
Rapid Sampling of Escherichia coli After Changing Oxygen Conditions Reveals Transcriptional Dynamics

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain and Growth Conditions
2.2. RNA Sequencing
2.3. Principal Component Analysis
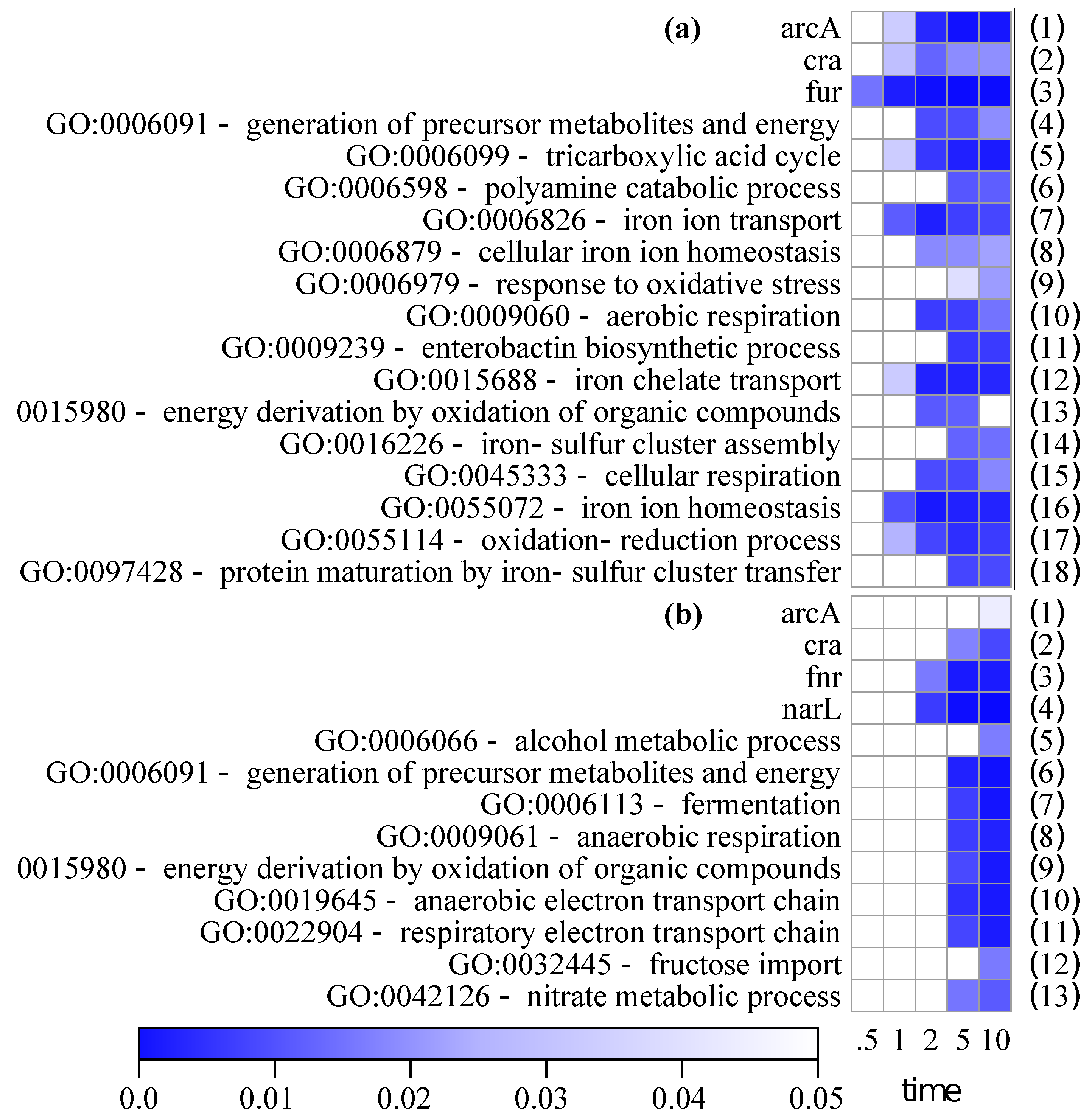
2.4. Regulon and Gene Ontology Enrichments
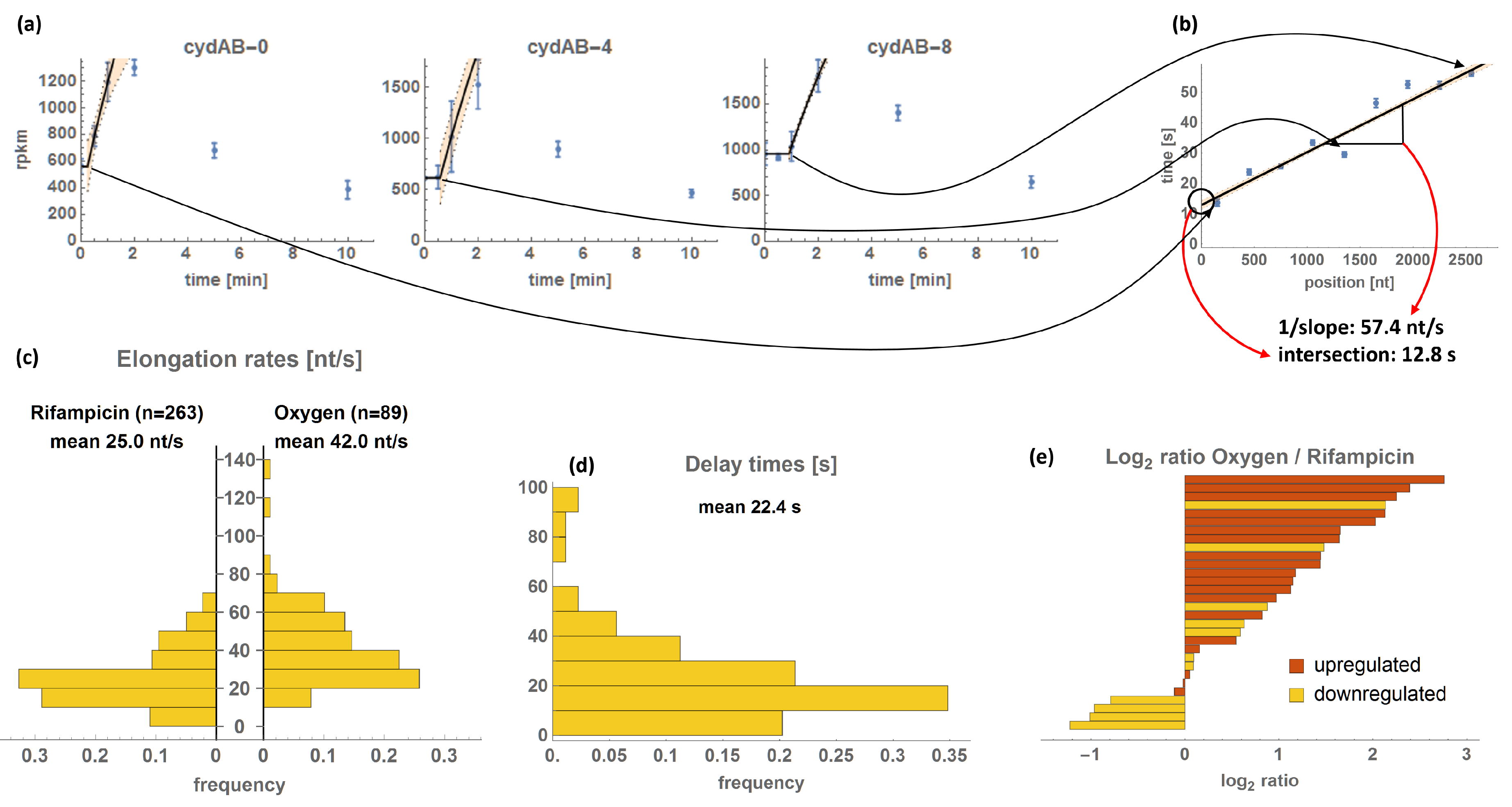
2.5. TER Estimation
3. Results
3.1. General Properties of the Dataset
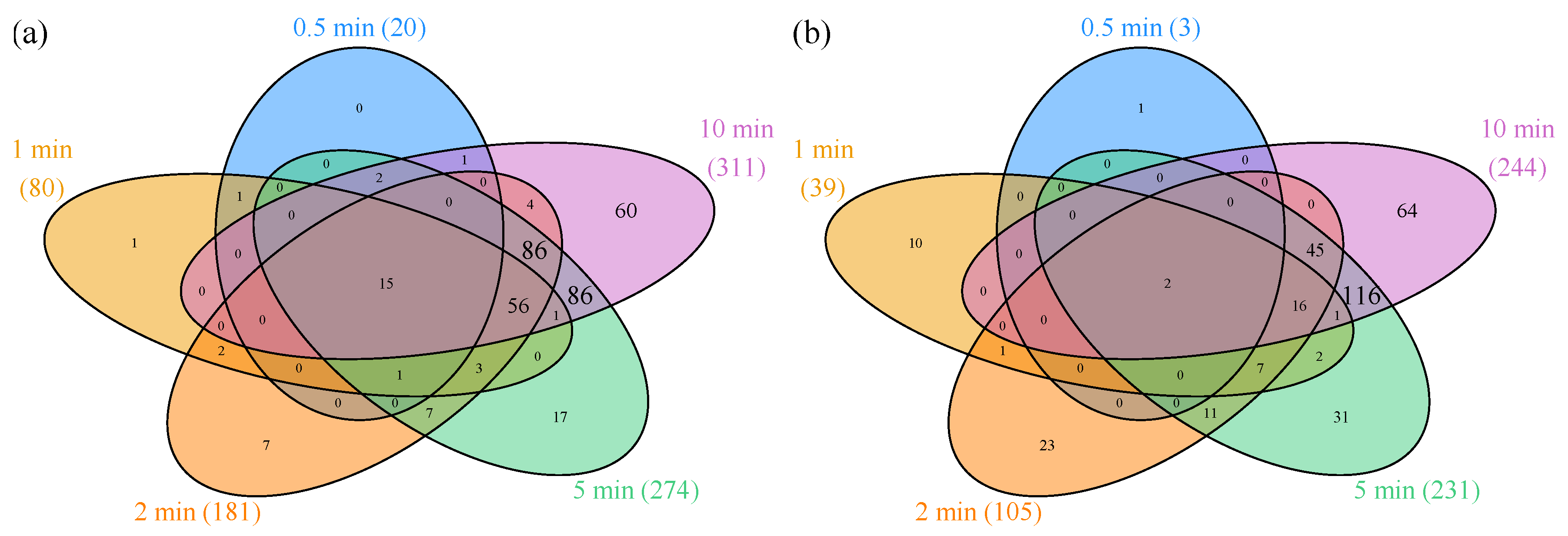
3.1.1. Differentially Expressed Genes
3.1.2. Principal Component Analysis
3.1.3. Regulon and Gene Ontology Enrichments
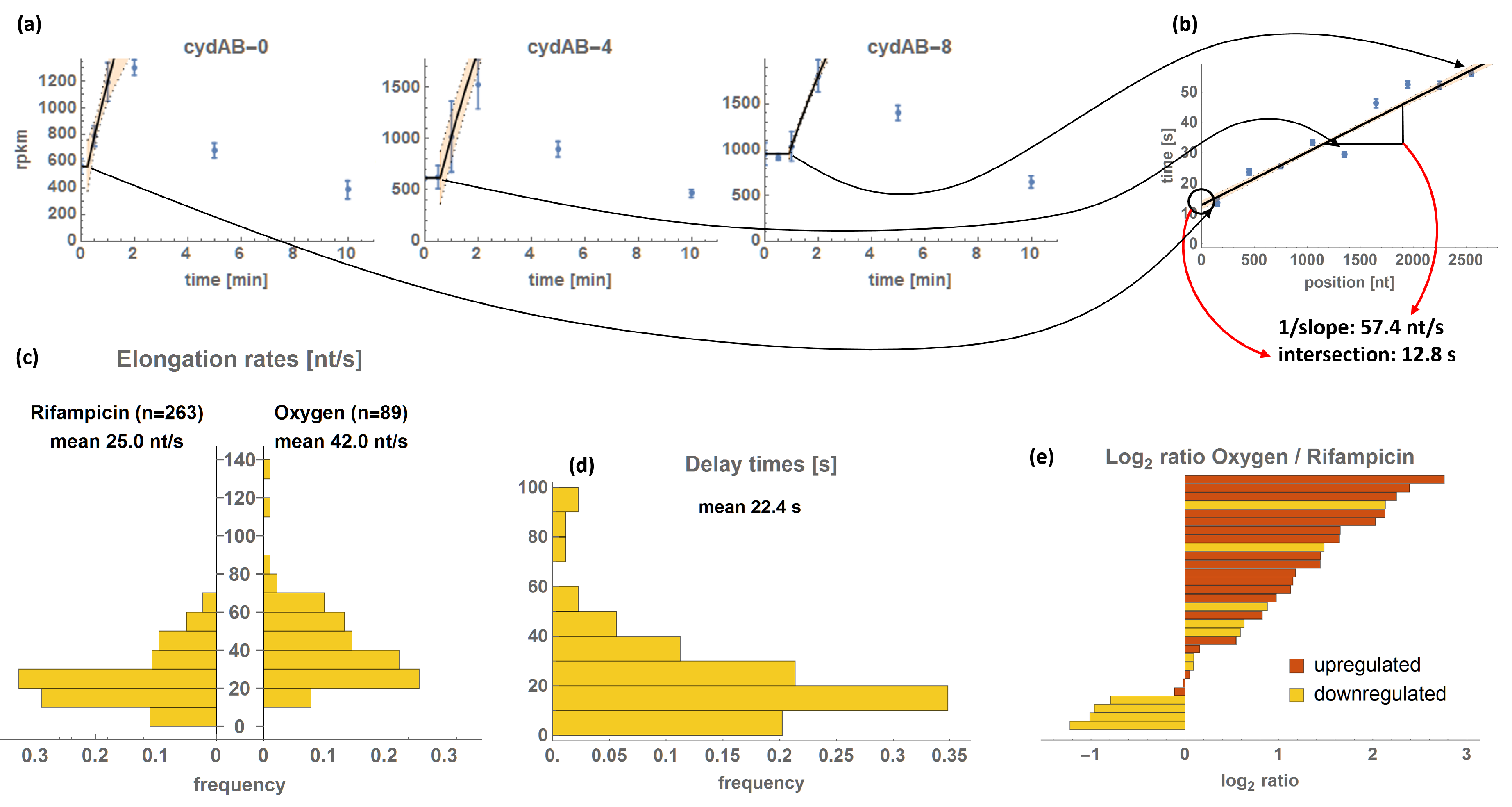
3.2. Transcriptional Elongation Rates
3.2.1. Correlation Analyses of TERs and Delay Times
4. Discussion
4.1. Gene Expression Patterns
4.1.1. Iron Balance
4.1.2. Expression of Oxidases
4.2. Transcriptional Elongation Rates
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Junker, B.H. Scale-Up Methodologies for Escherichia coli and Yeast Fermentation Processes. J. Biosci. Bioeng. 2004, 97, 347–364. [Google Scholar] [CrossRef]
- Takors, R. Scale-up of microbial processes: Impacts, tools and open questions. J. Biotechnol. 2012, 160, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lara, A.R.; Galindo, E.; Ramírez, O.T.; Palomares, L.A. Living With Heterogeneities in Bioreactors: Understanding the Effects of Environmental Gradients on Cells. Mol. Biotechnol. 2006, 34, 355–382. [Google Scholar] [CrossRef]
- Partridge, J.D.; Scott, C.; Tang, Y.; Poole, R.K.; Green, J. Escherichia coli Transcriptome Dynamics during the Transition from Anaerobic to Aerobic Conditions. J. Biol. Chem. 2006, 281, 27806–27815. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, M.D.; Ocone, A.; Stapleton, M.R.; Hall, S.; Trotter, E.W.; Poole, R.K.; Sanguinetti, G.; Green, J. Systems analysis of transcription factor activities in environments with stable and dynamic oxygen concentrations. Open Biol. 2012, 2, 120091. [Google Scholar] [CrossRef] [PubMed]
- Trotter, E.W.; Rolfe, M.D.; Hounslow, A.M.; Craven, C.J.; Williamson, M.P.; Sanguinetti, G.; Poole, R.K.; Green, J. Reprogramming of Escherichia coli K-12 Metabolism during the Initial Phase of Transition from an Anaerobic to a Micro-Aerobic Environment. PLoS ONE 2011, 6, e25501. [Google Scholar] [CrossRef] [PubMed]
- Ederer, M.; Steinsiek, S.; Stagge, S.; Rolfe, M.D.; Ter Beek, A.; Knies, D.; Teixeira de Mattos, M.J.; Sauter, T.; Green, J.; Poole, R.K.; et al. A mathematical model of metabolism and regulation provides a systems-level view of how Escherichia coli responds to oxygen. Front. Microbiol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- von Wulffen, J.; Sawodny, O.; Feuer, R. Transition of an Anaerobic Escherichia coli Culture to Aerobiosis: Balancing mRNA and Protein Levels in a Demand-Directed Dynamic Flux Balance Analysis. PLoS ONE 2016, 11, e0158711. [Google Scholar] [CrossRef] [PubMed]
- Bettenbrock, K.; Bai, H.; Ederer, M.; Green, J.; Hellingwerf, K.J.; Holcombe, M.; Kunz, S.; Rolfe, M.D.; Sanguinetti, G.; Sawodny, O.; et al. Towards a Systems Level Understanding of the Oxygen Response of Escherichia coli. Adv. Microb. Physiol. 2014, 64, 65–114. [Google Scholar] [PubMed]
- Löffler, M.; Simen, J.D.; Jäger, G.; Schäferhoff, K.; Freund, A.; Takors, R. Engineering E. coli for Large-Scale Production—Strategies Considering ATP Expenses and Transcriptional Responses. Metab. Eng. 2016, 38, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Kaleta, C.; Schäuble, S.; Rinas, U.; Schuster, S. Metabolic costs of amino acid and protein production in Escherichia coli. Biotechnol. J. 2013, 8, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.E.; Hixson, K.K.; Conrad, T.M.; Lerman, J.A.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.N.; Schramm, G.; Purvine, S.O.; Lopez-Ferrer, D.; et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shiroguchi, K.; Ge, H.; Xie, X.S. Genome-wide study of mRNA degradation and transcript elongation in Escherichia coli. Mol. Syst. Biol. 2015, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Miller, O.L.; Hamkalo, B.; Thomas, C. Visualization of bacterial genes in action. Science 1970, 169, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Proshkin, S.; Rahmouni, A.R.; Mironov, A.; Nudler, E. Cooperation Between Translating Ribosomes and RNA Polymerase in Transcription Elongation. Science 2010, 328, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Burmann, B.M.; Schweimer, K.; Luo, X.; Wahl, M.C.; Stitt, B.L.; Gottesman, M.E.; Rösch, P. A NusE:NusG complex links transcription and translation. Science 2010, 328, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.A. Antitermination mechanisms in rRNA operons of Escherichia coli. J. Bacteriol. 1986, 168, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rutberg, B. Antitermination of transcription of catabolic operons. Mol. Microbiol. 1997, 23, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Washburn, R.S.; Gottesman, M.E. Regulation of transcription elongation and termination. Biomolecules 2015, 5, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.; Haverkorn van Rijsewijk, B.R.; Sauer, U.; Bettenbrock, K. A role for EIIANtr in controlling fluxes in the central metabolism of E. coli K12. Biochim. Biophys. Acta 2013, 1833, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, T.; Larisch, C.; Lengeler, J.W.; Jahreis, K. Glucose Transporter Mutants of Escherichia coli K-12 with Changes in Substrate Recognition of IICBGlc and Induction Behavior of the ptsG Gene. J. Bacteriol. 2000, 182, 4443–4452. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 26 February 2015).
- Schneider, K.L.; Pollard, K.S.; Baertsch, R.; Pohl, A.; Lowe, T.M. The UCSC Archaeal Genome Browser. Nucleic Acids Res. 2006, 34, D407–D410. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Mardia, K.V.; Kent, J.T.; Bibby, J.M. Multivariate Analysis; Academic Press: New York, NY, USA, 1979. [Google Scholar]
- Karp, P.D.; Paley, S.; Romero, P. The Pathway Tools software. Bioinformatics 2002, 18, S225–S232. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Paley, S.M.; Krummenacker, M.; Latendresse, M.; Dale, J.M.; Lee, T.J.; Kaipa, P.; Gilham, F.; Spaulding, A.; Popescu, L.; et al. Pathway Tools version 19.0: Integrated Software for Pathway/Genome Informatics and Systems Biology. Brief. Bioinform. 2010, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.A.; Khodursky, A.B.; Lin, P.H.; Lin-Chao, S.; Cohen, S.N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. USA 2002, 99, 9697–9702. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by simulated annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from the international DNA sequence databases; its status 1999. Nucleic Acids Res. 1999, 27, 292. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. A major controversy in codon–anticodon adaptation resolved by a new codon usage index. Genetics 2015, 199, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatshefte Chem. Chem. Mon. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Mackie, A.; Peralta-Gil, M.; Santos-Zavaleta, A.; Gama-Castro, S.; Bonavides-Martinez, C.; Fulcher, C.; Huerta, A.M.; Kothari, A.; Krummenacker, M.; et al. EcoCyc: Fusing model organism databases with systems biology. Nucleic Acids Res. 2013, 41, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Alexeeva, S.; Hellingwerf, K.J.; Teixeira de Mattos, M.J.; Teixeira de, M.J. Quantitative Assessment of Oxygen Availability: Perceived Aerobiosis and Its Effect on Flux Distribution in the Respiratory Chain of Escherichia coli. J. Bacteriol. 2002, 184, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- De Vos, D.; Bruggeman, F.J.; Westerhoff, H.V.; Bakker, B.M. How molecular competition influences fluxes in gene expression networks. PLoS ONE 2011, 6, e28494. [Google Scholar] [CrossRef] [PubMed]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.Y.; Moody, D.B.; Murray, M.; Galagan, J.E. Interpreting expression data with metabolic flux models: Predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Membrillo-Hernández, J.; Poole, R.K.; Cook, G.M. Roles of respiratory oxidases in protecting Escherichia coli K12 from oxidative stress. Antonie van Leeuwenhoek 2000, 78, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.P.; Vargo, C.J.; Cui, X.; Kurtz, D.M. A [2Fe-2S] protein encoded by an open reading frame upstream of the Escherichia coli bacterioferritin gene. Biochemistry 1996, 35, 6297–6301. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M. Iron storage in bacteria. Nature 1979, 279, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Govantes, F.; Orjalo, A.V.; Gunsalus, R.P. Interplay between three global regulatory proteins mediates oxygen regulation of the Escherichia coli cytochrome d oxidase (cydAB) operon. Mol. Microbiol. 2002, 38, 1061–1073. [Google Scholar] [CrossRef]
- Iuchi, S.; Chepuri, V.; Fu, H.A.; Gennis, R.B.; Lin, E.C. Requirement for terminal cytochromes in generation of the aerobic signal for the arc regulatory system in Escherichia coli: Study utilizing deletions and lac fusions of cyo and cyd. J. Bacteriol. 1990, 172, 6020–6025. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, S.; Weiner, L. Cellular and molecular physiology of Escherichia coli in the adaptation to aerobic environments. J. Biochem. 1996, 120, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.F.; Rodriguez, C.; Georgellis, D. Ubiquinone and Menaquinone Electron Carriers Represent the Yin and Yang in the Redox Regulation of the ArcB Sensor Kinase. J. Bacteriol. 2013, 195, 3054–3061. [Google Scholar] [CrossRef] [PubMed]
- Bremer, H.; Yuan, D. RNA chain growth-rate in Escherichia coli. J. Mol. Biol. 1968, 38, 163–180. [Google Scholar] [CrossRef]
- Vogel, U.; Jensen, K.F. The RNA chain elongation rate in Escherichia coli depends on the growth rate. J. Bacteriol. 1994, 176, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Reid, P.; Speyer, J. Rifampicin inhibition of ribonucleic acid and protein synthesis in normal and ethylenediaminetetraacetic acid-treated Escherichia coli. J. Bacteriol. 1970, 104, 376–389. [Google Scholar] [PubMed]
- Epshtein, V.; Nudler, E. Cooperation between RNA polymerase molecules in transcription elongation. Science 2003, 300, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [PubMed]
- Lithwick, G.; Margalit, H. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res. 2003, 13, 2665–2673. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, R.; Beyer, A.; Heinisch, J.J.; Wilhelm, T. Posttranscriptional expression regulation: What determines translation rates? PLoS Comput. Biol. 2007, 3, 0531–0539. [Google Scholar] [CrossRef] [PubMed]
- Salmon, K.; Hung, S.p.; Mekjian, K.; Baldi, P.; Hatfield, G.W.; Gunsalus, R.P. Global Gene Expression Profiling in Escherichia coli K12: The Effects of Oxygen Availability and FNR. J. Biol. Chem. 2003, 278, 29837–29855. [Google Scholar] [CrossRef] [PubMed]
- Khoroshilova, N.; Popescu, C.; Munck, E.; Beinert, H.; Kiley, P.J. Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc. Natl. Acad. Sci. USA 1997, 94, 6087–6092. [Google Scholar] [CrossRef] [PubMed]
- Jervis, A.J.; Crack, J.C.; White, G.; Artymiuk, P.J.; Cheesman, M.R.; Thomson, A.J.; Le Brun, N.E.; Green, J. The O2 sensitivity of the transcription factor FNR is controlled by Ser24 modulating the kinetics of [4Fe-4S] to [2Fe-2S] conversion. Proc. Natl. Acad. Sci. USA 2009, 106, 4659–4664. [Google Scholar] [CrossRef] [PubMed]
- Ramseier, T. Cra and the control of carbon flux via metabolic pathways. Res. Microbiol. 1996, 147, 489–493. [Google Scholar] [CrossRef]
- Kochanowski, K.; Volkmer, B.; Gerosa, L.; Haverkorn van Rijsewijk, B.R.; Schmidt, A.; Heinemann, M. Functioning of a metabolic flux sensor in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Sauer, U. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 2003, 270, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Battesti, A.; Majdalani, N.; Gottesman, S. The RpoS-Mediated General Stress Response in Escherichia coli. Annu. Rev. Microbiol. 2011, 65, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Hantke, K. Iron and metal regulation in bacteria. Curr. Opin. Microbiol. 2001, 4, 172–177. [Google Scholar] [CrossRef]
- Sober, H.; Loach, P. Handbook of Biochemistry, Selected Data for Molecular Biology; The Chemical Rubber Co.: Cleveland, OH, USA, 1968; pp. J27–J34. [Google Scholar]
- Metzler, D.; Metzler, C. Biochemistry: The Chemical Reactions of Living Cells; Academic Press: New York, NY, USA, 2003; Volume 1, p. 301. [Google Scholar]
- Varghese, S.; Wu, A.; Park, S.; Imlay, K.R.C.; Imlay, J.A. Submicromolar hydrogen peroxide disrupts the ability of Fur protein to control free-iron levels in Escherichia coli. Mol. Microbiol. 2007, 64, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.A.; Marletta, M.A. Metal Binding Characteristics and Role of Iron Oxidation in the Ferric Uptake Regulator from Escherichia coli. Biochemistry 2005, 44, 13553–13559. [Google Scholar] [CrossRef] [PubMed]
- Bremer, H.; Dennis, P.; Ehrenberg, M. Free RNA polymerase and modeling global transcription in Escherichia coli. Biochimie 2003, 85, 597–609. [Google Scholar] [CrossRef]
- Saecker, R.M.; Record, M.T.; Dehaseth, P.L. Mechanism of bacterial transcription initiation: RNA polymerase—Promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011, 412, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, V.A.; Montelongo, L.R.; Farias, R.N.; Massa, E.M. Characterization of an NADH-Linked Cupric Reductase Activity from the Escherichia coli Respiratory Chain. Arch. Biochem. Biophys. 1999, 370, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, M.W.; Gennis, R.B. Demonstration of separate genetic loci encoding distinct membrane-bound respiratory NADH dehydrogenases in Escherichia coli. J. Bacteriol. 1993, 175, 3013–3019. [Google Scholar] [CrossRef] [PubMed]
- Bylund, F.; Collet, E.; Enfors, S.O.; Larsson, G. Substrate gradient formation in the large-scale bioreactor lowers cell yield and increases by-product formation. Bioprocess Eng. 1998, 18, 171–180. [Google Scholar] [CrossRef]
- Hardiman, T.; Lemuth, K.; Keller, M.A.; Reuss, M.; Siemann-Herzberg, M. Topology of the global regulatory network of carbon limitation in Escherichia coli. J. Biotechnol. 2007, 132, 359–374. [Google Scholar] [CrossRef] [PubMed]
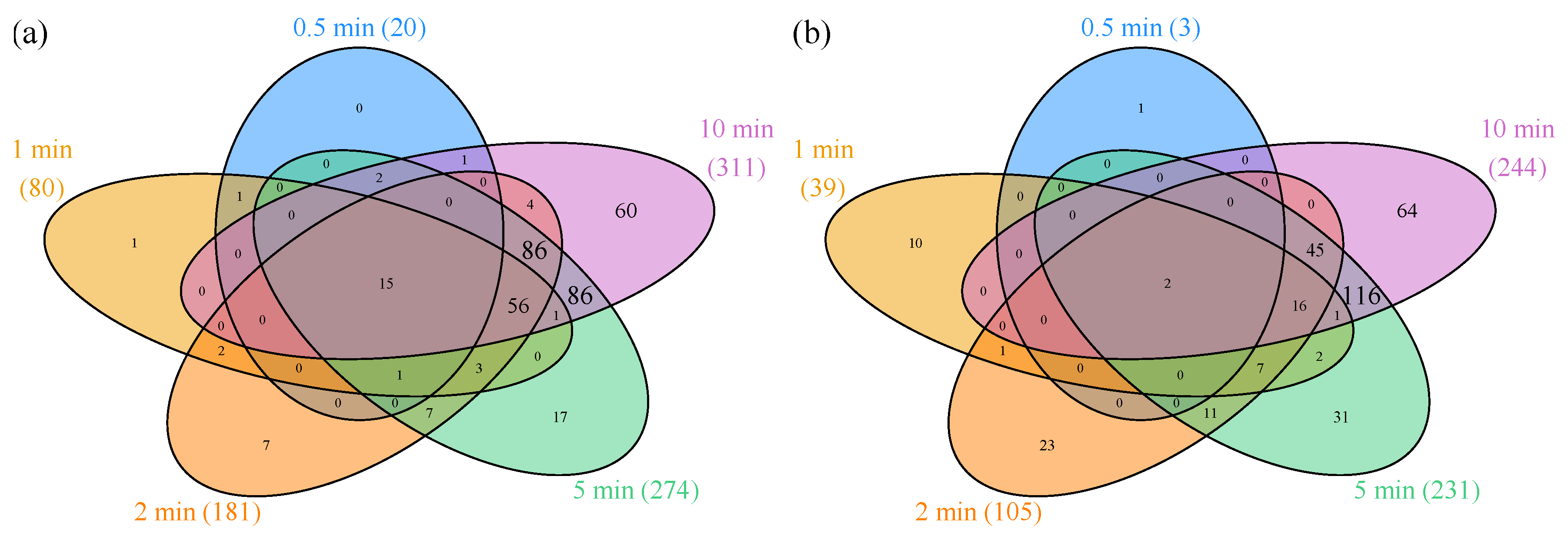
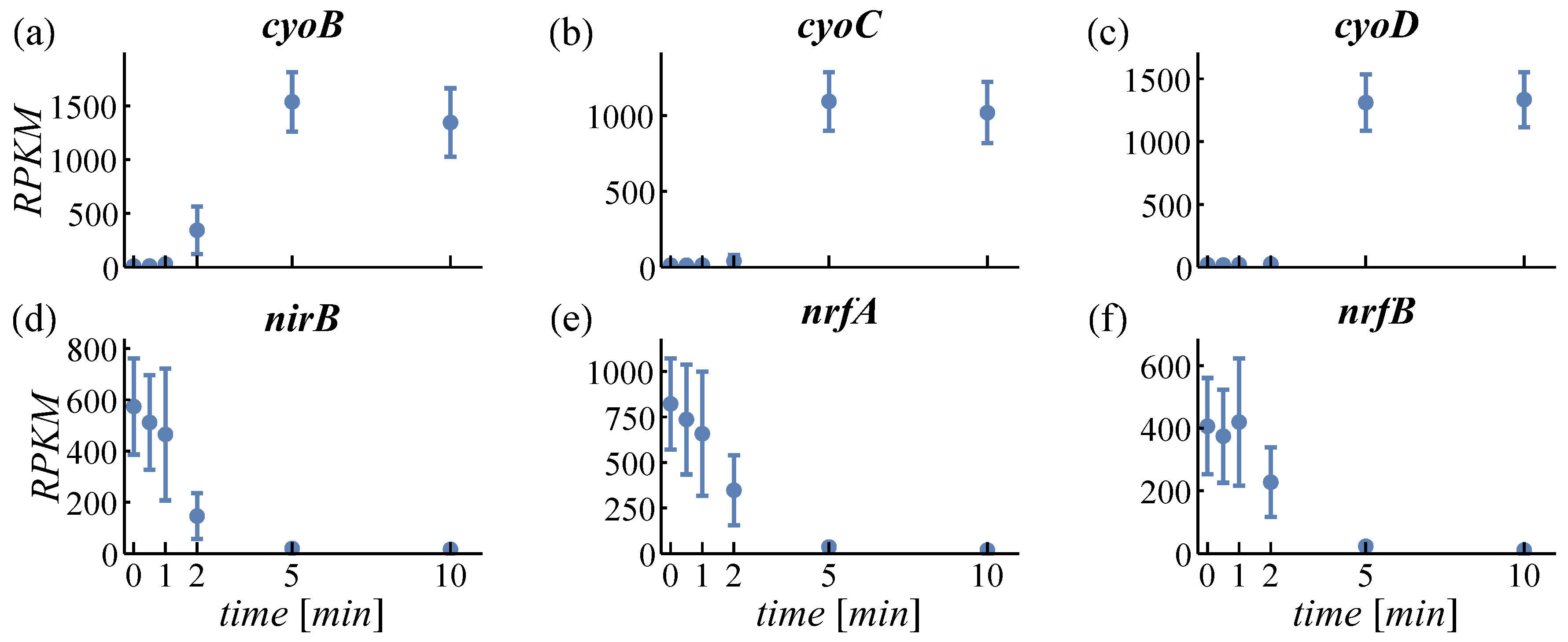
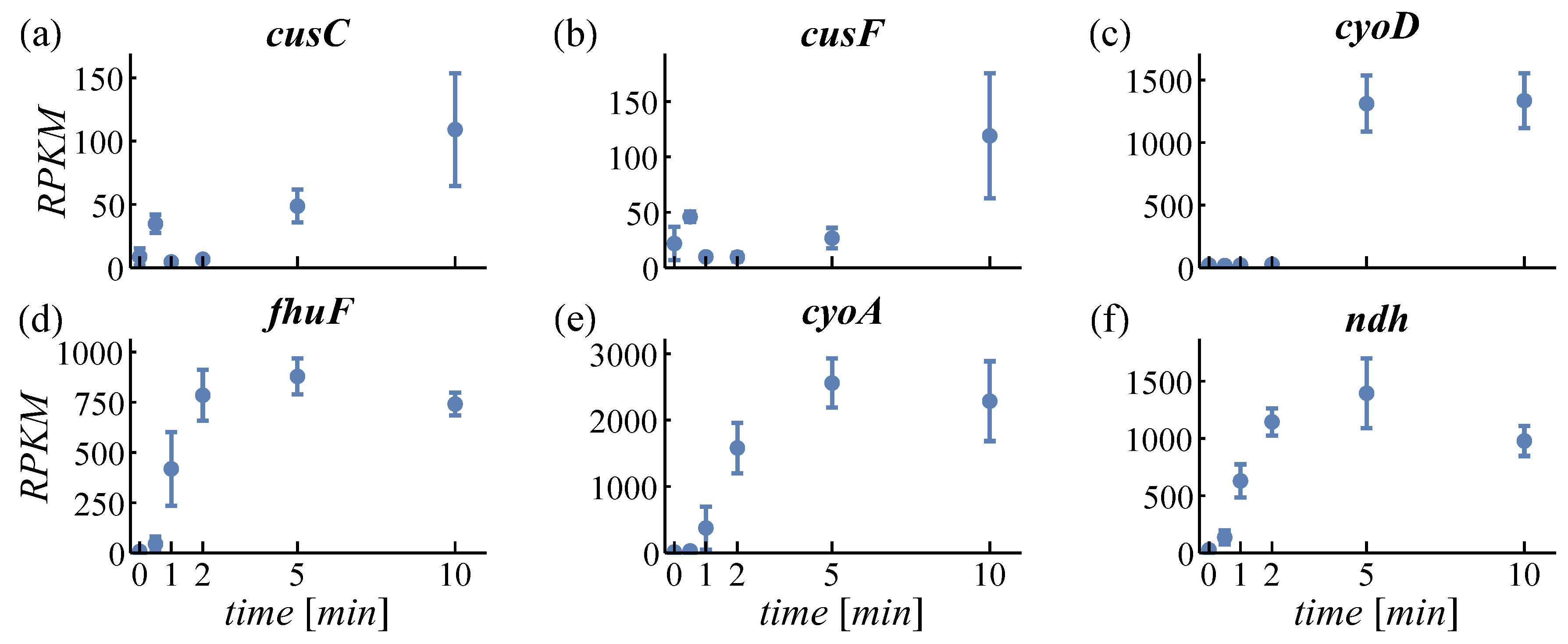
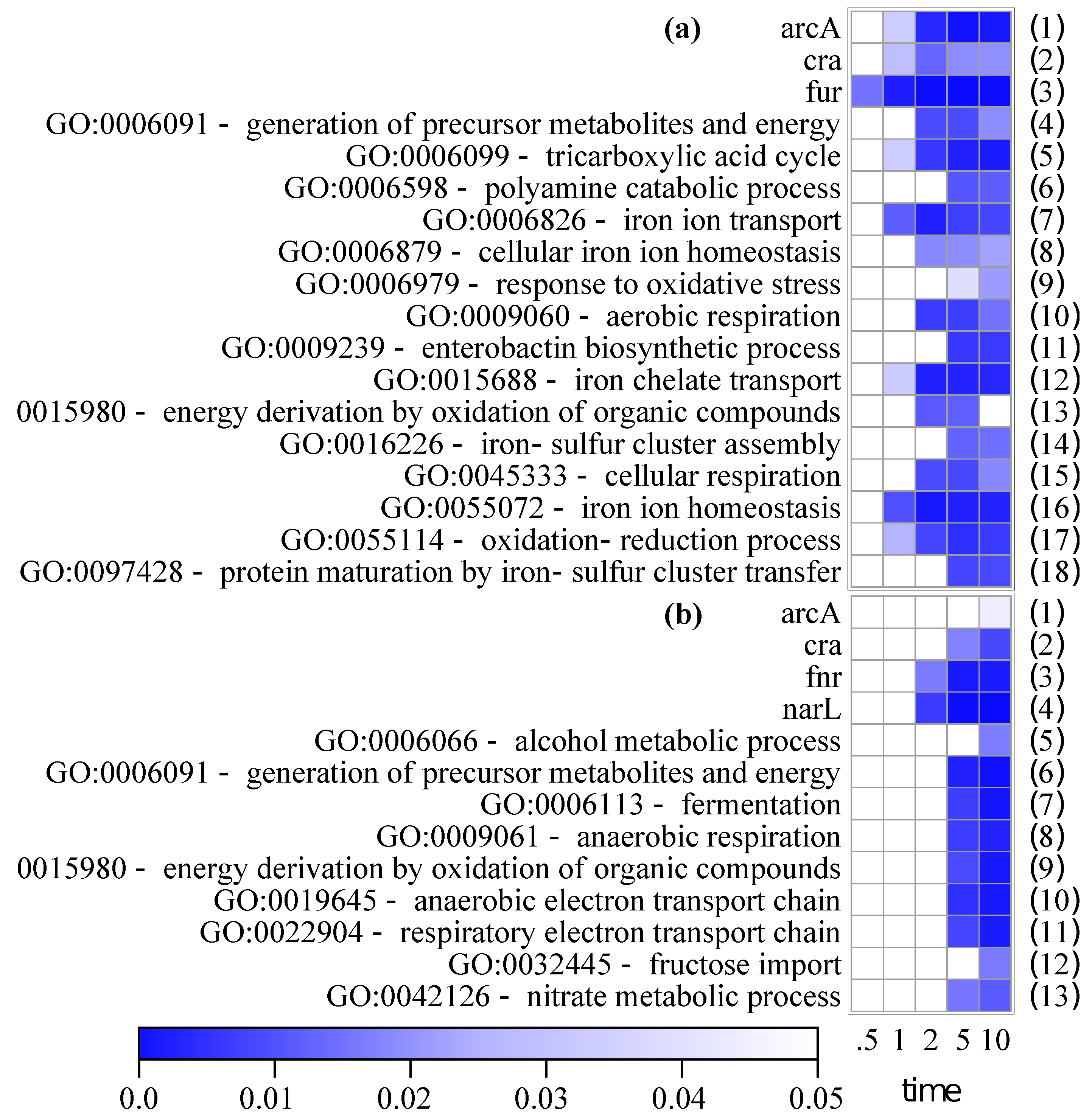

{kind=link}
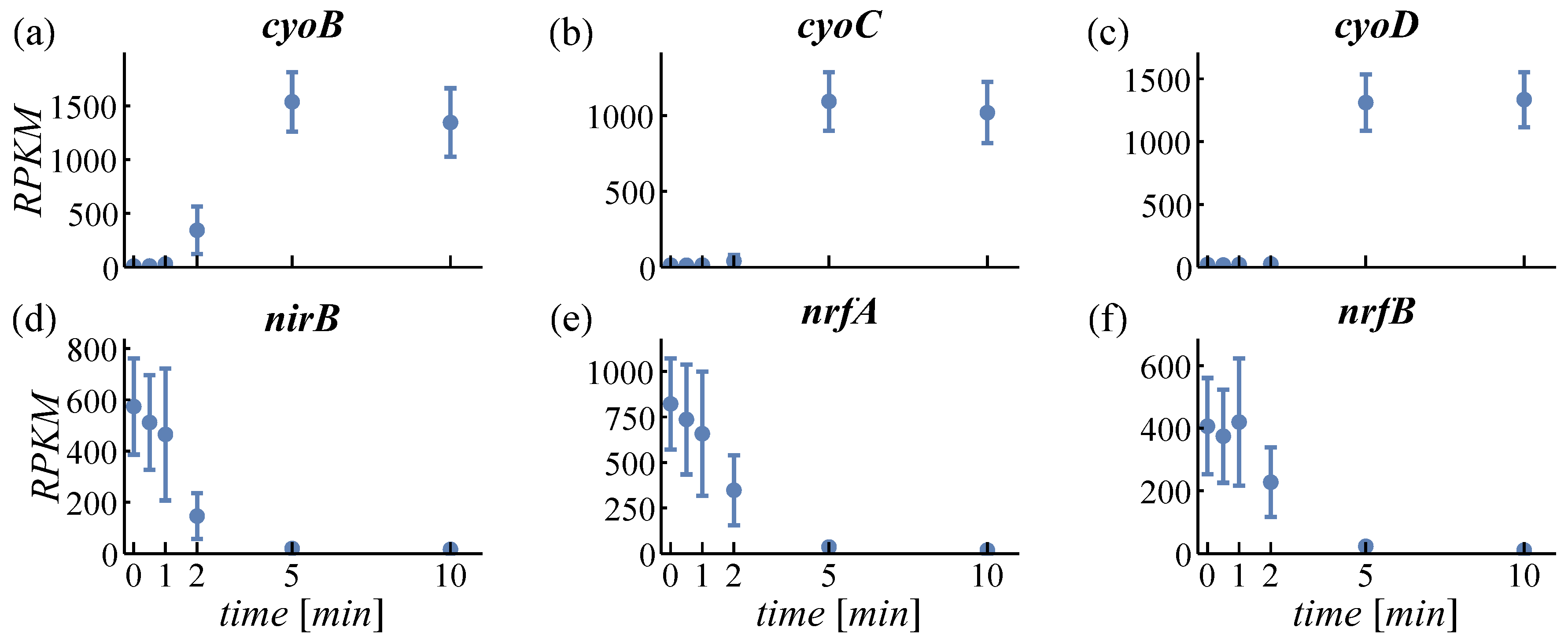{kind=link}
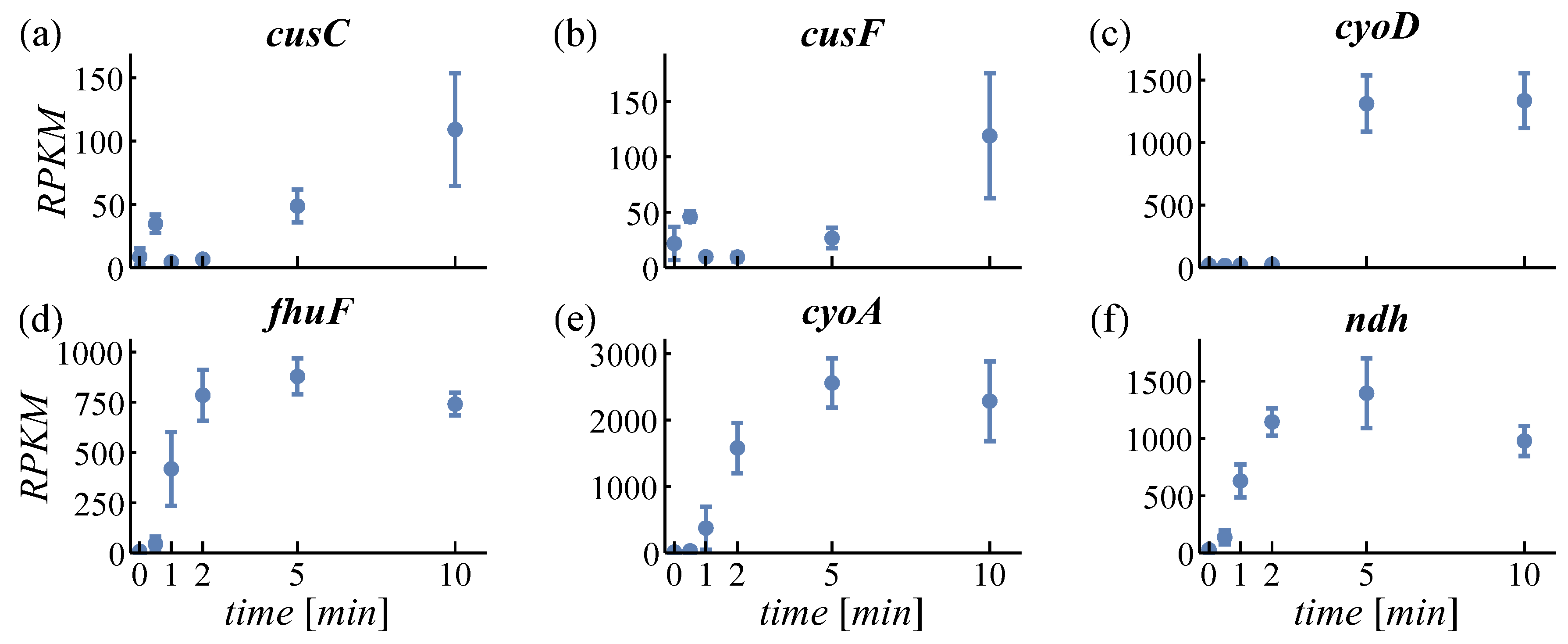{kind=link}
{kind=link}
{kind=link}
| ρ of TER | p Value | ρ of Delay Time | p Value | |
|---|---|---|---|---|
| upregulation | −0.084 | 0.43 | 0.37 | 2.7 × 10−4 |
| maximal FC | −0.17 | 0.10 | 0.416 | 7.2 × 10−6 |
| maximal RPKM | −0.10 | 0.36 | 0.05 | 0.61 |
| GC content | −0.11 | 0.32 | −0.04 | 0.73 |
| CAI | −0.23 | 0.03 | −0.10 | 0.35 |
| ITE | −0.25 | 0.02 | −0.15 | 0.14 |
| RNA folding energy | 0.17 | 0.10 | 0.12 | 0.25 |
| regulated by Cra * | −0.03 | 0.75 | −0.29 | 0.01 |
| regulated by FNR * | −0.13 | 0.21 | −0.21 | 0.04 |
| regulated by Fur-Fe * | −0.12 | 0.28 | 0.22 | 0.04 |
| controlled by sigma S | −0.15 | 0.15 | −0.22 | 0.04 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Von Wulffen, J.; Ulmer, A.; Jäger, G.; Sawodny, O.; Feuer, R. Rapid Sampling of Escherichia coli After Changing Oxygen Conditions Reveals Transcriptional Dynamics. Genes 2017, 8, 90. https://doi.org/10.3390/genes8030090
Von Wulffen J, Ulmer A, Jäger G, Sawodny O, Feuer R. Rapid Sampling of Escherichia coli After Changing Oxygen Conditions Reveals Transcriptional Dynamics. Genes. 2017; 8(3):90. https://doi.org/10.3390/genes8030090
Chicago/Turabian StyleVon Wulffen, Joachim, Andreas Ulmer, Günter Jäger, Oliver Sawodny, and Ronny Feuer. 2017. "Rapid Sampling of Escherichia coli After Changing Oxygen Conditions Reveals Transcriptional Dynamics" Genes 8, no. 3: 90. https://doi.org/10.3390/genes8030090
APA StyleVon Wulffen, J., Ulmer, A., Jäger, G., Sawodny, O., & Feuer, R. (2017). Rapid Sampling of Escherichia coli After Changing Oxygen Conditions Reveals Transcriptional Dynamics. Genes, 8(3), 90. https://doi.org/10.3390/genes8030090