 2006, 442, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, H.; Hunter, T. P27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Ebus, M.E.; van Sluis, P.; Westerhout, E.M.; de Preter, K.; Gisselsson, D.; Ora, I.; Speleman, F.; Caron, H.N.; et al. Copy number defects of G1-cell cycle genes in neuroblastoma are frequent and correlate with high expression of E2F target genes and a poor prognosis. Genes Chromosomes Cancer 2012, 51, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Boldrini, R.; Dominici, C.; Donfrancesco, A.; Yokota, Y.; Inserra, A.; Iavarone, A. Id2 is critical for cellular proliferation and is the oncogenic effector of N-myc in human neuroblastoma. Cancer Res. 2002, 62, 301–306. [Google Scholar] [PubMed]
- Ham, J.; Costa, C.; Sano, R.; Lochmann, T.L.; Sennott, E.M.; Patel, N.U.; Dastur, A.; Gomez-Caraballo, M.; Krytska, K.; Hata, A.N.; et al. Exploitation of the apoptosis-primed state of MYCN-amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell 2016, 29, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Iraci, N.; Gherardi, S.; Gamble, L.D.; Wood, K.M.; Perini, G.; Lunec, J.; Tweddle, D.A. P53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 2010, 70, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (Deltapsi(m)) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.; Chen, L.; Liu, T.; Marshall, G.M.; Lunec, J.; Tweddle, D.A. MYCN oncoprotein targets and their therapeutic potential. Cancer Lett. 2010, 293, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Kozma, S.C.; Tauler, A.; Ambrosio, S. MYCN concurrence with SAHA-induced cell death in human neuroblastoma cells. Cell. Oncol. 2015, 38, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Mehta, P.; Chen, Z.; Zhang, L.; Slack, A.; Berg, S.; Shohet, J.M. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol. Cancer Ther. 2006, 5, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Barker, K.; Sikka, A.; Almeida, G.S.; Hallsworth, A.; Smith, L.M.; Jamin, Y.; Ruddle, R.; Koers, A.; Webber, H.T.; et al. P53 loss in MYC-driven neuroblastoma leads to metabolic adaptations supporting radioresistance. Cancer Res. 2016, 76, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.L.; Cobrinik, D. MDM2 but not MDM4 promotes retinoblastoma cell proliferation through p53-independent regulation of mycn translation. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Valsesia-Wittmann, S.; Magdeleine, M.; Dupasquier, S.; Garin, E.; Jallas, A.C.; Combaret, V.; Krause, A.; Leissner, P.; Puisieux, A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 2004, 6, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Selmi, A.; de Saint-Jean, M.; Jallas, A.C.; Garin, E.; Hogarty, M.D.; Benard, J.; Puisieux, A.; Marabelle, A.; Valsesia-Wittmann, S. Twist1 is a direct transcriptional target of MYCN and MYC in neuroblastoma. Cancer Lett. 2015, 357, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.; Premkumar, R.; Carr, J.; Lu, X.; Lovat, P.E.; Kees, U.R.; Lunec, J.; Tweddle, D.A. The role of MYCN in the failure of MYCN amplified neuroblastoma cell lines to G1 arrest after DNA damage. Cell Cycle 2006, 5, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Diers, A.R.; Broniowska, K.A.; Chang, C.F.; Hogg, N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: Effect of monocarboxylate transporter inhibition. Biochem. J. 2012, 444, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerstrom, L.; Westermark, U.K.; Larsson, K.; Munksgaard Persson, M.; Hultenby, K.; Lehtio, J.; Einvik, C.; et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.R.; Sutton, S.K.; Pajic, M.; Murray, J.; Sekyere, E.O.; Fletcher, J.; Beckers, A.; De Preter, K.; Speleman, F.; George, R.E.; et al. Glutathione biosynthesis is upregulated at the initiation of MYCN-driven neuroblastoma tumorigenesis. Mol. Oncol. 2016, 10, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Remke, M.; Kool, M.; Hielscher, T.; Northcott, P.A.; Williamson, D.; Pfaff, E.; Witt, H.; Jones, D.T.; Ryzhova, M.; et al. Biological and clinical heterogeneity of MYCN-amplified medulloblastoma. Acta Neuropathol. 2012, 123, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Yue, M.; Xiao, D.; Xiu, R.; Gan, L.; Liu, H.; Qing, G. Atf4 and N-Myc coordinate glutamine metabolism in mycn-amplified neuroblastoma cells through ASCT2 activation. J. Pathol. 2015, 235, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, T.; Henriksson, M.A. Impact of MYC in regulation of tumor cell metabolism. Biochim. Biophys. Acta 2015, 1849, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuchner, J.; Kuznetsov, A.V.; Obexer, P.; Ausserlechner, M.J. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2013, 32, 4748–4757. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Cossins, L.R.; Hatzinisiriou, I.; Haber, M.; Nagley, P. Lack of correlation between MYCN expression and the Warburg effect in neuroblastoma cell lines. BMC Cancer 2008, 8, 259. [Google Scholar] [CrossRef] [PubMed]
- Giedt, R.J.; Pfeiffer, D.R.; Matzavinos, A.; Kao, C.Y.; Alevriadou, B.R. Mitochondrial dynamics and motility inside living vascular endothelial cells: Role of bioenergetics. Ann Biomed. Eng. 2012, 40, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hajnoczky, G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Casinelli, G.; LaRosa, J.; Sharma, M.; Cherok, E.; Banerjee, S.; Branca, M.; Edmunds, L.; Wang, Y.; Sims-Lucas, S.; Churley, L.; et al. N-Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Discov. 2016, 2, 16082. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Hsieh, A.L.; Sengupta, A.; Krishnanaiah, S.Y.; Stine, Z.E.; Walton, Z.E.; Gouw, A.M.; Venkataraman, A.; Li, B.; Goraksha-Hicks, P.; et al. MYC disrupts the circadian clock and metabolism in cancer cells. Cell Metab. 2015, 22, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Stringari, C.; Wang, H.; Geyfman, M.; Crosignani, V.; Kumar, V.; Takahashi, J.S.; Andersen, B.; Gratton, E. In vivo single-cell detection of metabolic oscillations in stem cells. Cell Rep. 2015, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.R.; Perkins, A.S.; Tessarollo, L.; Sassoon, D.A.; Parada, L.F. Loss of n-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 1992, 6, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Grady, E.F.; Schwab, M.; Rosenau, W. Expression of N-myc and c-src during the development of fetal human brain. Cancer Res. 1987, 47, 2931–2936. [Google Scholar] [PubMed]
- Hirvonen, H.; Makela, T.P.; Sandberg, M.; Kalimo, H.; Vuorio, E.; Alitalo, K. Expression of the myc proto-oncogenes in developing human fetal brain. Oncogene 1990, 5, 1787–1797. [Google Scholar] [PubMed]
- Hirvonen, H.; Sandberg, M.; Kalimo, H.; Hukkanen, V.; Vuorio, E.; Salmi, T.T.; Alitalo, K. The N-myc proto-oncogene and IGF-II growth factor mrnas are expressed by distinct cells in human fetal kidney and brain. J. Cell Biol. 1989, 108, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Charron, J.; Malynn, B.A.; Fisher, P.; Stewart, V.; Jeannotte, L.; Goff, S.P.; Robertson, E.J.; Alt, F.W. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992, 6, 2248–2257. [Google Scholar] [CrossRef] [PubMed]
- Sawai, S.; Shimono, A.; Wakamatsu, Y.; Palmes, C.; Hanaoka, K.; Kondoh, H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development 1993, 117, 1445–1455. [Google Scholar] [PubMed]
- Knoepfler, P.S.; Cheng, P.F.; Eisenman, R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002, 16, 2699–2712. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, Y.; Watanabe, Y.; Nakamura, H.; Kondoh, H. Regulation of the neural crest cell fate by N-myc: Promotion of ventral migration and neuronal differentiation. Development 1997, 124, 1953–1962. [Google Scholar] [PubMed]
- Alam, G.; Cui, H.; Shi, H.; Yang, L.; Ding, J.; Mao, L.; Maltese, W.A.; Ding, H.F. MYCN promotes the expansion of Phox2b-positive neuronal progenitors to drive neuroblastoma development. Am. J. Pathol. 2009, 175, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Mobley, B.C.; Kwon, M.; Kraemer, B.R.; Hickman, F.E.; Qiao, J.; Chung, D.H.; Carter, B.D. Expression of MYCN in multipotent sympathoadrenal progenitors induces proliferation and neural differentiation, but is not sufficient for tumorigenesis. PLoS ONE 2015, 10, e0133897. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Cotterman, R.; Knoepfler, P.S. N-myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PLoS ONE 2009, 4, e5799. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and n-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alboran, I.M.; Olson, J.M.; Eisenman, R.N. N-myc is an essential downstream effector of shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Varlakhanova, N.V.; Cotterman, R.F.; deVries, W.N.; Morgan, J.; Donahue, L.R.; Murray, S.; Knowles, B.B.; Knoepfler, P.S. Myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation 2010, 80, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.N.; Singh, A.M.; Dalton, S. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell 2010, 7, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F.; Tyler, M.S.; Kozlowski, R.N. Developmental Biology, 6th ed.; Sinauer Associates: Sunderland, MA, USA; Macmillan: Basingstoke, UK, 2000; p. 749. [Google Scholar]
- Noisa, P.; Raivio, T. Neural crest cells: From developmental biology to clinical interventions. Birth Defects Res. C Embryo Today 2014, 102, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Shtukmaster, S.; Schier, M.C.; Huber, K.; Krispin, S.; Kalcheim, C.; Unsicker, K. Sympathetic neurons and chromaffin cells share a common progenitor in the neural crest in vivo. Neural Dev. 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Krispin, S.; Nitzan, E.; Kalcheim, C. The dorsal neural tube: A dynamic setting for cell fate decisions. Dev. Neurobiol. 2010, 70, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, M.R.; Weiss, W.A. Childhood tumors of the nervous system as disorders of normal development. Curr. Opin. Pediatr. 2006, 18, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Scotting, P.J.; Walker, D.A.; Perilongo, G. Childhood solid tumours: A developmental disorder. Nat. Rev. Cancer 2005, 5, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Hickman, J.A. Why does stage 4s neuroblastoma regress spontaneously? Lancet 1994, 344, 869–870. [Google Scholar] [CrossRef]
- Nicolai, S.; Pieraccioli, M.; Peschiaroli, A.; Melino, G.; Raschella, G. Neuroblastoma: Oncogenic mechanisms and therapeutic exploitation of necroptosis. Cell Death Dis. 2015, 6, e2010. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, D.A.; Mele, E.; Ceccanti, S.; Natale, F.; Clerico, A.; Schiavetti, A.; Dominici, C. Long-term follow-up of the “wait and see” approach to localized perinatal adrenal neuroblastoma. World J. Surg. 2013, 37, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.H.; Cheung, N.K.; LaQuaglia, M.P.; Ambros, P.F.; Ambros, I.M.; Bonilla, M.A.; Gerald, W.L.; Ladanyi, M.; Gilbert, F.; Rosenfield, N.S.; et al. Survival from locally invasive or widespread neuroblastoma without cytotoxic therapy. J. Clin. Oncol. 1996, 14, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Bagatell, R. Mechanisms of neuroblastoma regression. Nat. Rev. Clin. Oncol. 2014, 11, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, J.B.; Perrin, E.V. In situ neuroblastomas: A contribution to the natural history of neural crest tumors. Am. J. Pathol. 1963, 43, 1089–1104. [Google Scholar] [PubMed]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frebourg, T.; Munnich, A.; et al. Germline mutations of the paired-like homeobox 2b (PHOX2B) gene in neuroblastoma. Am. J Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Van Limpt, V.; Schramm, A.; van Lakeman, A.; Sluis, P.; Chan, A.; van Noesel, M.; Baas, F.; Caron, H.; Eggert, A.; Versteeg, R. The Phox2b homeobox gene is mutated in sporadic neuroblastomas. Oncogene 2004, 23, 9280–9288. [Google Scholar] [CrossRef] [PubMed]
- Caren, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- De Mariano, M.; Gallesio, R.; Chierici, M.; Furlanello, C.; Conte, M.; Garaventa, A.; Croce, M.; Ferrini, S.; Tonini, G.P.; Longo, L. Identification of GALNT14 as a novel neuroblastoma predisposition gene. Oncotarget 2015, 6, 26335–26346. [Google Scholar] [CrossRef] [PubMed]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef]
- Schwab, M. Amplification of N-myc as a prognostic marker for patients with neuroblastoma. Semin. Cancer Biol 1993, 4, 13–18. [Google Scholar] [PubMed]
- Wang, L.L.; Teshiba, R.; Ikegaki, N.; Tang, X.X.; Naranjo, A.; London, W.B.; Hogarty, M.D.; Gastier-Foster, J.M.; Look, A.T.; Park, J.R.; et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: A children’s oncology group study. Br. J. Cancer 2015, 113, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Park, J.R. Neuroblastoma: Paradigm for precision medicine. Pediatr. Clin. N. Am. 2015, 62, 225–256. [Google Scholar] [CrossRef] [PubMed]
- Hara, J. Development of treatment strategies for advanced neuroblastoma. Int. J. Clin. Oncol. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of mycn causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.A.; Hoppe, P.; Brilliant, M.; Chikaraishi, D.M. 5’ flanking sequences of the rat tyrosine hydroxylase gene target accurate tissue-specific, developmental, and transsynaptic expression in transgenic mice. J. Neurosci. 1992, 12, 4460–4467. [Google Scholar] [PubMed]
- Chesler, L.; Weiss, W.A. Genetically engineered murine models--contribution to our understanding of the genetics, molecular pathology and therapeutic targeting of neuroblastoma. Semin. Cancer Biol. 2011, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, C.A.; Cheng, A.J.; Madafiglio, J.; Kavallaris, M.; Mili, M.; Marshall, G.M.; Weiss, W.A.; Khachigian, L.M.; Norris, M.D.; Haber, M. Effects of MYCN antisense oligonucleotide administration on tumorigenesis in a murine model of neuroblastoma. J. Natl. Cancer Inst. 2003, 95, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Althoff, K.; Beckers, A.; Bell, E.; Nortmeyer, M.; Thor, T.; Sprussel, A.; Lindner, S.; De Preter, K.; Florin, A.; Heukamp, L.C.; et al. A cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 2015, 34, 3357–3368. [Google Scholar] [CrossRef] [PubMed]
- Fredlund, E.; Ringner, M.; Maris, J.M.; Pahlman, S. High myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc. Natl. Acad. Sci. USA 2008, 105, 14094–14099. [Google Scholar] [CrossRef] [PubMed]
- Schweigerer, L.; Breit, S.; Wenzel, A.; Tsunamoto, K.; Ludwig, R.; Schwab, M. Augmented mycn expression advances the malignant phenotype of human neuroblastoma cells: Evidence for induction of autocrine growth factor activity. Cancer Res. 1990, 50, 4411–4416. [Google Scholar] [PubMed]
- Gogolin, S.; Dreidax, D.; Becker, G.; Ehemann, V.; Schwab, M.; Westermann, F. Mycn/Myc-mediated drug resistance mechanisms in neuroblastoma. Int. J. Clin. Pharmacol. Ther. 2010, 48, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Slack, A.; Chen, Z.; Tonelli, R.; Pule, M.; Hunt, L.; Pession, A.; Shohet, J.M. The p53 regulatory gene MDM2 is a direct transcriptional target of mycn in neuroblastoma. Proc. Natl. Acad. Sci. USA 2005, 102, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Chrochemore, C.; Cambuli, F.; Iraci, N.; Contestabile, A.; Perini, G. Nitric oxide control of MYCN expression and multi drug resistance genes in tumours of neural origin. Curr. Pharm. Des. 2010, 16, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Thiele, C.J.; Deutsch, L.A.; Israel, M.A. The expression of multiple proto-oncogenes is differentially regulated during retinoic acid induced maturation of human neuroblastoma cell lines. Oncogene 1988, 3, 281–288. [Google Scholar] [PubMed]
- Guglielmi, L.; Cinnella, C.; Nardella, M.; Maresca, G.; Valentini, A.; Mercanti, D.; Felsani, A.; D’Agnano, I. MYCN gene expression is required for the onset of the differentiation programme in neuroblastoma cells. Cell Death Dis. 2014, 5, e1081. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, M.V.; Sanchez-Jimenez, F.; Alonso, F.J.; Segura, J.A.; Marquez, J.; Medina, M.A. Glutamine, glucose and other fuels for cancer. Curr. Pharm. Des. 2014, 20, 2557–2579. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.D.; Al-Saadi, R.; Natrajan, R.; Mackay, A.; Chagtai, T.; Little, S.; Hing, S.N.; Fenwick, K.; Ashworth, A.; Grundy, P.; et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in wilms tumor. Genes Chromosomes Cancer 2011, 50, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Treger, T.D.; Brok, J.; Pritchard-Jones, K. Biology and treatment of wilms’ tumours in childhood. Revue d’Oncologie Hématologie Pédiatrique 2016, 4, 170–181. [Google Scholar] [CrossRef]
- Rivera, M.N.; Haber, D.A. Wilms’ tumour: Connecting tumorigenesis and organ development in the kidney. Nat. Rev. Cancer 2005, 5, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.D.; Al-Saadi, R.; Chagtai, T.; Popov, S.; Messahel, B.; Sebire, N.; Gessler, M.; Wegert, J.; Graf, N.; Leuschner, I.; et al. Subtype-specific FBXW7 mutation and MYCN copy number gain in wilms’ tumor. Clin. Cancer Res. 2010, 16, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.D.; Chagtai, T.; Alcaide-German, M.; Apps, J.; Wegert, J.; Popov, S.; Vujanic, G.; van Tinteren, H.; van den Heuvel-Eibrink, M.M.; Kool, M.; et al. Multiple mechanisms of MYCN dysregulation in wilms tumour. Oncotarget 2015, 6, 7232–7243. [Google Scholar] [CrossRef] [PubMed]
- Micale, M.A.; Embrey, B.T.; Macknis, J.K.; Harper, C.E.; Aughton, D.J. Constitutional 560.49 kb chromosome 2p24.3 duplication including the MYCN gene identified by snp chromosome microarray analysis in a child with multiple congenital anomalies and bilateral wilms tumor. Eur. J. Med. Genet. 2016, 59, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W. Role of MYCN in retinoblastoma. Lancet Oncol. 2013, 14, 270–271. [Google Scholar] [CrossRef]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Theriault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, A.; Luna, M.; Rucker, N.; Wong, S.; Lederman, A.; Kim, J.; Gomer, C. Targeting survivin enhances chemosensitivity in retinoblastoma cells and orthotopic tumors. PLoS ONE 2016, 11, e0153011. [Google Scholar] [CrossRef] [PubMed]
- Li, W.L.; Buckley, J.; Sanchez-Lara, P.A.; Maglinte, D.T.; Viduetsky, L.; Tatarinova, T.V.; Aparicio, J.G.; Kim, J.W.; Au, M.; Ostrow, D.; et al. A rapid and sensitive next-generation sequencing method to detect RB1 mutations improves care for retinoblastoma patients and their families. J. Mol. Diagn. 2016, 18, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to n-myc. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Gessi, M.; von Bueren, A.; Treszl, A.; zur Muhlen, A.; Hartmann, W.; Warmuth-Metz, M.; Rutkowski, S.; Pietsch, T. MYCN amplification predicts poor outcome for patients with supratentorial primitive neuroectodermal tumors of the central nervous system. Neuro Oncol. 2014, 16, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Jasnos, L.; Gil, V.; Howell, L.; Hallsworth, A.; Petrie, K.; Sawado, T.; Chesler, L. Molecular and in vivo characterization of cancer-propagating cells derived from MYCN-dependent medulloblastoma. PLoS ONE 2015, 10, e0119834. [Google Scholar] [CrossRef] [PubMed]
- Aldosari, N.; Bigner, S.H.; Burger, P.C.; Becker, L.; Kepner, J.L.; Friedman, H.S.; McLendon, R.E. Mycc and mycn oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the children’s oncology group. Arch. Pathol. Lab. Med. 2002, 126, 540–544. [Google Scholar] [PubMed]
- Massimino, M.; Biassoni, V.; Gandola, L.; Garre, M.L.; Gatta, G.; Giangaspero, F.; Poggi, G.; Rutkowski, S. Childhood medulloblastoma. Crit. Rev. Oncol. Hematol. 2016, 105, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Driman, D.; Thorner, P.S.; Greenberg, M.L.; Chilton-MacNeill, S.; Squire, J. MYCN gene amplification in rhabdomyosarcoma. Cancer 1994, 73, 2231–2237. [Google Scholar] [CrossRef]
- Xia, S.J.; Pressey, J.G.; Barr, F.G. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol. Ther. 2002, 1, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, D.A.; Anderson, J. MYCN deregulation as a potential target for novel therapies in rhabdomyosarcoma. Expert Rev. Anticancer Ther. 2006, 6, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Toffolatti, L.; Frascella, E.; Ninfo, V.; Gambini, C.; Forni, M.; Carli, M.; Rosolen, A. Mycn expression in human rhabdomyosarcoma cell lines and tumour samples. J. Pathol. 2002, 196, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.; Lu, Y.J.; Gordon, T.; Sciot, R.; Kelsey, A.; Fisher, C.; Poremba, C.; Anderson, J.; Pritchard-Jones, K.; Shipley, J. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J. Clin. Oncol. 2005, 23, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor activity of sustained N-myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Cognet, M.; Nougayrede, A.; Malan, V.; Callier, P.; Cretolle, C.; Faivre, L.; Genevieve, D.; Goldenberg, A.; Heron, D.; Mercier, S.; et al. Dissection of the MYCN locus in feingold syndrome and isolated oesophageal atresia. Eur. J. Hum. Genet. 2011, 19, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Fiori, E.; Babicola, L.; Andolina, D.; Coassin, A.; Pascucci, T.; Patella, L.; Han, Y.C.; Ventura, A.; Ventura, R. Neurobehavioral alterations in a genetic murine model of Feingold syndrome 2. Behav. Genet. 2015, 45, 547–559. [Google Scholar] [CrossRef] [PubMed]
- van Bokhoven, H.; Celli, J.; van Reeuwijk, J.; Rinne, T.; Glaudemans, B.; van Beusekom, E.; Rieu, P.; Newbury-Ecob, R.A.; Chiang, C.; Brunner, H.G. MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in feingold syndrome. Nat. Genet. 2005, 37, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Petroni, M.; Sardina, F.; Heil, C.; Sahun-Roncero, M.; Colicchia, V.; Veschi, V.; Albini, S.; Fruci, D.; Ricci, B.; Soriani, A.; et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. 2016, 23, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.M.; Beltran, H.; Park, K.; MacDonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Krishna, N.S.; Witton, C.J.; Bartlett, J.M. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin. Cancer Res. 2003, 9, 5271–5281. [Google Scholar] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC-aurora kinase a protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Freier, K.; Flechtenmacher, C.; Devens, F.; Hartschuh, W.; Hofele, C.; Lichter, P.; Joos, S. Recurrent NMYC copy number gain and high protein expression in basal cell carcinoma. Oncol. Rep. 2006, 15, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant Hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Kieran, M.W. Targeted treatment for sonic hedgehog-dependent medulloblastoma. Neuro Oncol. 2014, 16, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Rohner, A.; Spilker, M.E.; Lam, J.L.; Pascual, B.; Bartkowski, D.; Li, Q.J.; Yang, A.H.; Stevens, G.; Xu, M.; Wells, P.A.; et al. Effective targeting of Hedgehog signaling in a medulloblastoma model with pf-5274857, a potent and selective smoothened antagonist that penetrates the blood-brain barrier. Mol. Cancer Ther. 2012, 11, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Stano-Kozubik, K.; Malcikova, J.; Tichy, B.; Kotaskova, J.; Borsky, M.; Hrabcakova, V.; Francova, H.; Valaskova, I.; Bourkova, L.; Smardova, J.; et al. Inactivation of p53 and amplification of MYCN gene in a terminal lymphoblastic relapse in a chronic lymphocytic leukemia patient. Cancer Genet. Cytogenet. 2009, 189, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Vendemini, F.; Urbini, M.; Melchionda, F.; Masetti, R.; Franzoni, M.; Libri, V.; Serravalle, S.; Togni, M.; Paone, G.; et al. MYCN is a novel oncogenic target in pediatric t-cell acute lymphoblastic leukemia. Oncotarget 2014, 5, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Schwaenen, C.; Nessling, M.; Wessendorf, S.; Salvi, T.; Wrobel, G.; Radlwimmer, B.; Kestler, H.A.; Haslinger, C.; Stilgenbauer, S.; Dohner, H.; et al. Automated array-based genomic profiling in chronic lymphocytic leukemia: Development of a clinical tool and discovery of recurrent genomic alterations. Proc. Natl. Acad. Sci. USA 2004, 101, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Zwaans, B.M.; Lombard, D.B. Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Dis. Models Mech. 2014, 7, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef]
- Hirvonen, H.; Hukkanen, V.; Salmi, T.T.; Makela, T.P.; Pelliniemi, T.T.; Knuutila, S.; Alitalo, R. Expression of L-myc and N-myc proto-oncogenes in human leukemias and leukemia cell lines. Blood 1991, 78, 3012–3020. [Google Scholar] [PubMed]
- Hirvonen, H.; Hukkanen, V.; Salmi, T.T.; Pelliniemi, T.T.; Alitalo, R. L-myc and N-myc in hematopoietic malignancies. Leuk Lymphoma 1993, 11, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Lu, P.; Sun, G.; Yang, L.; Wang, Z.; Wang, Z. MiR-34a sensitizes lung cancer cells to cisplatin via p53/mir-34a/mycn axis. Biochem. Biophys. Res. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wang, S.; Liu, Y.; Gu, J.; Gu, S.; Xu, Z.; Zhang, R.; Wang, Z.; Ma, H.; Chen, Y.; et al. Overexpression of MYCN promotes proliferation of non-small cell lung cancer. Tumour Biol. 2016, 37, 12855–12866. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Dietlein, F.; Peifer, M.; Schottle, J.; Balke-Want, H.; Muller, C.; Koker, M.; Richters, A.; Heynck, S.; Malchers, F.; et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 17034–17039. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Girard, L.; Giacomini, C.P.; Wang, P.; Hernandez-Boussard, T.; Tibshirani, R.; Minna, J.D.; Pollack, J.R. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of myc family gene amplification. Oncogene 2006, 25, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a Myc/MYCN-activated microrna, regulates e-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Marrano, P.; Irwin, M.S.; Thorner, P.S. Heterogeneity of MYCN amplification in neuroblastoma at diagnosis, treatment, relapse, and metastasis. Genes Chromosomes Cancer 2017, 56, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Benabou, S.; Ferreira, R.; Avino, A.; Gonzalez, C.; Lyonnais, S.; Sola, M.; Eritja, R.; Jaumot, J.; Gargallo, R. Solution equilibria of cytosine- and guanine-rich sequences near the promoter region of the N-myc gene that contain stable hairpins within lateral loops. Biochim. Biophys. Acta 2014, 1840, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Hurley, L.H. Structure of the biologically relevant g-quadruplex in the c-myc promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, T.; Grotzer, M.A. MYC as therapeutic target for embryonal tumors: Potential and challenges. Curr. Cancer Drug Targets 2016, 16, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. Small molecules targeting c-Myc oncogene: Promising anti-cancer therapeutics. Int. J. Biol. Sci. 2014, 10, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, H.; Zhou, J.; Yuan, G. Exploration of the selective recognition of the G-quadruplex in the N-myc oncogene by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2015, 29, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016, 106, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Barton, M.C. Bromodomain histone readers and cancer. J. Mol. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of bet bromodomain inhibition. Mol Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting bet bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in neuroblastoma by bet bromodomain inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Ganji, G.; Smitheman, K.N.; Chung, C.W.; Korenchuk, S.; Bai, Y.; Barbash, O.; Le, B.; Craggs, P.D.; McCabe, M.T.; et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE 2013, 8, e72967. [Google Scholar] [CrossRef] [PubMed]
- Henssen, A.; Althoff, K.; Odersky, A.; Beckers, A.; Koche, R.; Speleman, F.; Schafers, S.; Bell, E.; Nortmeyer, M.; Westermann, F.; et al. Targeting MYCN-driven transcription by bet-bromodomain inhibition. Clin. Cancer Res. 2016, 22, 2470–2481. [Google Scholar] [CrossRef] [PubMed]
- Ikegaki, N.; Bukovsky, J.; Kennett, R.H. Identification and characterization of the nmyc gene product in human neuroblastoma cells by monoclonal antibodies with defined specificities. Proc. Natl. Acad. Sci. USA 1986, 83, 5929–5933. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small molecule inhibitors of Aurora-A induce proteasomal degradation of N-Myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Romain, C.; Paul, P.; Kim, K.W.; Lee, S.; Qiao, J.; Chung, D.H. Targeting aurora kinase-a downregulates cell proliferation and angiogenesis in neuroblastoma. J. Pediatr. Surg. 2014, 49, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Marachelian, A.; Fox, E.; Kudgus, R.A.; Reid, J.M.; Groshen, S.; Malvar, J.; Bagatell, R.; Wagner, L.; Maris, J.M.; et al. Phase I study of the aurora a kinase inhibitor alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma: A nant (new approaches to neuroblastoma therapy) trial. J. Clin. Oncol. 2016, 34, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Calero, R.; Morchon, E.; Johnsen, J.I.; Serrano, R. Sunitinib suppress neuroblastoma growth through degradation of mycn and inhibition of angiogenesis. PLoS ONE 2014, 9, e95628. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Segerstrom, L.; Orrego, A.; Elfman, L.; Henriksson, M.; Kagedal, B.; Eksborg, S.; Sveinbjornsson, B.; Kogner, P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 2008, 27, 2910–2922. [Google Scholar] [CrossRef] [PubMed]
- Segerstrom, L.; Baryawno, N.; Sveinbjornsson, B.; Wickstrom, M.; Elfman, L.; Kogner, P.; Johnsen, J.I. Effects of small molecule inhibitors of PI3K/AKT/mTOR signaling on neuroblastoma growth in vitro and in vivo. Int. J. Cancer 2011, 129, 2958–2965. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, L.; Clarke, P.A.; Barker, K.; Chanthery, Y.; Gustafson, C.W.; Tucker, E.; Renshaw, J.; Raynaud, F.; Li, X.; Burke, R.; et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget 2016, 7, 57525–57544. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.H.; Cheung, N.V.; Modak, S.; Becher, O.J.; Basu, E.M.; Roberts, S.S.; Kramer, K.; Dunkel, I.J. A phase I/Ib trial targeting the PI3K/AKT pathway using perifosine: Long-term progression-free survival of patients with resistant neuroblastoma. Int. J. Cancer 2017, 140, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Henriksson, M. Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proc. Natl. Acad. Sci. USA 2006, 103, 6344–6349. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.; Larsson, K.; Frenzel, A.; Oliynyk, G.; Zirath, H.; Prochownik, E.V.; Westwood, N.J.; Henriksson, M.A. Targeting of the MYCN protein with small molecule c-MYC inhibitors. PLoS ONE 2014, 9, e97285. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Vita, M.; Crespin, M.; Henriksson, M. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle 2006, 5, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Parise, R.A.; Joseph, E.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-f4 [z,e]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother. Pharmacol. 2009, 63, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Clausen, D.M.; Guo, J.; Parise, R.A.; Beumer, J.H.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J. Pharmacol. Exp. Ther. 2010, 335, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.B. Potent Analogues of the C-Myc Inhibitor 10074-G5 with Improved Cell Permeability. US Patent 9242944, 26 January 2016. [Google Scholar]
- Ioannides, C.G.; Ioannides, M.G.; O’Brian, C.A. T-cell recognition of oncogene products: A new strategy for immunotherapy. Mol. Carcinog. 1992, 6, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.K.; Nuchtern, J.G. Lysis of MYCN-amplified neuroblastoma cells by mycn peptide-specific cytotoxic t lymphocytes. Cancer Res. 2000, 60, 1908–1913. [Google Scholar] [PubMed]
- Stermann, A.; Huebener, N.; Seidel, D.; Fest, S.; Eschenburg, G.; Stauder, M.; Schramm, A.; Eggert, A.; Lode, H.N. Targeting of mycn by means of DNA vaccination is effective against neuroblastoma in mice. Cancer Immunol. Immunother. 2015, 64, 1215–1227. [Google Scholar] [PubMed]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [PubMed]
- Saulnier Sholler, G.L.; Gerner, E.W.; Bergendahl, G.; MacArthur, R.B.; VanderWerff, A.; Ashikaga, T.; Bond, J.P.; Ferguson, W.; Roberts, W.; Wada, R.K.; et al. A phase I trial of DFMO targeting polyamine addiction in patients with relapsed/refractory neuroblastoma. PLoS ONE 2015, 10, e0127246. [Google Scholar] [CrossRef] [PubMed]
- Bassiri, H.; Benavides, A.; Haber, M.; Gilmour, S.K.; Norris, M.D.; Hogarty, M.D. Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Transl. Pediatr. 2015, 4, 226–238. [Google Scholar] [PubMed]
- Cole, K.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. Rnai screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Ebus, M.E.; Geerts, D.; Koster, J.; Lamers, F.; Valentijn, L.J.; Westerhout, E.M.; Versteeg, R.; Caron, H.N. Inactivation of CDK2 is synthetically lethal to mycn over-expressing cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12968–12973. [Google Scholar] [CrossRef] [PubMed]
- Chayka, O.; D’Acunto, C.W.; Middleton, O.; Arab, M.; Sala, A. Identification and pharmacological inactivation of the MYCN gene network as a therapeutic strategy for neuroblastic tumor cells. J. Biol. Chem. 2015, 290, 2198–2212. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Chromosomal Location | |||
|---|---|---|---|
| Gene | Human | Mouse | References |
| MYCN/Mycn | 2p24.3 | 12-A3.B | [7,8] |
| MYCL/Mycl | 1p34.2 | 4-D2.2 | [8,9] |
| MYC/c-myc | 8q24.21 | 15-D1 | [10,11] |
| Disease | Onset | Primary Location | MYCN Aberration | % of Patients Showing MYCN Aberration |
|---|---|---|---|---|
| Neuroblastoma | Childhood | Peripheral nervous system | Amplification/overexpression | 25% |
| Medulloblastoma | Childhood | Cerebellum | Amplification | 7–10% |
| Retinoblastoma | Childhood | Retina | Amplification | 2% |
| Rabdomyosarcoma (ARMS/ERMS) | Childhood | Skeletal muscle | Copy number gain/amplification/Increased stability | 25% of ARMS 16% of ERMS |
| Wilms’ tumor | Childhood | Kidney | Copy number gain/Overexpression/mutation | 13% 9% 4% |
| Feingold syndrome | Childhood | Physical and learning developmental disorders | Amplification/ deletion/loss of function | 50% |
| Leukemia (CLL/ALL) | Childhood/adulthood | White blood cells | Copy number gain/amplification/Increased expression | 7% of ALL 35% of CLL |
| Prostate cancer | Adulthood | Prostate | Amplification | 14% |
| Basal cell carcinoma | Adulthood | Skin | Copy number gain/increased expression | 18% 73% |
| Lung cancer | Adulthood | Lung | Copy number gain | 6% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Pérez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. https://doi.org/10.3390/genes8040113
Ruiz-Pérez MV, Henley AB, Arsenian-Henriksson M. The MYCN Protein in Health and Disease. Genes. 2017; 8(4):113. https://doi.org/10.3390/genes8040113
Chicago/Turabian StyleRuiz-Pérez, María Victoria, Aine Brigette Henley, and Marie Arsenian-Henriksson. 2017. "The MYCN Protein in Health and Disease" Genes 8, no. 4: 113. https://doi.org/10.3390/genes8040113
APA StyleRuiz-Pérez, M. V., Henley, A. B., & Arsenian-Henriksson, M. (2017). The MYCN Protein in Health and Disease. Genes, 8(4), 113. https://doi.org/10.3390/genes8040113