
The Intra-S Checkpoint Responses to DNA Damage


Abstract
:1. Introduction
2. Source of Damage
2.1. Intrinsic Sources of Damage
2.2. Extrinsic Sources of Damage
3. The Intra-S Checkpoint
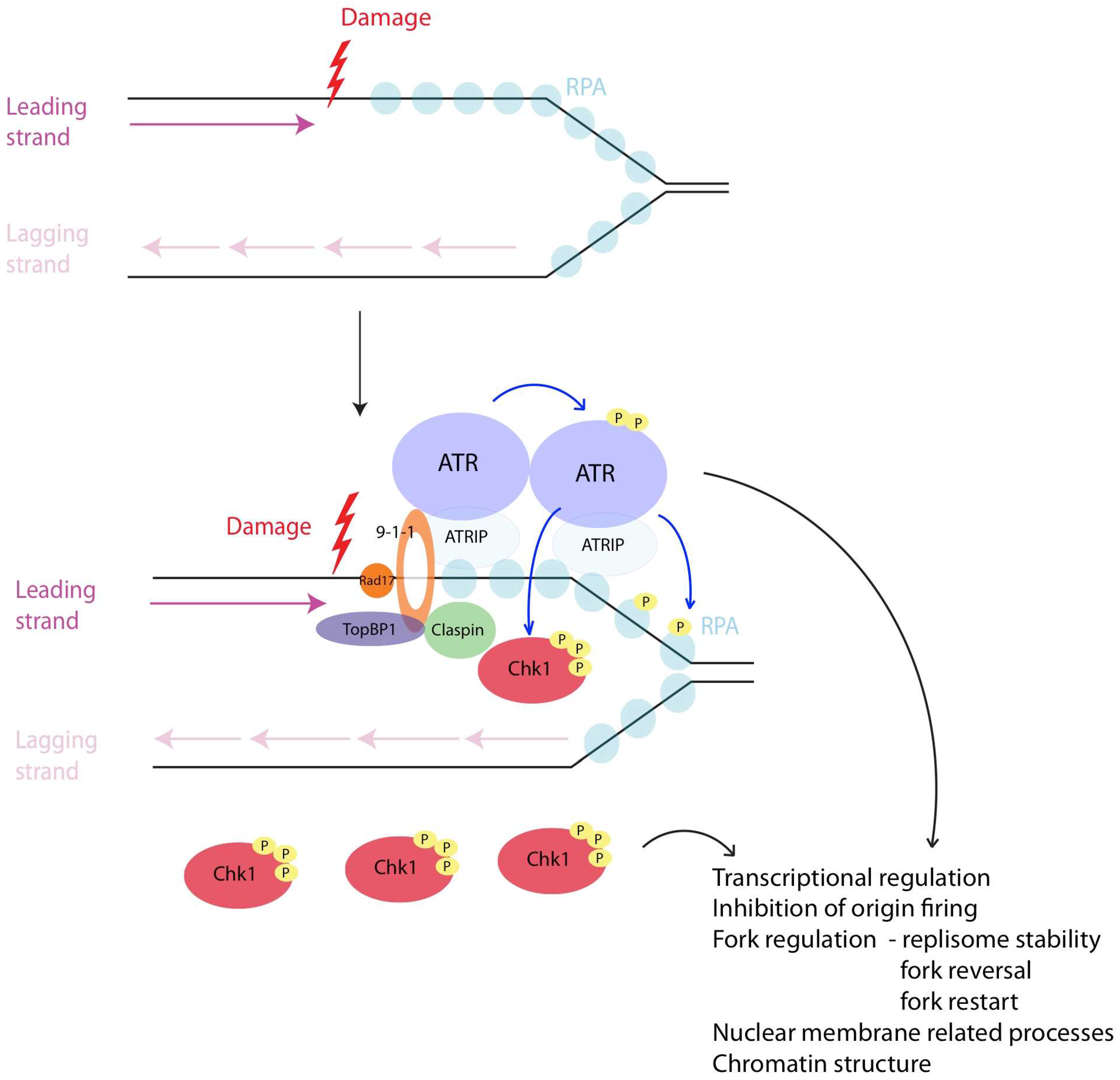
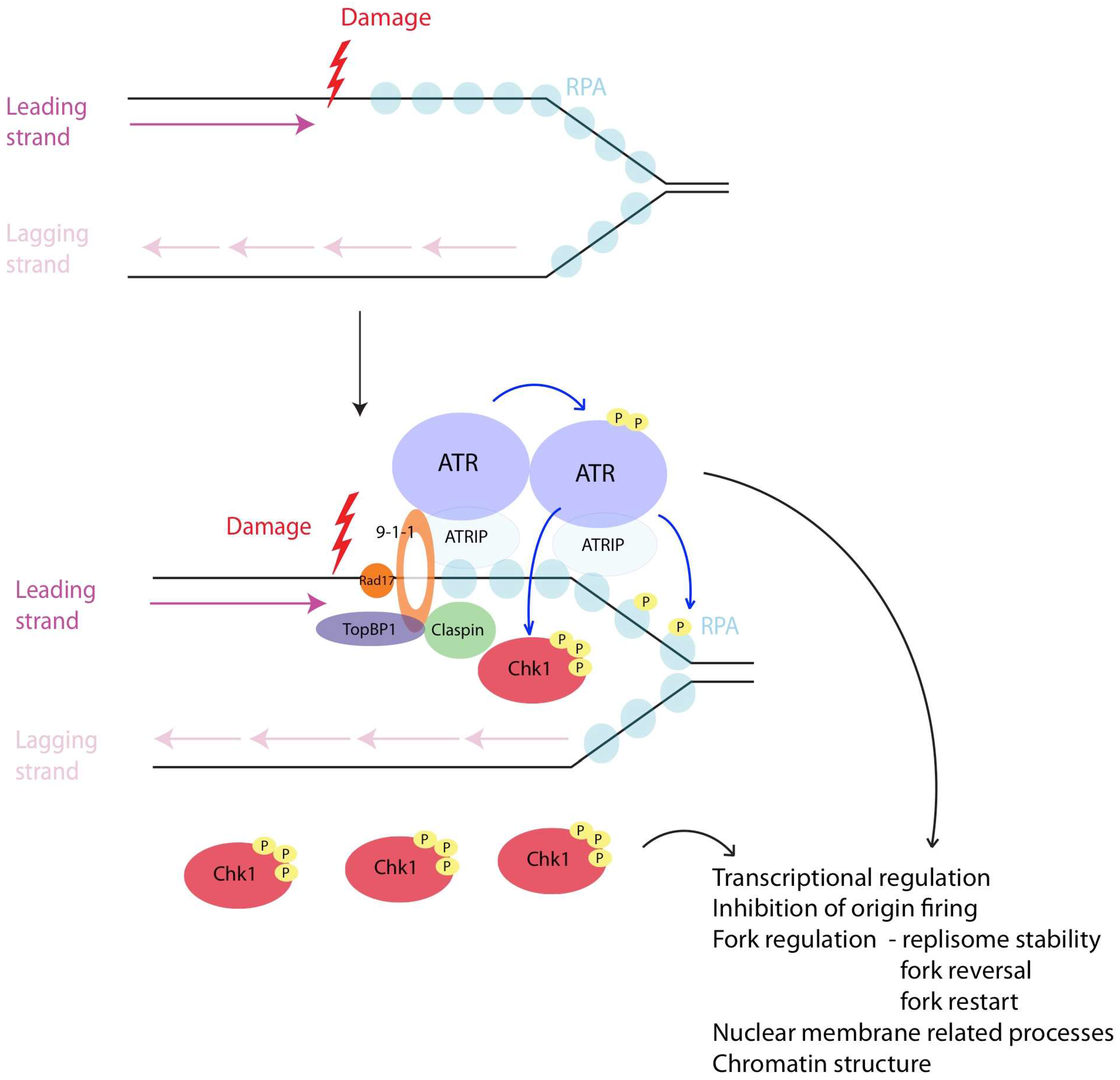
4. Detection of Lesion During S Phase
5. Intra S-Checkpoint Activation
5.1. The Structure Necessary for Checkpoint Activation
5.2. The Factors Necessary for Checkpoint Activation
5.3. Downstream Effectors of Checkpoint Activation
6. Strength of Checkpoint Activation
7. Downstream Targets
8. Transcriptional Regulation by the Checkpoint
8.1. G1/S Regulation
8.2. RNR Regulation
9. Regulation of Replication Kinetics by the Checkpoint
10. Inhibition of Origin Firing
10.1. Activation of Dormant Origins
11. Fork Regulation
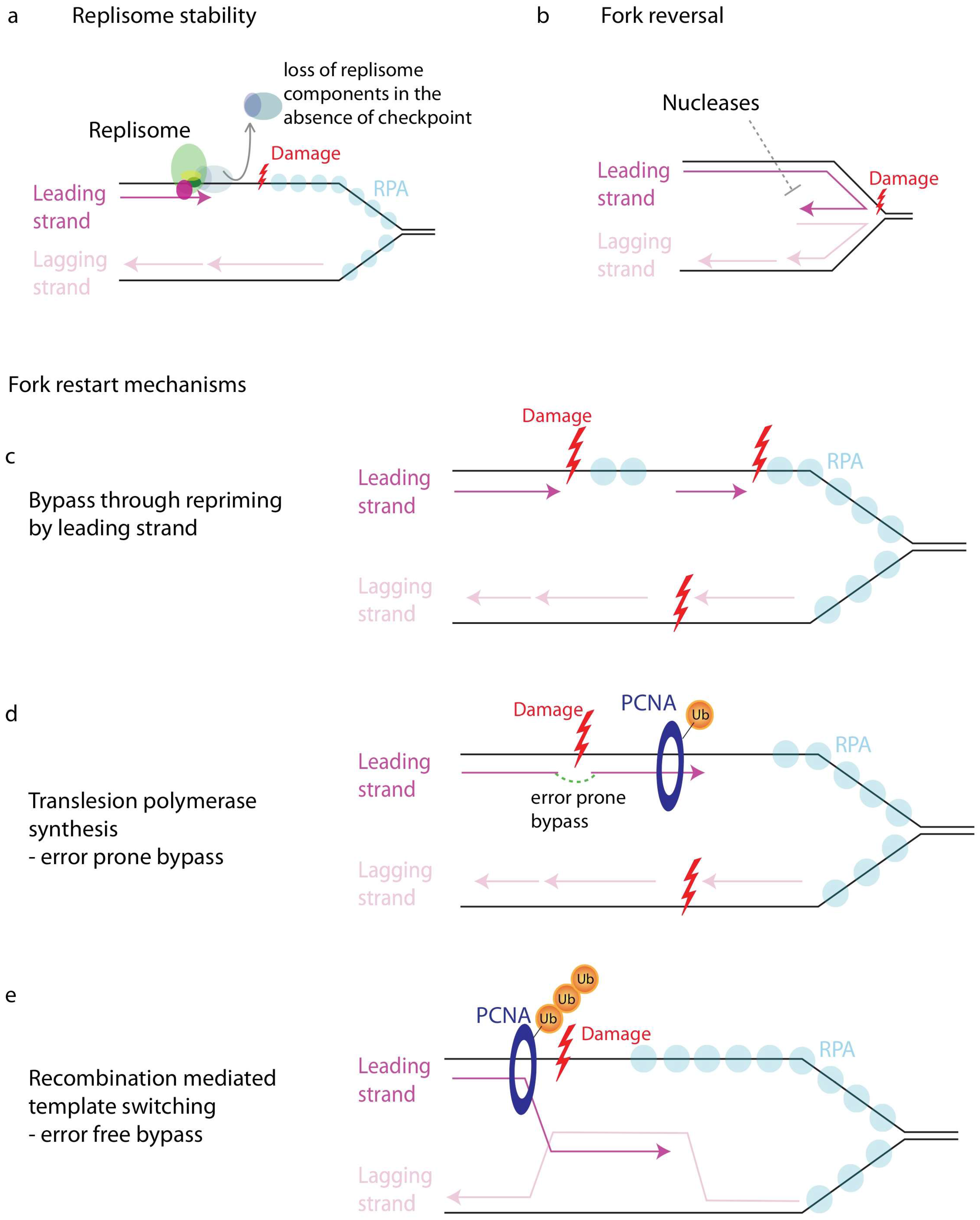
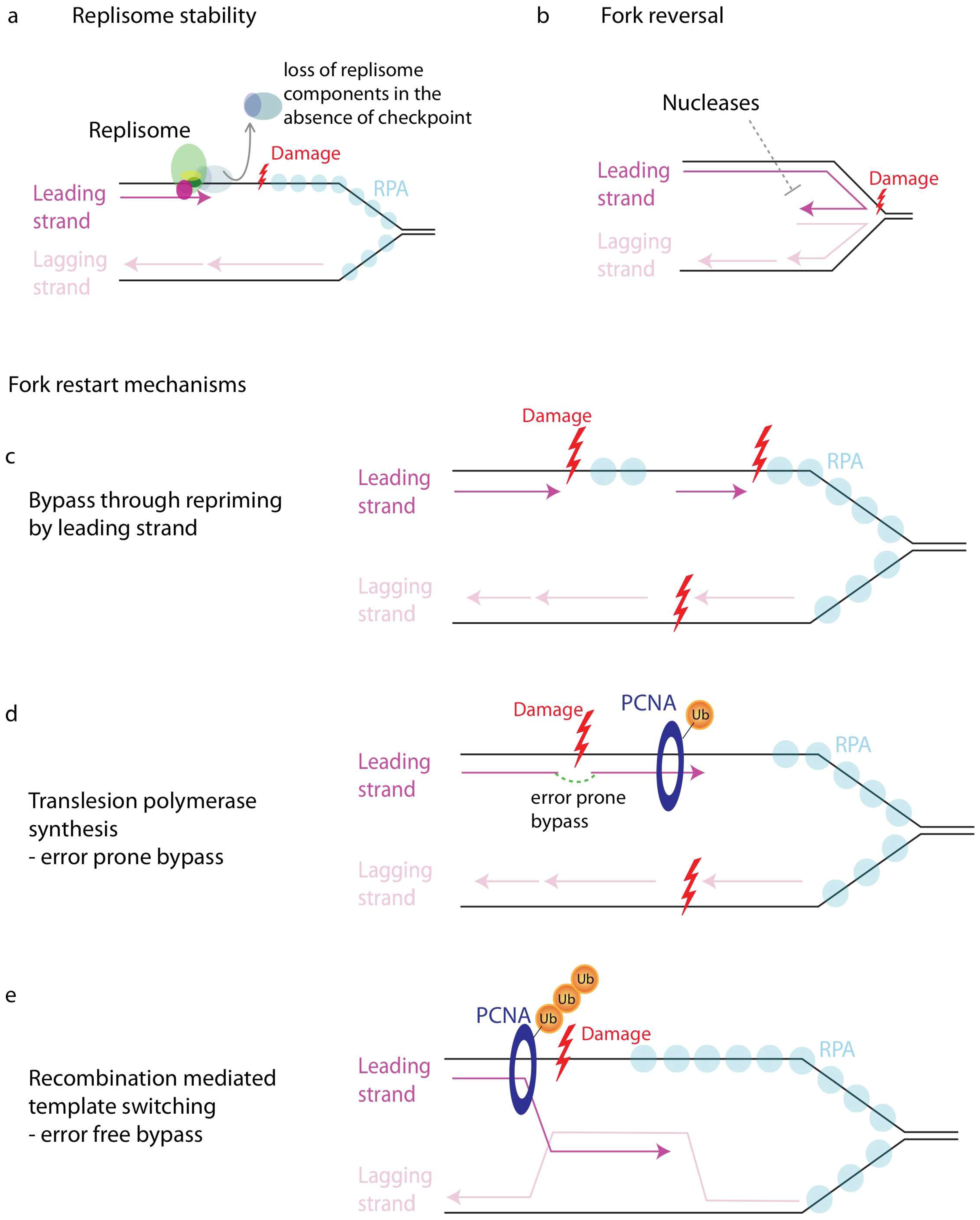
11.1. Importance of Fork Regulation
11.2. Regulation of Number of Forks
11.3. Maintenance of Replisome Stability
11.4. Fork Reversal
11.5. Regulation of Nucleases
11.6. Restart of Stalled Forks
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Valton, A.L.; Prioleau, M.N. G-Quadruplexes in DNA Replication: A Problem or a Necessity. Trends Genet. 2016, 32, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, M.; Noguchi, E. Regulation of Replication through Natural Impediments in the Eukaryotic Genome. Genes. in press.
- Hartwell, L.H.; Weinert, T.A. Checkpoints: controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell. Biol. 2004, 5, 792–804. [Google Scholar] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Livneh, Z.; Cohen, I.S.; Paz-Elizur, T.; Davidovsky, D.; Carmi, D.; Swain, U.; Mirlas-Neisberg, N. High-resolution genomic assays provide insight into the division of labor between TLS and HDR in mammalian replication of damaged DNA. DNA Repair (Amst.) 2016, 44, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Burcham, P.C. Internal hazards: baseline DNA damage by endogenous products of normal metabolism. Mutat. Res. 1999, 443, 11–36. [Google Scholar] [CrossRef]
- Brooks, P.J.; Theruvathu, J.A. DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol 2005, 35, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Dalgaard, J.Z. Causes and consequences of ribonucleotide incorporation into nuclear DNA. Trends Genet. 2012, 28, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Watts, B.E.; Kumar, D.; Watt, D.L.; Lundström, E.B.; Burgers, P.M.; Johansson, E.; Chabes, A.; Kunkel, T.A. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 4949–4954. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, F.; Novarina, D.; Amara, F.; Watt, D.L.; Stone, J.E.; Costanzo, V.; Burgers, P.M.; Kunkel, T.A.; Plevani, P.; Muzi-Falconi, M. RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol. Cell 2012, 45, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Mirkin, S.M. The balancing act of DNA repeat expansions. Curr. Opin. Genet. Dev. 2013, 23, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Lai, M.S.; Foiani, M. Preventing replication stress to maintain genome stability: Resolving conflicts between replication and transcription. Mol. Cell 2012, 45, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Sage, E.; Douki, T. Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res. 2005, 571, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Desai, S.D.; Li, T.K.; Mao, Y.; Sun, M.; Sim, S.P. Mechanism of action of camptothecin. Ann. N Y Acad. Sci. 2000, 922, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Wood, R.D. Quality control by DNA repair. Science 1999, 286, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell. Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold. Spring. Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Costanzo, V.; Gautier, J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell. Biol. 2004, 6, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Syljuåsen, R.G.; Sørensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef]
- Sørensen, C.S.; Syljuåsen, R.G.; Lukas, J.; Bartek, J. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell. Cycle 2004, 3, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Marheineke, K.; Hyrien, O. Control of replication origin density and firing time in Xenopus egg extracts: Role of a caffeine-sensitive, ATR-dependent checkpoint. J. Biol. Chem. 2004, 279, 28071–28081. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A. Probing ATR activation with model DNA templates. Cell. Cycle 2007, 6, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Cimprich, K.A. The ATR pathway: Fine-tuning the fork. DNA Repair (Amst.) 2007, 6, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Unwind and slow down: Checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005, 19, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring. Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Costanzo, V.; Gautier, J. Regulation of DNA replication by ATR: Signaling in response to DNA intermediates. DNA Repair (Amst.) 2004, 3, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Côté, J. Cellular machineries for chromosomal DNA repair. Genes Dev. 2004, 18, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Dutta, A. DNA replication and progression through S phase. Oncogene 2005, 24, 2827–2843. [Google Scholar] [CrossRef] [PubMed]
- Tercero, J.A.; Diffley, J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001, 412, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Byun, T.; Yee, M.C.; Hekmat-Nejad, M.; Cimprich, K.A. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 2002, 16, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Van Hatten, R.; Lindsay, H.D.; Michael, W.M. DNA replication is required for the checkpoint response to damaged DNA in Xenopus egg extracts. J. Cell. Biol. 2002, 158, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Callegari, A.J.; Clark, E.; Pneuman, A.; Kelly, T.J. Postreplication gaps at UV lesions are signals for checkpoint activation. Proc. Natl. Acad. Sci. USA 2010, 107, 8219–8224. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Minn, K.; Chen, J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J. Biol. Chem. 2004, 279, 9677–9680. [Google Scholar] [CrossRef] [PubMed]
- Pellicioli, A.; Lucca, C.; Liberi, G.; Marini, F.; Lopes, M.; Plevani, P.; Romano, A.; Di Fiore, P.P.; Foiani, M. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 1999, 18, 6561–6572. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M.; Breeden, L.L. Rad53-dependent phosphorylation of Swi6 and down-regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae. Genes Dev. 1997, 11, 3032–3045. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fay, D.S.; Marini, F.; Foiani, M.; Stern, D.F. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev. 1996, 10, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Nardo, T.; Giannattasio, M.; Minuzzo, M.; Stefanini, M.; Plevani, P.; Muzi Falconi, M. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc. Natl. Acad. Sci. USA 2006, 103, 17325–17330. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Follonier, C.; Tourrière, H.; Puddu, F.; Lazzaro, F.; Pasero, P.; Lopes, M.; Plevani, P.; Muzi-Falconi, M. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol. Cell. 2010, 40, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Hanasoge, S.; Ljungman, M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis 2007, 28, 2298–2304. [Google Scholar] [CrossRef] [PubMed]
- Longhese, M.P.; Neecke, H.; Paciotti, V.; Lucchini, G.; Plevani, P. The 70 kDa subunit of replication protein A is required for the G1/S and intra-S DNA damage checkpoints in budding yeast. Nucleic Acids Res. 1996, 24, 3533–3537. [Google Scholar] [CrossRef] [PubMed]
- Garvik, B.; Carson, M.; Hartwell, L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 1995, 15, 6128–6138. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Brill, S.J. Rfc4 interacts with Rpa1 and is required for both DNA replication and DNA damage checkpoints in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 3725–3737. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, V.; Shechter, D.; Lupardus, P.J.; Cimprich, K.A.; Gottesman, M.; Gautier, J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell. 2003, 11, 203–213. [Google Scholar] [CrossRef]
- Ball, H.L.; Ehrhardt, M.R.; Mordes, D.A.; Glick, G.G.; Chazin, W.J.; Cortez, D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell. Biol. 2007, 27, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.; Toczyski, D. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell. Biol. 2002, 14, 237–245. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell. Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Paciotti, V.; Clerici, M.; Lucchini, G.; Longhese, M.P. The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev. 2000, 14, 2046–2059. [Google Scholar] [PubMed]
- Rouse, J.; Jackson, S.P. Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol. Cell 2002, 9, 857–869. [Google Scholar] [CrossRef]
- MacDougall, C.A.; Byun, T.S.; Van, C.; Yee, M.C.; Cimprich, K.A. The structural determinants of checkpoint activation. Genes Dev. 2007, 21, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Liu, D.; Elledge, S.J. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 13827–13832. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, C.Y.; Melo, J.A.; Toczyski, D.P. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell 2008, 30, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Navadgi-Patil, V.M.; Burgers, P.M. The unstructured C-terminal tail of the 9–1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol. Cell 2009, 36, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Majka, J.; Niedziela-Majka, A.; Burgers, P.M. The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol. Cell 2006, 24, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.P.; Lindsey-Boltz, L.A.; Cesare, A.J.; Maniwa, Y.; Griffith, J.D.; Hurwitz, J.; Sancar, A. Loading of the human 9–1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Parrilla-Castellar, E.R.; Karnitz, L.M. Cut5 is required for the binding of Atr and DNA polymerase alpha to genotoxin-damaged chromatin. J. Biol. Chem. 2003, 278, 45507–45511. [Google Scholar] [CrossRef] [PubMed]
- Ellison, V.; Stillman, B. Biochemical characterization of DNA damage checkpoint complexes: Clamp loader and clamp complexes with specificity for 5’ recessed DNA. PLoS Biol. 2003, 1, E33. [Google Scholar] [CrossRef] [PubMed]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008, 22, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9–1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Foss, E.J. Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae. Genetics 2001, 157, 567–577. [Google Scholar] [PubMed]
- Chou, D.M.; Elledge, S.J. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl. Acad. Sci. USA 2006, 103, 18143–18147. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, E.; Noguchi, C.; Du, L.L.; Russell, P. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 2003, 23, 7861–7874. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, E.; Noguchi, C.; McDonald, W.H.; Yates, J.R.; Russell, P. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 2004, 24, 8342–8355. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Osborn, A.J.; Elledge, S.J. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003, 17, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004, 117, 575–588. [Google Scholar] [CrossRef]
- Liu, S.; Bekker-Jensen, S.; Mailand, N.; Lukas, C.; Bartek, J.; Lukas, J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol. Cell. Biol. 2006, 26, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa-Sugata, N.; Masai, H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J. Biol. Chem. 2007, 282, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Kaçmaz, K.; Chastain, P.D.; Qu, P.P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 2007, 27, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Jossen, R.; Colosio, A.; Frattini, C.; Carotenuto, W.; Cocito, A.; Doksani, Y.; Klein, H.; Gómez-González, B.; et al. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 2011, 146, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Kumar, A.; Foiani, M. Preserving the genome by regulating chromatin association with the nuclear envelope. Trends Cell. Biol. 2012, 22, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.H.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 10364–10369. [Google Scholar] [CrossRef] [PubMed]
- Randell, J.C.; Fan, A.; Chan, C.; Francis, L.I.; Heller, R.C.; Galani, K.; Bell, S.P. Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol. Cell 2010, 40, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Albuquerque, C.P.; Liang, J.; Suhandynata, R.T.; Zhou, H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 2010, 285, 12803–12812. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Tsukiyama, T. ATR-like kinase Mec1 facilitates both chromatin accessibility at DNA replication forks and replication fork progression during replication stress. Genes Dev. 2013, 27, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.A.; Zhou, C.; Elia, A.E.; Murray, J.M.; Carr, A.M.; Elledge, S.J.; Rhind, N. Identification of S-phase DNA damage-response targets in fission yeast reveals conservation of damage-response networks. Proc. Natl. Acad. Sci. USA 2016, 113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer. 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N. Changing of the guard: how ATM hands off DNA double-strand break signaling to ATR. Mol. Cell 2009, 33, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr. Opin. Cell. Biol. 2001, 13, 738–747. [Google Scholar] [CrossRef]
- Bartek, J.; Falck, J.; Lukas, J. CHK2 kinase—A busy messenger. Nat. Rev. Mol. Cell. Biol. 2001, 2, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and ATR: networking cellular responses to DNA damage. Curr. Opin. Genet. Dev. 2001, 11, 71–77. [Google Scholar] [CrossRef]
- Rhind, N.; Russell, P. Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J. Cell. Sci. 2000, 113, 3889–3896. [Google Scholar] [PubMed]
- Sanchez, Y.; Bachant, J.; Wang, H.; Hu, F.; Liu, D.; Tetzlaff, M.; Elledge, S.J. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 1999, 286, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, H.D.; Griffiths, D.J.; Edwards, R.J.; Christensen, P.U.; Murray, J.M.; Osman, F.; Walworth, N.; Carr, A.M. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998, 12, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N.; Russell, P. The Schizosaccharomyces pombe S-phase checkpoint differentiates between different types of DNA damage. Genetics 1998, 149, 1729–1737. [Google Scholar] [PubMed]
- Boddy, M.N.; Russell, P. DNA replication checkpoint control. Front. Biosci. 1999, 4, D841–D848. [Google Scholar] [CrossRef] [PubMed]
- Pichierri, P.; Rosselli, F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004, 23, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yehoyada, M.; Wang, L.C.; Kozekov, I.D.; Rizzo, C.J.; Gottesman, M.E.; Gautier, J. Checkpoint signaling from a single DNA interstrand crosslink. Mol. Cell 2009, 35, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Newport, J.W. Regulation of replicon size in Xenopus egg extracts. Science 1997, 275, 993–995. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Pasero, P.; Gasser, S.M. ORC and the intra-S-phase checkpoint: A threshold regulates Rad53p activation in S phase. Genes Dev. 2002, 16, 3236–3252. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell. Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.S.; Cortez, D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.E.; Medhurst, A.L.; Dart, D.A.; Lakin, N.D. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene 2006, 25, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; Clerici, M.; Lucchini, G.; Longhese, M.P. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007, 8, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Moore, J.K.; Holmes, A.; Umezu, K.; Kolodner, R.D.; Haber, J.E. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998, 94, 399–409. [Google Scholar] [CrossRef]
- Nakada, D.; Hirano, Y.; Sugimoto, K. Requirement of the Mre11 complex and exonuclease 1 for activation of the Mec1 signaling pathway. Mol. Cell. Biol. 2004, 24, 10016–10025. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Klier, S.; McGowan, C.; Wittenberg, C.; de Bruin, R.A. Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell-cycle transcription. Curr. Biol. 2013, 23, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.A.; Kalashnikova, T.I.; Chahwan, C.; McDonald, W.H.; Wohlschlegel, J.; Yates, J.; Russell, P.; Wittenberg, C. Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol. Cell 2006, 23, 483–496. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.A.; Kalashnikova, T.I.; Aslanian, A.; Wohlschlegel, J.; Chahwan, C.; Yates, J.R.; Russell, P.; Wittenberg, C. DNA replication checkpoint promotes G1-S transcription by inactivating the MBF repressor Nrm1. Proc. Natl Acad. Sci. USA 2008, 105, 11230–11235. [Google Scholar] [CrossRef] [PubMed]
- Dutta, C.; Patel, P.K.; Rosebrock, A.; Oliva, A.; Leatherwood, J.; Rhind, N. The DNA replication checkpoint directly regulates MBF-dependent G1/S transcription. Mol. Cell. Biol. 2008, 28, 5977–5985. [Google Scholar] [CrossRef] [PubMed]
- Travesa, A.; Kuo, D.; de Bruin, R.A.; Kalashnikova, T.I.; Guaderrama, M.; Thai, K.; Aslanian, A.; Smolka, M.B.; Yates, J.R.; Ideker, T.; Wittenberg, C. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012, 31, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P.; Huang, M.; Metzner, S.; Botstein, D.; Elledge, S.J.; Brown, P.O. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol. Biol. Cell. 2001, 12, 2987–3003. [Google Scholar] [CrossRef] [PubMed]
- Jaehnig, E.J.; Kuo, D.; Hombauer, H.; Ideker, T.G.; Kolodner, R.D. Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell. Rep. 2013, 4, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Putnam, C.D.; Jaehnig, E.J.; Kolodner, R.D. Perspectives on the DNA damage and replication checkpoint responses in Saccharomyces cerevisiae. DNA Repair (Amst.) 2009, 8, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Birrell, G.W.; Brown, J.A.; Wu, H.I.; Giaever, G.; Chu, A.M.; Davis, R.W.; Brown, J.M. Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. Proc. Natl Acad. Sci. USA 2002, 99, 8778–8783. [Google Scholar] [CrossRef] [PubMed]
- Dutta, C.; Rhind, N. The role of specific checkpoint-induced S-phase transcripts in resistance to replicative stress. PLoS ONE 2009, 4, e6944. [Google Scholar] [CrossRef] [PubMed]
- Christensen, P.U.; Bentley, N.J.; Martinho, R.G.; Nielsen, O.; Carr, A.M. Mik1 levels accumulate in S phase and may mediate an intrinsic link between S phase and mitosis. Proc. Natl Acad. Sci. USA 2000, 97, 2579–2584. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhou, Z.; Elledge, S.J. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 1998, 94, 595–605. [Google Scholar] [CrossRef]
- Tsaponina, O.; Barsoum, E.; Aström, S.U.; Chabes, A. Ixr1 is required for the expression of the ribonucleotide reductase Rnr1 and maintenance of dNTP pools. PLoS Genet. 2011, 7, e1002061. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Lancaster, C.S.; Shi, B.; Guo, H.; Thimmaiah, P.; Bjornsti, M.A. TOR signaling is a determinant of cell survival in response to DNA damage. Mol. Cell. Biol. 2007, 27, 7007–7017. [Google Scholar] [CrossRef] [PubMed]
- Tomar, R.S.; Zheng, S.; Brunke-Reese, D.; Wolcott, H.N.; Reese, J.C. Yeast Rap1 contributes to genomic integrity by activating DNA damage repair genes. EMBO J. 2008, 27, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.; Kersey, P.J.; McInerny, C.J.; Fantes, P.A. Cell cycle, DNA damage and heat shock regulate suc22+ expression in fission yeast. Mol. Gen. Genet. 1996, 252, 284–291. [Google Scholar] [PubMed]
- Zhang, Y.W.; Jones, T.L.; Martin, S.E.; Caplen, N.J.; Pommier, Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. J. Biol. Chem. 2009, 284, 18085–18095. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Pastushok, L.; Xiao, W. DNA damage-induced gene expression in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2008, 32, 908–926. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Bastos de Oliveira, F.M.; Harris, M.R.; de Bruin, R.A. The checkpoint transcriptional response: Make sure to turn it off once you are satisfied. Cell. Cycle 2012, 11, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Guarino, E.; Salguero, I.; Kearsey, S.E. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell. Dev. Biol. 2014, 30, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.D.; Wang, J.; Stubbe, J.; Elledge, S.J. Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol. Cell 2008, 32, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, M. Dif1 controls subcellular localization of ribonucleotide reductase by mediating nuclear import of the R2 subunit. Mol. Cell. Biol. 2008, 28, 7156–7167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rothstein, R. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc. Natl Acad. Sci USA 2002, 99, 3746–3751. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, P.; Hofer, A.; Thelander, L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem. 2006, 281, 7834–7841. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, P.; Dahl, L.; Chilkova, O.; Domkin, V.; Thelander, L. The Schizosaccharomyces pombe replication inhibitor Spd1 regulates ribonucleotide reductase activity and dNTPs by binding to the large Cdc22 subunit. J. Biol. Chem. 2006, 281, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Powell, K.A.; Mundt, K.; Wu, L.; Carr, A.M.; Caspari, T. Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev. 2003, 17, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Nestoras, K.; Mohammed, A.H.; Schreurs, A.S.; Fleck, O.; Watson, A.T.; Poitelea, M.; O’Shea, C.; Chahwan, C.; Holmberg, C.; Kragelund, B.B.; et al. Regulation of ribonucleotide reductase by Spd1 involves multiple mechanisms. Genes Dev. 2010, 24, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Poitelea, M.; Watson, A.; Yoshida, S.H.; Shimoda, C.; Holmberg, C.; Nielsen, O.; Carr, A.M. Transactivation of Schizosaccharomyces pombe cdt2+ stimulates a Pcu4-Ddb1-CSN ubiquitin ligase. EMBO J. 2005, 24, 3940–3951. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, C.; Fleck, O.; Hansen, H.A.; Liu, C.; Slaaby, R.; Carr, A.M.; Nielsen, O. Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev. 2005, 19, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Vejrup-Hansen, R.; Fleck, O.; Landvad, K.; Fahnøe, U.; Broendum, S.S.; Schreurs, A.S.; Kragelund, B.B.; Carr, A.M.; Holmberg, C.; Nielsen, O. Spd2 assists Spd1 in the modulation of ribonucleotide reductase architecture but does not regulate deoxynucleotide pools. J. Cell. Sci. 2014, 127, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Arnaoutov, A.; Dasso, M. Enzyme regulation. IRBIT is a novel regulator of ribonucleotide reductase in higher eukaryotes. Science 2014, 345, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.; Georgieva, B.; Domkin, V.; Zhao, X.; Rothstein, R.; Thelander, L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 2003, 112, 391–401. [Google Scholar] [CrossRef]
- Ord, M.G.; Stocken, L.A. The effects of x- and gamma-radiation on nucleic acid metabolism in the rat in vivo and in vitro. Biochem. J. 1956, 63, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ord, M.G.; Stocken, L.A. Studies in synthesis of deoxyribonucleic acid; radiobiochemical lesion in animal cells. Nature 1958, 182, 1787–1788. [Google Scholar] [CrossRef] [PubMed]
- Lajtha, L.G.; Oliver, R.; Berry, R.; Noyes, W.D. Mechanism of radiation effect on the process of synthesis of deoxyribonucleic acid. Nature 1958, 182, 1788–1790. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.B. Thymidine incorporation as a measure of DNA-synthesis in irradiated cell cultures. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1967, 13, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.B.; Young, B.R. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc. Natl Acad. Sci. USA 1980, 77, 7315–7317. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.B. Radioresistant DNA synthesis: an intrinsic feature of ataxia telangiectasia. Mutat. Res. 1981, 84, 183–190. [Google Scholar] [CrossRef]
- Houldsworth, J.; Lavin, M.F. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980, 8, 3709–3720. [Google Scholar] [CrossRef] [PubMed]
- Young, B.R.; Painter, R.B. Radioresistant DNA synthesis and human genetic diseases. Hum. Genet. 1989, 82, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Gatti, R.A.; Becker-Catania, S.; Chun, H.H.; Sun, X.; Mitui, M.; Lai, C.H.; Khanlou, N.; Babaei, M.; Cheng, R.; Clark, C.; et al. The pathogenesis of ataxia-telangiectasia. Learning from a Rosetta Stone. Clin. Rev. Allergy Immunol. 2001, 20, 87–108. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Walker, G.C.; Siede, W. DNA Repair and Mutagenesis, 1st ed.; ASM Press: Washington, DC, USA, 1995; p. 698. [Google Scholar]
- Paulovich, A.G.; Hartwell, L.H. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 1995, 82, 841–847. [Google Scholar] [CrossRef]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [PubMed]
- Santocanale, C.; Diffley, J.F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 1998, 395, 615–618. [Google Scholar] [PubMed]
- Shirahige, K.; Hori, Y.; Shiraishi, K.; Yamashita, M.; Takahashi, K.; Obuse, C.; Tsurimoto, T.; Yoshikawa, H. Regulation of DNA-replication origins during cell-cycle progression. Nature 1998, 395, 618–621. [Google Scholar] [PubMed]
- Kaufmann, W.K.; Cleaver, J.E.; Painter, R.B. Ultraviolet radiation inhibits replicon initiation in S phase human cells. Biochim. Biophys. Acta 1980, 608, 191–195. [Google Scholar] [CrossRef]
- Merrick, C.J.; Jackson, D.; Diffley, J.F. Visualization of altered replication dynamics after DNA damage in human cells. J. Biol. Chem. 2004, 279, 20067–20075. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Petrini, J.H.; Williams, B.R.; Lukas, J.; Bartek, J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat. Genet. 2002, 30, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Chastain, P.D.; Heffernan, T.P.; Nevis, K.R.; Lin, L.; Kaufmann, W.K.; Kaufman, D.G.; Cordeiro-Stone, M. Checkpoint regulation of replication dynamics in UV-irradiated human cells. Cell. Cycle. 2006, 5, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.A.; Conti, C.; Syed, A.; Aladjem, M.I.; Pommier, Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007, 27, 5806–5818. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Huberman, J.A. Checkpoint-dependent regulation of origin firing and replication fork movement in response to DNA damage in fission yeast. Mol. Cell. Biol. 2009, 29, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Luciani, M.G.; Oehlmann, M.; Blow, J.J. Characterization of a novel ATR-dependent, Chk1-independent, intra-S-phase checkpoint that suppresses initiation of replication in Xenopus. J. Cell. Sci. 2004, 117, 6019–6030. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mosqueda, J.; Maas, N.L.; Jonsson, Z.O.; Defazio-Eli, L.G.; Wohlschlegel, J.; Toczyski, D.P. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 2010, 467, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, Y.; Tak, Y.S.; Sugino, A.; Araki, H. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J. 2001, 20, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Kanemaki, M.; Labib, K. Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks. EMBO J. 2006, 25, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Pahl, P.M.; Harrison, K.; Rosamond, J.; Sclafani, R.A. Cell cycle regulation of the yeast Cdc7 protein kinase by association with the Dbf4 protein. Mol. Cell. Biol. 1993, 13, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Kumagai, A.; Schlacher, K.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol. Cell. 2015, 57, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, T.P.; Unsal-Kaçmaz, K.; Heinloth, A.N.; Simpson, D.A.; Paules, R.S.; Sancar, A.; Cordeiro-Stone, M.; Kaufmann, W.K. Cdc7-Dbf4 and the human S checkpoint response to UVC. J. Biol. Chem. 2007, 282, 9458–9468. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Yekezare, M.; Gómez-González, B.; Diffley, J.F. Controlling DNA replication origins in response to DNA damage - inhibit globally, activate locally. J. Cell. Sci. 2013, 126, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Dutta, A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell. Biol. 2005, 6, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F. Quality control in the initiation of eukaryotic DNA replication. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 3545–3553. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Araki, H. Multiple regulatory mechanisms to inhibit untimely initiation of DNA replication are important for stable genome maintenance. PLoS Genet. 2011, 7, e1002136. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. Eukaryotic chromosome DNA replication: Where, when, and how. Annu. Rev. Biochem. 2010, 79, 89–130. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell. Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.G.; Winter, S.L.; Zaika, E.; Cao, T.V.; Oguz, U.; Koomen, J.M.; Hamlin, J.L.; Alexandrow, M.G. Cdc45 limits replicon usage from a low density of preRCs in mammalian cells. PLoS ONE 2011, 6, e17533. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Göhler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell. Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Anglana, M.; Apiou, F.; Bensimon, A.; Debatisse, M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 2003, 114, 385–394. [Google Scholar] [CrossRef]
- Paciotti, V.; Clerici, M.; Scotti, M.; Lucchini, G.; Longhese, M.P. Characterization of mec1 kinase-deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Mol. Cell. Biol. 2001, 21, 3913–3925. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; Cortez, D. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; Donehower, L.A.; Elledge, S.J. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Jossen, R.; Bermejo, R. The DNA damage checkpoint response to replication stress: A Game of Forks. Front. Genet. 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Preventing replication fork collapse to maintain genome integrity. DNA Repair (Amst.) 2015, 32, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; Bjergbaek, L.; Shimada, K.; Frei, C.; Gasser, S.M. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003, 22, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; Schleker, T.; Rojas, V.; Bjergbaek, L.; Tercero, J.A.; Gasser, S.M. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005, 19, 3055–3069. [Google Scholar] [CrossRef] [PubMed]
- Cotta-Ramusino, C.; Fachinetti, D.; Lucca, C.; Doksani, Y.; Lopes, M.; Sogo, J.; Foiani, M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell. 2005, 17, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lucca, C.; Vanoli, F.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Haber, J.; Foiani, M. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 2004, 23, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.L.; Li, J.M.; Osborn, A.J.; Elledge, S.J. Mrc1 phosphorylation in response to DNA replication stress is required for Mec1 accumulation at the stalled fork. Proc. Natl Acad. Sci. USA 2009, 106, 12765–12770. [Google Scholar] [CrossRef] [PubMed]
- Trenz, K.; Smith, E.; Smith, S.; Costanzo, V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006, 25, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Ragland, R.L.; Patel, S.; Rivard, R.S.; Smith, K.; Peters, A.A.; Bielinsky, A.K.; Brown, E.J. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev. 2013, 27, 2259–2273. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Puddu, F.; Costanzo, V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2011, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol. Cell. 2012, 45, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell. Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell. Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell. Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Froget, B.; Blaisonneau, J.; Lambert, S.; Baldacci, G. Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol. Biol. Cell. 2008, 19, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Rossi, M.J.; Mazin, A.V. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011, 39, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Bétous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Bétous, R.; Couch, F.B.; Mason, A.C.; Eichman, B.F.; Manosas, M.; Cortez, D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell. Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Yusufzai, T.; Kadonaga, J.T. HARP is an ATP-driven annealing helicase. Science 2008, 322, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Yusufzai, T.; Kadonaga, J.T. Annealing helicase 2 (AH2), a DNA-rewinding motor with an HNH motif. Proc. Natl Acad. Sci. USA 2010, 107, 20970–20973. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; Décaillet, C.; Delannoy, M.; Wu, L.; Constantinou, A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl Acad. Sci. USA 2008, 105, 16107–16112. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Blastyák, A.; Pintér, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell. 2007, 28, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Blastyák, A.; Hajdú, I.; Unk, I.; Haracska, L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Kile, A.C.; Chavez, D.A.; Bacal, J.; Eldirany, S.; Korzhnev, D.M.; Bezsonova, I.; Eichman, B.F.; Cimprich, K.A. HLTF’s Ancient HIRAN Domain Binds 3’ DNA Ends to Drive Replication Fork Reversal. Mol. Cell. 2015, 58, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Fugger, K.; Mistrik, M.; Neelsen, K.J.; Yao, Q.; Zellweger, R.; Kousholt, A.N.; Haahr, P.; Chu, W.K.; Bartek, J.; Lopes, M.; Hickson, I.D.; Sørensen, C.S. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Xiao, L.; Groden, J.; Orren, D.K. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry 2006, 45, 13939–13946. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Karale, R.; Xu, X.; Liu, Y.; Orren, D.K. The Werner and Bloom syndrome proteins help resolve replication blockage by converting (regressed) holliday junctions to functional replication forks. Biochemistry 2011, 50, 6774–6788. [Google Scholar] [CrossRef] [PubMed]
- Segurado, M.; Diffley, J.F. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008, 22, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Morin, I.; Ngo, H.P.; Greenall, A.; Zubko, M.K.; Morrice, N.; Lydall, D. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008, 27, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; Cejka, P.; Stewart, S.; Lopes, M.; Vindigni, A. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell. Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Moore, H.R.; Sidorova, J.; Karanja, K.; Honaker, Y.; Dao, B.; Piwnica-Worms, H.; Campbell, J.L.; Monnat, R.J.; Stewart, S.A. Okazaki fragment processing-independent role for human Dna2 enzyme during DNA replication. J. Biol. Chem. 2012, 287, 21980–21991. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; Blanco, M.G.; Maslen, S.; Skehel, J.M.; West, S.C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011, 147, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; Blanco, M.G.; West, S.C. Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell. Rep. 2013, 4, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Szakal, B.; Branzei, D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013, 32, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Dehé, P.M.; Coulon, S.; Scaglione, S.; Shanahan, P.; Takedachi, A.; Wohlschlegel, J.A.; Yates, J.R.; Llorente, B.; Russell, P.; Gaillard, P.H. Regulation of Mus81-Eme1 Holliday junction resolvase in response to DNA damage. Nat. Struct. Mol. Biol. 2013, 20, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; West, S.C. MUS81-EME2 promotes replication fork restart. Cell. Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Whitby, M.C.; Osman, F.; Dixon, J. Cleavage of model replication forks by fission yeast Mus81-Eme1 and budding yeast Mus81-Mms4. J. Biol. Chem. 2003, 278, 6928–6935. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; West, S.C. Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 2014, 42, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Amangyeld, T.; Shin, Y.K.; Lee, M.; Kwon, B.; Seo, Y.S. Human MUS81-EME2 can cleave a variety of DNA structures including intact Holliday junction and nicked duplex. Nucleic Acids Res. 2014, 42, 5846–5862. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Torres, M.J.; Martin, M.M.; Rao, V.A.; Pommier, Y.; Katsura, M.; Miyagawa, K.; Aladjem, M.I. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J. Mol. Biol. 2008, 375, 1152–1164. [Google Scholar] [CrossRef] [PubMed]
- Regairaz, M.; Zhang, Y.W.; Fu, H.; Agama, K.K.; Tata, N.; Agrawal, S.; Aladjem, M.I.; Pommier, Y. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell. Biol. 2011, 195, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Fugger, K.; Chu, W.K.; Haahr, P.; Kousholt, A.N.; Beck, H.; Payne, M.J.; Hanada, K.; Hickson, I.D.; Sørensen, C.S. FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat. Commun. 2013, 4, 1423. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst.) 2016, 44, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair (Amst.) 2007, 6, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The checkpoint response to replication stress. DNA Repair (Amst.) 2009, 8, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D.; Walden, H. Ubiquitin signalling in DNA replication and repair. Nat. Rev. Mol. Cell. Biol. 2010, 11, 479–489. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, N.; Wong, R.P.; Ulrich, H.D. Functions of Ubiquitin and SUMO in DNA Replication and Replication Stress. Front. Genet. 2016, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E. Competition, collaboration and coordination--determining how cells bypass DNA damage. J. Cell. Sci. 2012, 125, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst.) 2009, 8, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.C.; Marians, K.J. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 2006, 439, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.; Marians, K.J. The Escherichia coli replisome is inherently DNA damage tolerant. Science 2011, 334, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; Doherty, A.J. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell. 2013, 52, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Mourón, S.; Rodriguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. PrimPol breaks replication barriers. Nat. Struct. Mol. Biol. 2013, 20, 1348–1350. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006, 21, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Bianchi, J.; Doherty, A.J. PrimPol-A new polymerase on the block. Mol. Cell. Oncol. 2014, 1, e960754. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.; Irmisch, A.; Green, C.M.; Neiss, A.; Trickey, M.; Ulrich, H.D.; Furuya, K.; Watts, F.Z.; Carr, A.M.; Lehmann, A.R. Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol. Biol. Cell 2006, 17, 2976–2985. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Myung, K. PCNA modifications for regulation of post-replication repair pathways. Mol. Cells 2008, 26, 5–11. [Google Scholar] [PubMed]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of Ubiquitin-Dependent DNA Damage Bypass Is Mediated by Replication Protein A. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D.; Jentsch, S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000, 19, 3388–3397. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Ulrich, H.D. Mechanistic analysis of PCNA poly-ubiquitylation by the ubiquitin protein ligases Rad18 and Rad5. EMBO J. 2009, 28, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lawrence, C.W. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc. Natl Acad. Sci. USA 2005, 102, 15954–15959. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Kubota, Y.; Carr, A.M.; Iwasaki, H. RAD6-RAD18-RAD5-pathway-dependent tolerance to chronic low-dose ultraviolet light. Nature 2009, 457, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Seki, M.; Enomoto, T. Rad18/Rad5/Mms2-mediated polyubiquitination of PCNA is implicated in replication completion during replication stress. Genes Cells 2004, 9, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.K.; Brun, J.; Ramaekers, C.; Theys, J.; Weng, L.; Lambin, P.; Gray, D.A.; Wouters, B.G. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2006, 2, e116. [Google Scholar] [CrossRef]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Cortez, D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 2012, 7, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Cortez, D. Purification of proteins on newly synthesized DNA using iPOND. Methods Mol. Biol. 2015, 1228, 123–131. [Google Scholar] [PubMed]
- Sirbu, B.M.; McDonald, W.H.; Dungrawala, H.; Badu-Nkansah, A.; Kavanaugh, G.M.; Chen, Y.; Tabb, D.L.; Cortez, D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013, 288, 31458–31467. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Title 1 | S. cerevisiae | S. pombe | Mammals |
|---|---|---|---|
| Checkpoint kinase | Mec1 Ddc2 Rad24 | Rad3 Rad26 Rad17 | ATR ATRIP Rad17 |
| Sensors | Ddc1 Mec3 Rad17 Dpb11 | Rad9 Hus1 Rad1 Cut5 | Rad9 Hus1 Rad1 TopBP1 |
| Adaptors | Mrc1 Tof1 Csm3 | Mrc1 Swi1 Swi3 | Claspin Tim Tipin |
| Effector kinase | Rad53 | Cds1 | Chk1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, D.R.; Rhind, N. The Intra-S Checkpoint Responses to DNA Damage. Genes 2017, 8, 74. https://doi.org/10.3390/genes8020074
Iyer DR, Rhind N. The Intra-S Checkpoint Responses to DNA Damage. Genes. 2017; 8(2):74. https://doi.org/10.3390/genes8020074
Chicago/Turabian StyleIyer, Divya Ramalingam, and Nicholas Rhind. 2017. "The Intra-S Checkpoint Responses to DNA Damage" Genes 8, no. 2: 74. https://doi.org/10.3390/genes8020074
APA StyleIyer, D. R., & Rhind, N. (2017). The Intra-S Checkpoint Responses to DNA Damage. Genes, 8(2), 74. https://doi.org/10.3390/genes8020074