
Systematic Identification and Assessment of Therapeutic Targets for Breast Cancer Based on Genome-Wide RNA Interference Transcriptomes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.1.1. Gene Expression and Methylation Data
2.1.2. Clinical Survival Data
2.1.3. LINCS Dataset
2.1.4. Cancer Gene Census
2.1.5. Drug Target
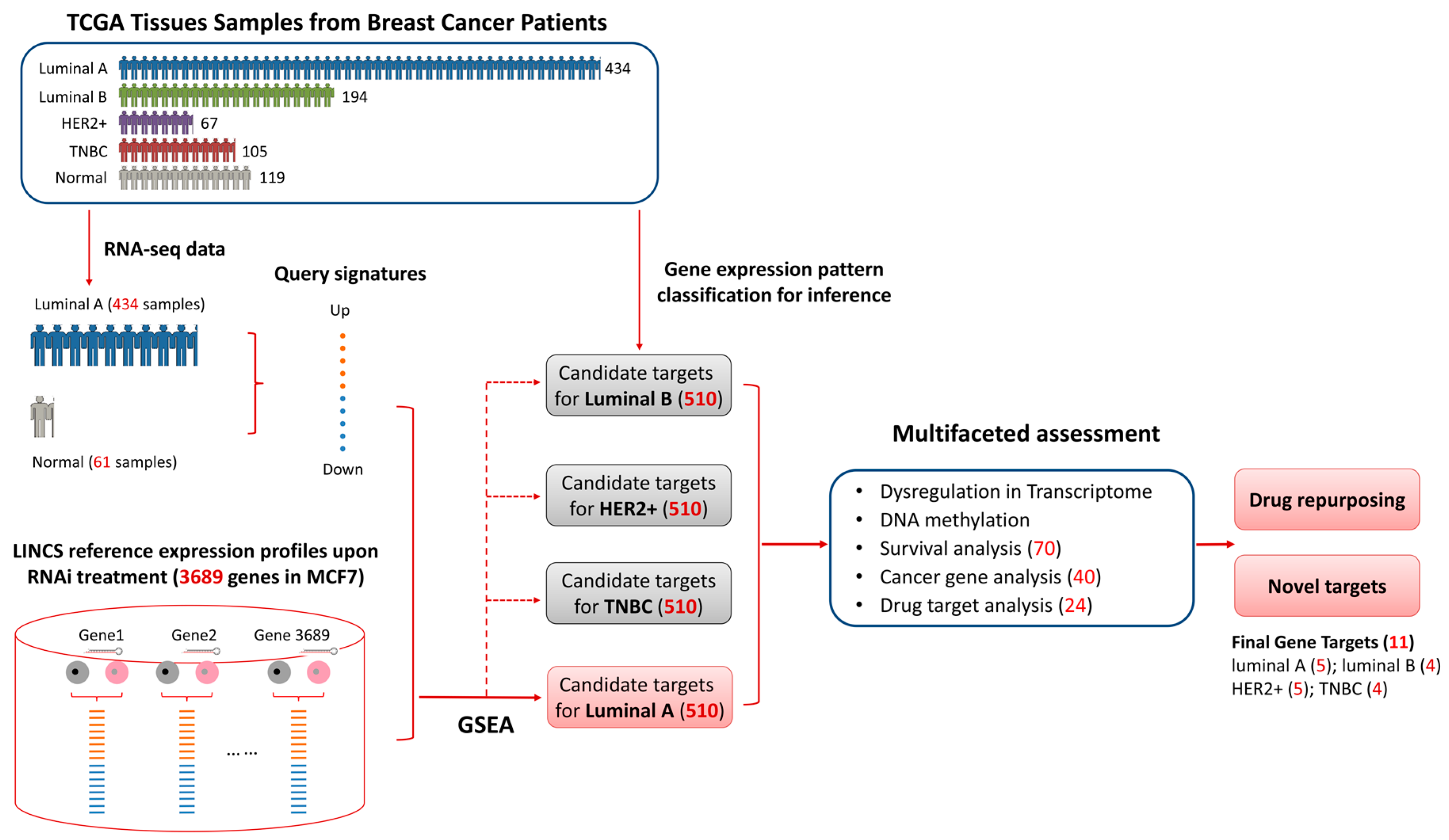
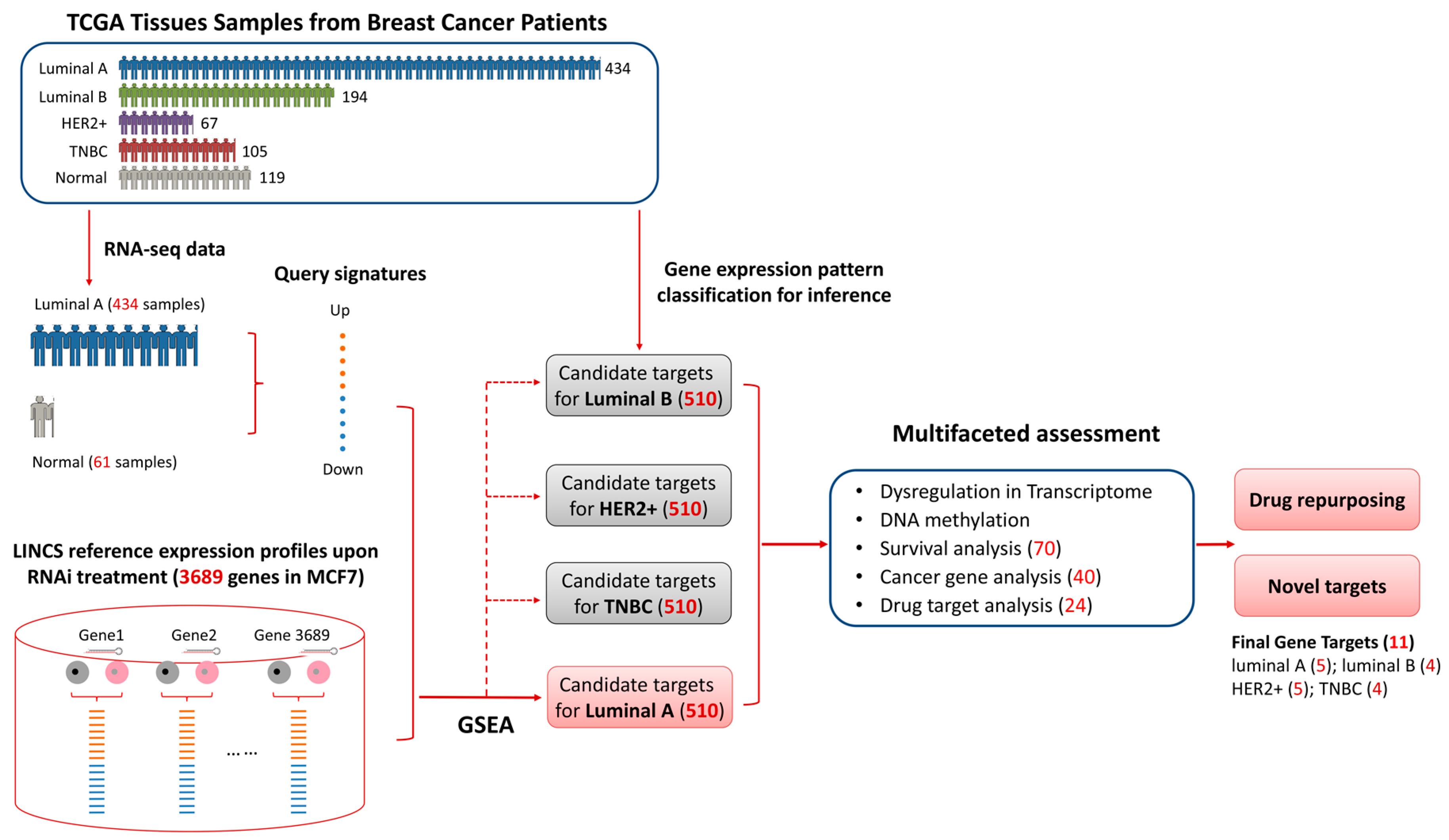
2.2. Candiate Target Idenfication Pipeline
2.3. Differential Expression Analayis
2.4. Methylation Analysis
2.5. Survival Analysis
2.6. Gene Functional Annotation
3. Results
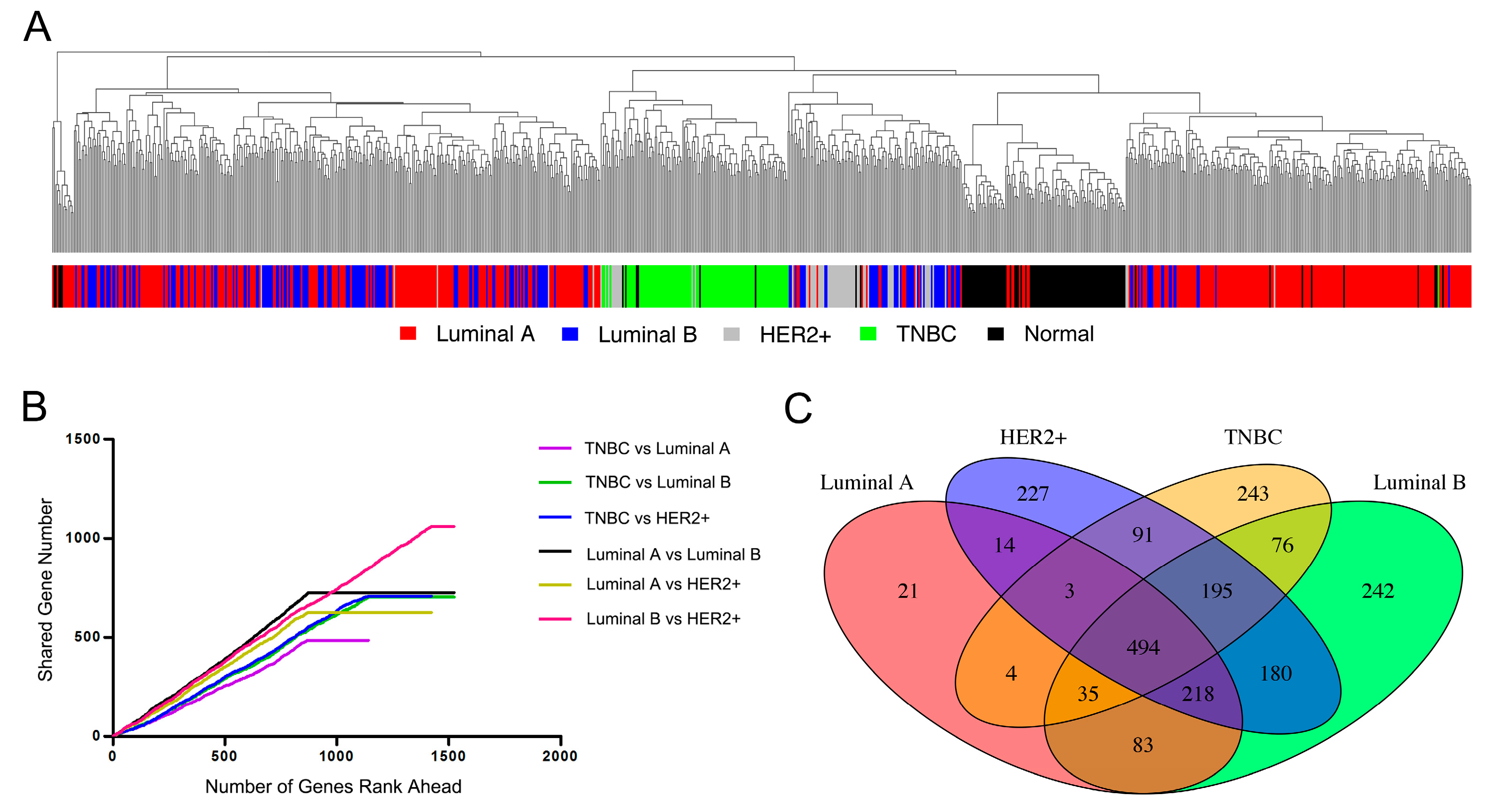
3.1. Characteristic Pattern of Gene Expression among Breast Cancer Subtypes
3.2. Identifying Candidate Gene Targets for Breast Cancer Luminal A Subtype
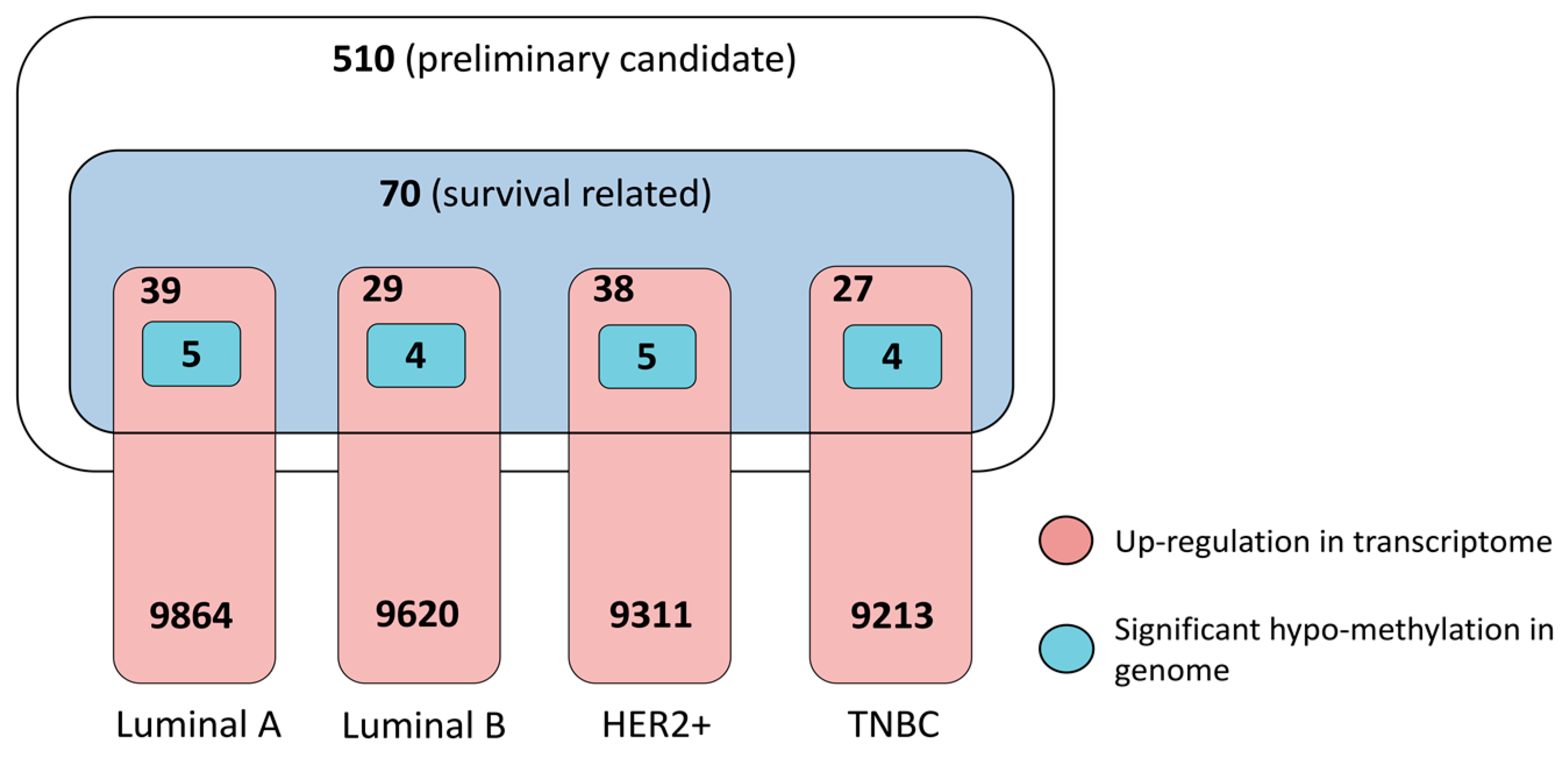
3.3. Multifaceted Assessment for Priotizing Theraperutic Targets for Four Breast Cancer Subtypes
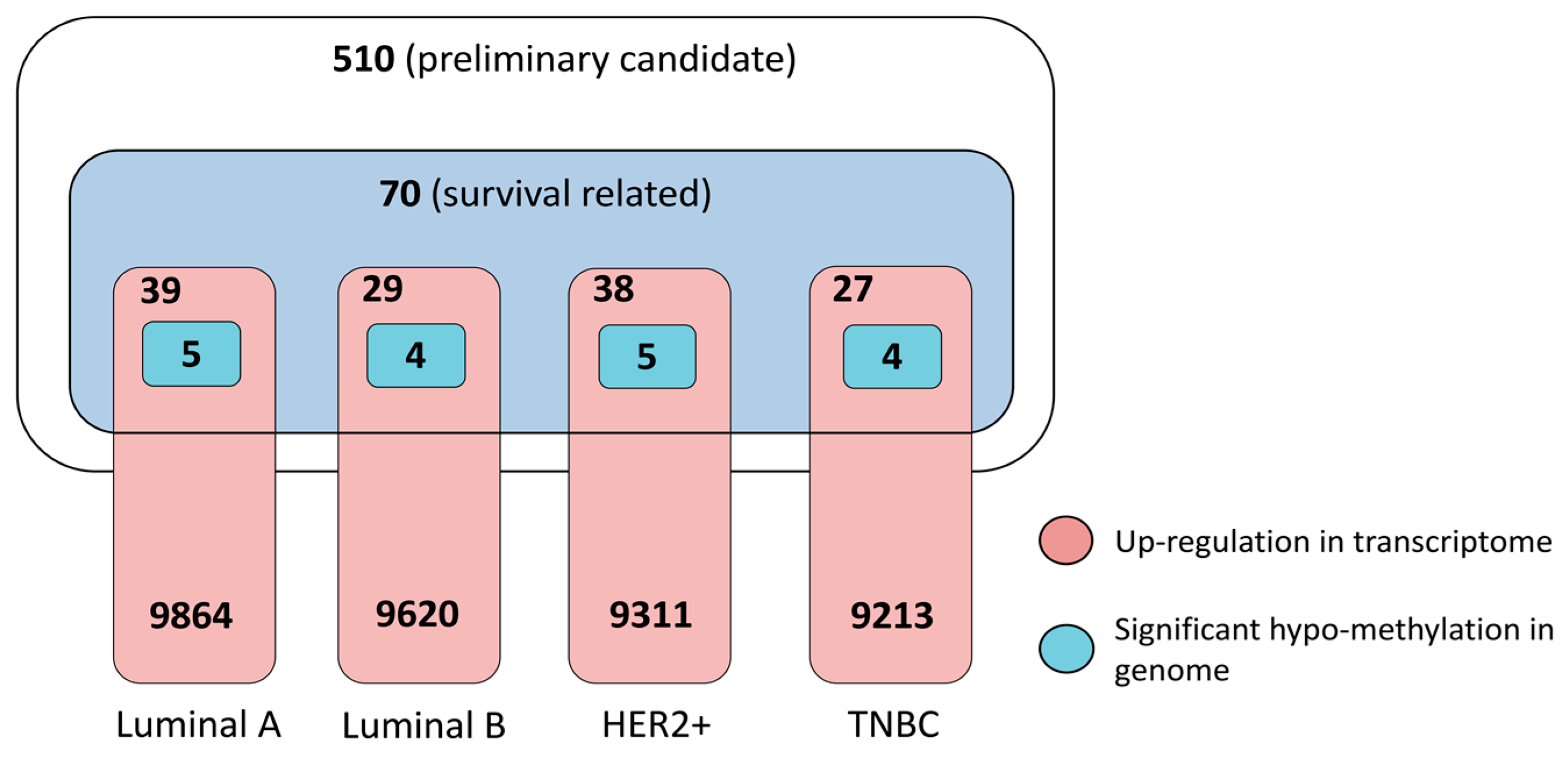
3.3.1. Dysregulation in Transcriptome and Determinant Roles in Survival
3.3.2. Pathogenic Importance in Cancer and Methylation at Genomic Level
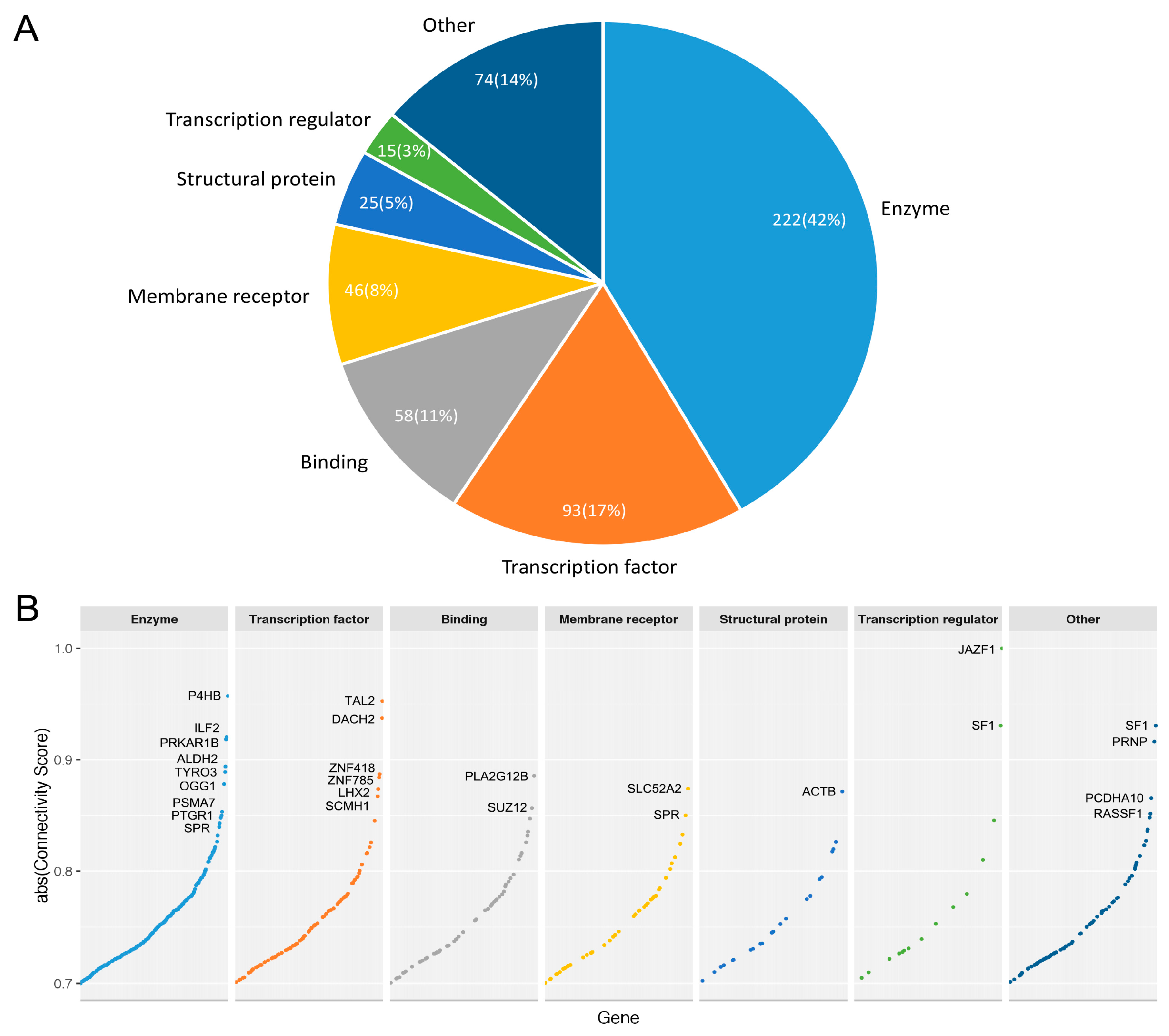
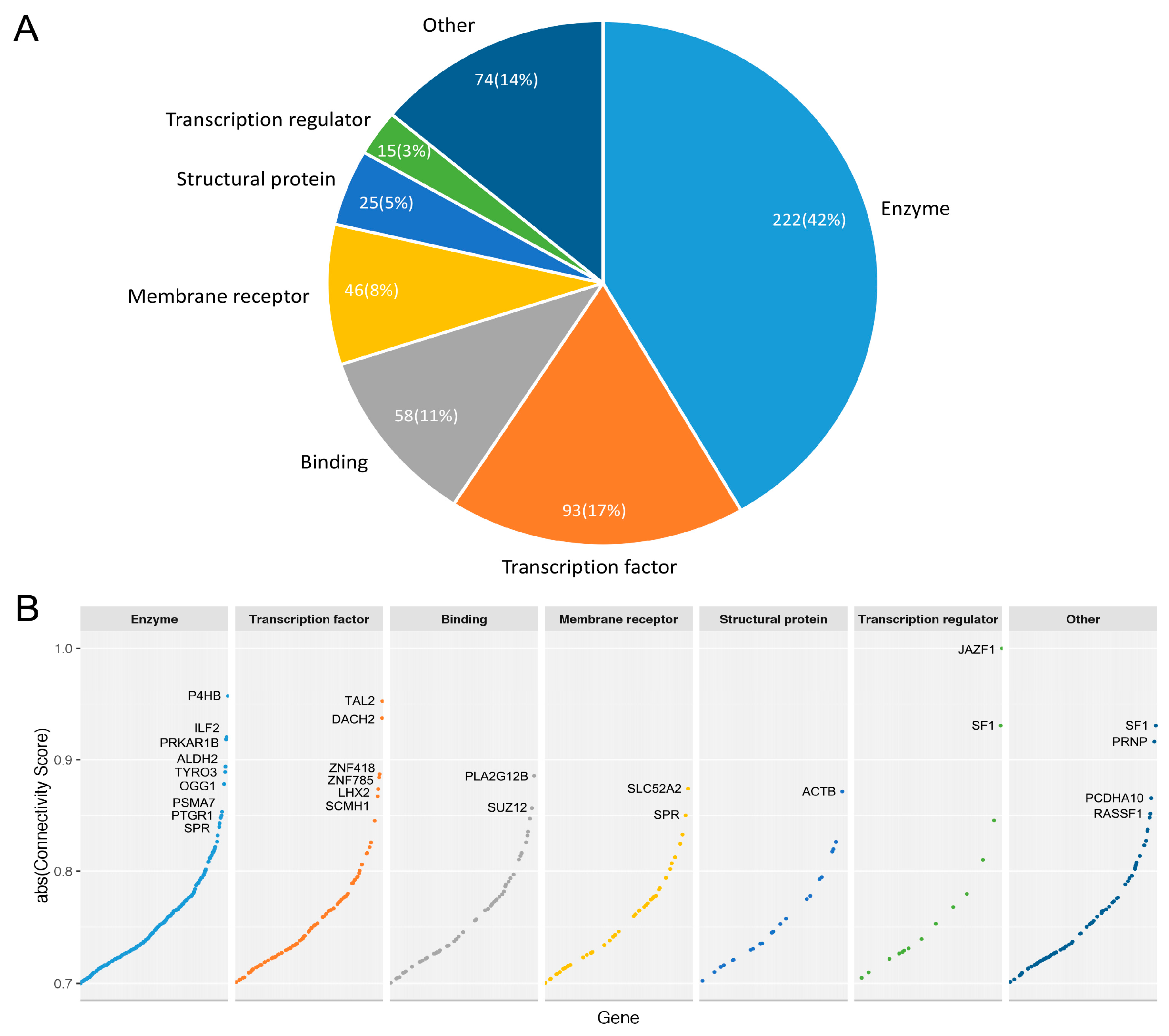
3.3.3. Druggable Candidate Genes
3.3.4. Prioritized Therapeutic Targets for Four Breast Cancer Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TNBC | Triple Negative Breast Cancer |
| LINCS | Library of Integrated Network-based Cellular Signatures |
| GSEA | Gene Set Enrichment Analysis |
| TCGA | The Cancer Genomics Atlas |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted therapies for triple-negative breast cancer: Combating a stubborn disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Untch, M.; Blohmer, J.U.; Costa, S.D.; Eidtmann, H.; Fasching, P.A.; Gerber, B.; Eiermann, W.; Hilfrich, J.; Huober, J.; et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 2012, 30, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Eirew, P.; Mullaly, S.C.; Aparicio, S. The omics of triple-negative breast cancers. Clin. Chem. 2014, 60, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Speers, C.; Tsimelzon, A.; Sexton, K.; Herrick, A.M.; Gutierrez, C.; Culhane, A.; Quackenbush, J.; Hilsenbeck, S.; Chang, J.; Brown, P. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin. Cancer Res. 2009, 15, 6327–6340. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Simpson, P.T.; Sanus, J.M.; Klein, K.; Kalimutho, M.; Shi, W.; Miranda, M.; Kutasovic, J.; Raghavendra, A.; Madore, J.; et al. Meta-analysis of the global gene expression profile of triple-negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis 2014, 3, e100. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.T.; Perez, E.M.; Hernandez, D.; Miller, C.P.; Haas, K.M.; Irie, H.Y.; Lee, S.I.; Blau, C.A.; Villen, J. The proteomic landscape of triple-negative breast cancer. Cell Rep. 2015, 11, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Osmanbeyoglu, H.U.; Pelossof, R.; Bromberg, J.F.; Leslie, C.S. Linking signaling pathways to transcriptional programs in breast cancer. Genome Res. 2014, 24, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hedayati, M.; Hobbs, R.F.; Shao, C.; Bruchertseifer, F.; Morgenstern, A.; Deweese, T.L.; Sgouros, G. Targeting aberrant DNA double-strand break repair in triple-negative breast cancer with alpha-particle emitter radiolabeled anti-egfr antibody. Mol. Cancer Ther. 2013, 12, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Ueno, N.T.; Zhang, D. Targeting egfr in triple negative breast cancer. J. Cancer 2011, 2, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Dent, S.F. The role of vegf in triple-negative breast cancer: Where do we go from here? Ann. Oncol. 2009, 20, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Furth, P.A. Stat signaling in different breast cancer sub-types. Mol. Cell. Endocrinol. 2014, 382, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.; Banerji, S. Molecular pathways: Pi3k pathway targets in triple-negative breast cancers. Clin. Cancer Res. 2013, 19, 3738–3744. [Google Scholar] [CrossRef] [PubMed]
- Yunokawa, M.; Koizumi, F.; Kitamura, Y.; Katanasaka, Y.; Okamoto, N.; Kodaira, M.; Yonemori, K.; Shimizu, C.; Ando, M.; Masutomi, K.; et al. Efficacy of everolimus, a novel mtor inhibitor, against basal-like triple-negative breast cancer cells. Cancer Sci. 2012, 103, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.R.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting akt3 signaling in triple-negative breast cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.C.; Esparis-Ogando, A.; Re-Louhau, M.F.; Seoane, S.; Abad, M.; Calero, R.; Ocana, A.; Pandiella, A. Active kinase profiling, genetic and pharmacological data define mtor as an important common target in triple-negative breast cancer. Oncogene 2014, 33, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant brca1 protein confers parp inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [PubMed]
- Crown, J.; O′Shaughnessy, J.; Gullo, G. Emerging targeted therapies in triple-negative breast cancer. Ann. Oncol. 2012, 23, vi56–vi65. [Google Scholar] [CrossRef] [PubMed]
- Jamdade, V.S.; Sethi, N.; Mundhe, N.A.; Kumar, P.; Lahkar, M.; Sinha, N. Therapeutic targets of triple-negative breast cancer: A review. Br. J. Pharmacol. 2015, 172, 4228–4237. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Nanda, R. Concepts and targets in triple-negative breast cancer: Recent results and clinical implications. Ther. Adv. Med. Oncol. 2016, 8, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Li, C.; Xing, Z.; Hu, Q.; Liang, K.; Han, L.; Wang, C.; Hawke, D.H.; Wang, S.; Zhang, Y.; et al. The link-a lncrna activates normoxic hif1alpha signalling in triple-negative breast cancer. Nat. Cell Biol. 2016, 18, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-negative breast cancer: Molecular subtypes and new targets for therapy. Am. Soc. Clin. Oncol. Educ. Book 2015. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lopez-Berestein, G.; Calin, G.A.; Sood, A.K. RNAi therapies: Drugging the undruggable. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Sandoghchian Shotorbani, S.; Baradaran, B. RNA interference and its role in cancer therapy. Adv. Pharm. Bull. 2014, 4, 313–321. [Google Scholar] [PubMed]
- Abdelrahim, M.; Safe, S.; Baker, C.; Abudayyeh, A. RNAi and cancer: Implications and applications. J. RNAi Gene Silencing 2006, 2, 136–145. [Google Scholar] [PubMed]
- Trevino, V.; Falciani, F.; Barrera-Saldana, H.A. DNA microarrays: A powerful genomic tool for biomedical and clinical research. Mol. Med. 2007, 13, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Peck, D.; Crawford, E.D.; Ross, K.N.; Stegmaier, K.; Golub, T.R.; Lamb, J. A method for high-throughput gene expression signature analysis. Genome Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sanborn, J.Z.; Benz, S.; Szeto, C.; Hsu, F.; Kuhn, R.M.; Karolchik, D.; Archie, J.; Lenburg, M.E.; Esserman, L.J.; et al. The ucsc cancer genomics browser. Nat. Methods 2009, 6, 239–240. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. Drugbank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Poudel, S.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Panther version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016, 44, D336–D342. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.B.; Prendes, M.J.; Sternberg, H.; Kidd, J.L.; Funk, W.D.; Wagner, J.; West, M.D. COL10A1 expression is elevated in diverse solid tumor types and is associated with tumor vasculature. Future Oncol. 2012, 8, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Yu, J.C.; Wang, H.W.; Huang, C.S.; Shieh, J.C.; Fu, Y.P.; Chang, C.W.; Wu, P.E.; Shen, C.Y. The clinical implications of MMP-11 and CK-20 expression in human breast cancer. Clin. Chim. Acta 2010, 411, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Blaser, H.; Gorrini, C.; Lin, D.C.; Elia, A.J.; Wakeham, A.; Haider, S.; Boutros, P.C.; Mason, J.M.; Miller, N.A.; et al. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene 2014, 33, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, M.M.; Benjamin, D.I.; Ji, X.; Le Scolan, E.; Louie, S.M.; Shieh, A.; Green, M.; Narasimhalu, T.; Morris, P.J.; Luo, K.; et al. Metabolic profiling reveals PAFAH1B3 as a critical driver of breast cancer pathogenicity. Chem. Biol. 2014, 21, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, C.; Li, Q.; Xie, J.; Chen, T.; Tan, Y.; Wu, C.; Jiang, J. The role of kif4a in doxorubicin-induced apoptosis in breast cancer cells. Mol. Cells 2014, 37, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Jones, P.A. DNA methylation and breast carcinogenesis. Oncogene 2002, 21, 5462–5482. [Google Scholar] [CrossRef] [PubMed]
- Pakneshan, P.; Szyf, M.; Farias-Eisner, R.; Rabbani, S.A. Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J. Biol. Chem. 2004, 279, 31735–31744. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of beta-value and m-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Gendler, S.J. MUC1, the renaissance molecule. J. Mammary Gland Biol. Neoplasia 2001, 6, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. Muc1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Pietersz, G.A.; McKenzie, I.F. Muc1 and breast cancer. Curr. Opin. Mol. Ther. 1999, 1, 98–103. [Google Scholar] [PubMed]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. Xbp1 promotes triple-negative breast cancer by controlling the hif1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J.; Cassetta, L.; Qian, B.Z.; Lewkowich, I.; Li, J.F.; Stefater, J.A.; Smith, A.N.; Wiechmann, L.S.; Wang, Y.; Pollard, J.W.; et al. Myeloid WNT7B mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014, 74, 2962–2973. [Google Scholar] [CrossRef] [PubMed]
- Rangel, L.B.; Agarwal, R.; Sherman-Baust, C.A.; Mello-Coelho, V.; Pizer, E.S.; Ji, H.; Taub, D.D.; Morin, P.J. Anomalous expression of the HLA-DR alpha and beta chains in ovarian and other cancers. Cancer Biol. Ther. 2004, 3, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Zhu, H.; Shao, L.; Chen, Y.W. Ankyrin-Tiam1 interaction promotes Rac1 signaling and metastatic breast tumor cell invasion and migration. J. Cell. Biol. 2000, 150, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, G.M.; Sizemore, S.T.; Seachrist, D.D.; Keri, R.A. GABA(A) receptor pi (GABRP) stimulates basal-like breast cancer cell migration through activation of extracellular-regulated kinase 1/2 (ERK1/2). J. Biol. Chem. 2014, 289, 24102–24113. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.R. Human cancer cell lines: Fact and fantasy. Nature Rev. Mol. Cell Biol. 2000, 1, 233–236. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Luminal A | Luminal B | HER2+ | TNBC | Protein Name | No. of Targeted Drugs | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CS | logFC | CS | logFC | CS | logFC | CS | logFC | ||||
| MUC1 | mucin 1, cell surface associated | −0.75 | 3.11 | −0.75 | 1.97 | −0.75 | 2.33 | −0.75 | 0.43 | Mucin-1 | 0 |
| HLA-DRA | major histocompatibility complex, class II, DR alpha | −0.73 | 0.31 | - | - | −0.73 | 0.8 | - | - | HLA class II histocompatibility antigen, DR alpha chain | 0 |
| WNT7B | Wnt family member 7B | −0.73 | 2.14 | −0.73 | 1.96 | - | - | - | - | Protein Wnt-7b | 0 |
| XBP1 | X-box binding protein 1 | −0.72 | 1.58 | −0.72 | 1.68 | −0.72 | 0.94 | - | - | X-box-binding protein 1 | 0 |
| EFCAB2 | EF-hand calcium binding domain 2 | −0.72 | 0.08 | - | - | - | - | - | - | EF-hand calcium-binding domain-containing protein 2 | 0 |
| ATG16L2 | autophagy related 16 like 2 | - | - | −0.77 | 0.19 | - | - | - | - | Autophagy-related protein 16-2 | 0 |
| C1QTNF6 | C1q and tumor necrosis factor related protein 6 | - | - | - | - | −0.77 | 2.23 | - | - | Complement C1q tumor necrosis factor-related protein 6 | 0 |
| NDUFS6 | NADH: ubiquinone oxidoreductase subunit S6 | - | - | - | - | −0.77 | 1.34 | - | - | NADH dehydrogenase [ubiquinone] iron-sulfur protein 6, mitochondrial | 1 |
| CHERP | calcium homeostasis endoplasmic reticulum protein | - | - | - | - | - | - | −0.77 | 0.46 | Calcium homeostasis endoplasmic reticulum protein | 0 |
| TIAM1 | T-cell lymphoma invasion and metastasis 1 | - | - | - | - | - | - | −0.71 | 0.68 | T-lymphoma invasion and metastasis-inducing protein 1 | 0 |
| GABRP | gamma-aminobutyric acid type A receptor pi subunit | - | - | - | - | - | - | −0.7 | 1.5 | Gamma-aminobutyric acid receptor subunit pi | 41 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Yin, X.; Zhong, J.; Guan, N.; Luo, Z.; Min, L.; Yao, X.; Bo, X.; Dai, L.; Bai, H. Systematic Identification and Assessment of Therapeutic Targets for Breast Cancer Based on Genome-Wide RNA Interference Transcriptomes. Genes 2017, 8, 86. https://doi.org/10.3390/genes8030086
Liu Y, Yin X, Zhong J, Guan N, Luo Z, Min L, Yao X, Bo X, Dai L, Bai H. Systematic Identification and Assessment of Therapeutic Targets for Breast Cancer Based on Genome-Wide RNA Interference Transcriptomes. Genes. 2017; 8(3):86. https://doi.org/10.3390/genes8030086
Chicago/Turabian StyleLiu, Yang, Xiaoyao Yin, Jing Zhong, Naiyang Guan, Zhigang Luo, Lishan Min, Xing Yao, Xiaochen Bo, Licheng Dai, and Hui Bai. 2017. "Systematic Identification and Assessment of Therapeutic Targets for Breast Cancer Based on Genome-Wide RNA Interference Transcriptomes" Genes 8, no. 3: 86. https://doi.org/10.3390/genes8030086
APA StyleLiu, Y., Yin, X., Zhong, J., Guan, N., Luo, Z., Min, L., Yao, X., Bo, X., Dai, L., & Bai, H. (2017). Systematic Identification and Assessment of Therapeutic Targets for Breast Cancer Based on Genome-Wide RNA Interference Transcriptomes. Genes, 8(3), 86. https://doi.org/10.3390/genes8030086