
Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis


{kind=link}
Abstract
:1. Introduction
2. T1D Pathogenesis
3. Expression of T1D Risk Genes in Islets
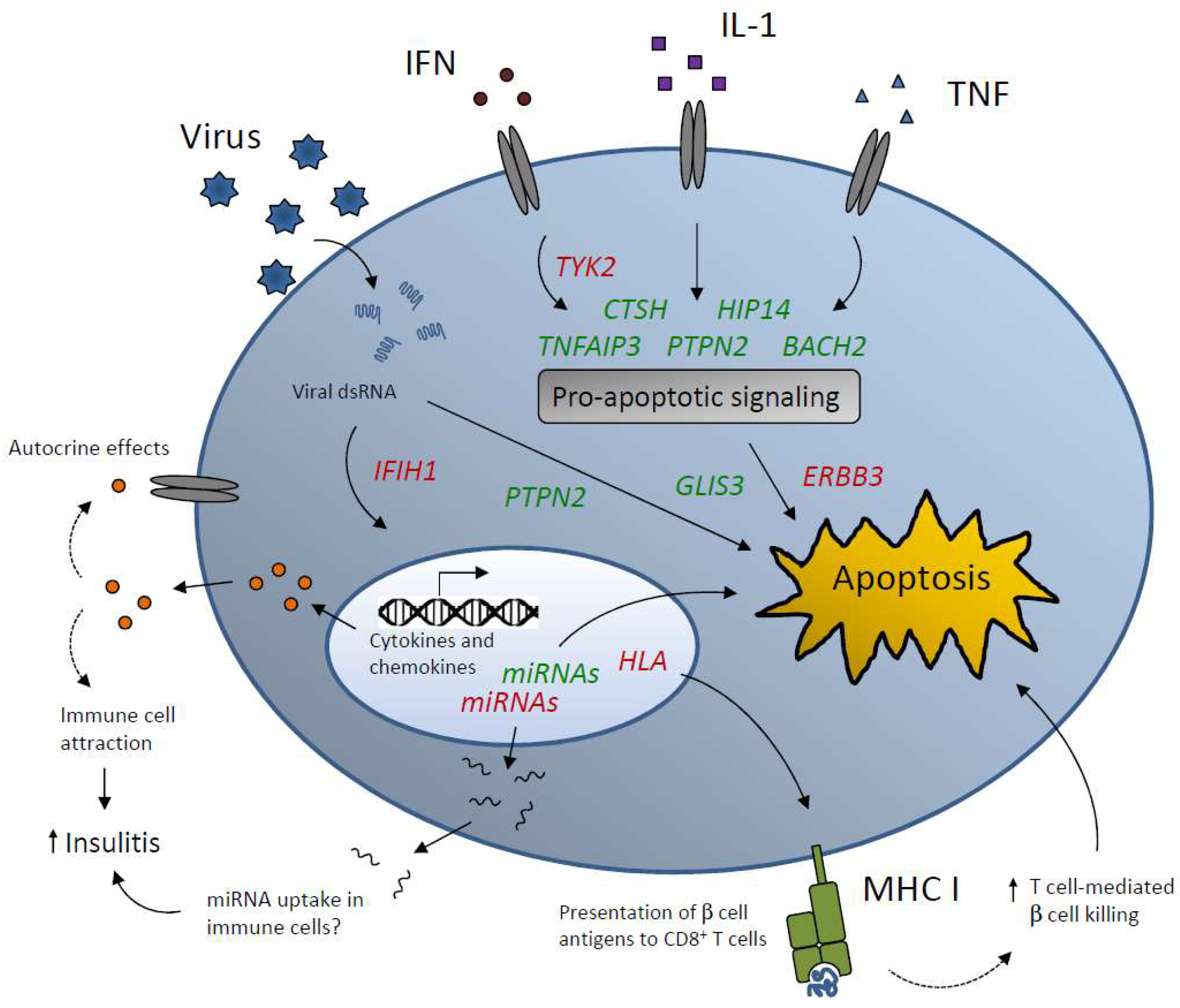
4. T1D Candidate Genes that Affect Islet Inflammation and Apoptosis
5. Genetic Risk-Score of Islet-Expressed T1D Candidate Genes as Predictor of Disease Progression
6. MHC and the β-Cell
7. Non-Coding RNAs Regulating the β-Cell Genome
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pociot, F.; Lernmark, A. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Bergholdt, R.; Brorsson, C.; Palleja, A.; Berchtold, L.A.; Floyel, T.; Bang-Berthelsen, C.H.; Frederiksen, K.S.; Jensen, L.J.; Storling, J.; Pociot, F. Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes 2012, 61, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Thomas, C.E.; Brunak, S. Network biology concepts in complex disease comorbidities. Nat. Rev. Genet. 2016, 17, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Floyel, T.; Kaur, S.; Pociot, F. Genes affecting beta-cell function in type 1 diabetes. Curr. Diabetes Rep. 2015, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Santin, I.; Dos Santos, R.S.; Eizirik, D.L. Pancreatic beta cell survival and signaling pathways: Effects of type 1 diabetes-associated genetic variants. Methods Mol. Biol. 2016, 1433, 21–54. [Google Scholar] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Akolkar, B.; Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nierras, C.R.; Todd, J.A.; Rich, S.S.; Nerup, J. Genetics of type 1 diabetes: What’s next? Diabetes 2010, 59, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Corradin, O.; Saiakhova, A.; Akhtar-Zaidi, B.; Myeroff, L.; Willis, J.; Cowper-Sal lari, R.; Lupien, M.; Markowitz, S.; Scacheri, P.C. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 Tcells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Nerup, J.; Mandrup-Poulsen, T.; Helqvist, S.; Andersen, H.U.; Pociot, F.; Reimers, J.I.; Cuartero, B.G.; Karlsen, A.E.; Bjerre, U.; Lorenzen, T. On the pathogenesis of IDDM. Diabetologia 1994, 37 (Suppl. S2), S82–S89. [Google Scholar] [CrossRef] [PubMed]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Analysis of islet inflammation in human type 1 diabetes. Clin. Exp. Immunol. 2009, 155, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, L.A.; Prause, M.; Storling, J.; Mandrup-Poulsen, T. Cytokines and pancreatic beta-cell apoptosis. Adv. Clin. Chem. 2016, 75, 99–158. [Google Scholar] [PubMed]
- Thomas, H.E.; Trapani, J.A.; Kay, T.W. The role of perforin and granzymes in diabetes. Cell Death Differ. 2010, 17, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Keenan, H.A.; Sun, J.K.; Levine, J.; Doria, A.; Aiello, L.P.; Eisenbarth, G.; Bonner-Weir, S.; King, G.L. Residual insulin production and pancreatic ss-cell turnover after 50 years of diabetes: Joslin medalist study. Diabetes 2010, 59, 2846–2853. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.H.; Digon, B.J., 3rd; Hirshberg, B.; Chang, R.; Wood, B.J.; Neeman, Z.; Kam, A.; Wesley, R.A.; Polly, S.M.; Hofmann, R.M.; et al. Pancreatic beta cell function persists in many patients with chronic type 1 diabetes, but is not dramatically improved by prolonged immunosuppression and euglycaemia from a beta cell allograft. Diabetologia 2009, 52, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Gianani, R.; Campbell-Thompson, M.; Sarkar, S.A.; Wasserfall, C.; Pugliese, A.; Solis, J.M.; Kent, S.C.; Hering, B.J.; West, E.; Steck, A.; et al. Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia 2010, 53, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Krogvold, L.; Skog, O.; Sundstrom, G.; Edwin, B.; Buanes, T.; Hanssen, K.F.; Ludvigsson, J.; Grabherr, M.; Korsgren, O.; Dahl-Jorgensen, K. Function of isolated pancreatic islets from patients at onset of type 1 diabetes: Insulin secretion can be restored after some days in a nondiabetogenic environment in vitro: Results from the divid study. Diabetes 2015, 64, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. Beta-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Van Belle, T.L.; Coppieters, K.T.; von Herrath, M.G. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.D.; Smyth, D.J.; Smiles, A.M.; Plagnol, V.; Walker, N.M.; Allen, J.E.; Downes, K.; Barrett, J.C.; Healy, B.C.; Mychaleckyj, J.C.; et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat. Genet. 2008, 40, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Bradfield, J.P.; Qu, H.Q.; Wang, K.; Zhang, H.; Sleiman, P.M.; Kim, C.E.; Mentch, F.D.; Qiu, H.; Glessner, J.T.; Thomas, K.A.; et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011, 7, e1002293. [Google Scholar] [CrossRef] [PubMed]
- Onengut-Gumuscu, S.; Chen, W.M.; Burren, O.; Cooper, N.J.; Quinlan, A.R.; Mychaleckyj, J.C.; Farber, E.; Bonnie, J.K.; Szpak, M.; Schofield, E.; et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015, 47, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Pociot, F. Genetics of diabetes—Are we missing the genes or the disease? Mol. Cell. Endocrinol. 2014, 382, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Yang, J.; Goddard, M.E.; Visscher, P.M. The genetic interpretation of area under the roc curve in genomic profiling. PLoS Genet. 2010, 6, e1000864. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.G. Prediction and interaction in complex disease genetics: Experience in type 1 diabetes. PLoS Genet. 2009, 5, e1000540. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A. Bringing genome-wide association findings into clinical use. Nat. Rev. Genet. 2013, 14, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.H.; Kaur, S.; Brorsson, C.A.; Pociot, F. Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS ONE 2014, 9, e105723. [Google Scholar] [CrossRef] [PubMed]
- Bergholdt, R.; Brorsson, C.; Lage, K.; Nielsen, J.H.; Brunak, S.; Pociot, F. Expression profiling of human genetic and protein interaction networks in type 1 diabetes. PLoS ONE 2009, 4, e6250. [Google Scholar] [CrossRef] [PubMed]
- Storling, J.; Brorsson, C.A. Candidate genes expressed in human islets and their role in the pathogenesis of type 1 diabetes. Curr. Diabetes Rep. 2013, 13, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Kutlu, B.; Miani, M.; Bang-Berthelsen, C.H.; Storling, J.; Pociot, F.; Goodman, N.; Hood, L.; Welsh, N.; Bontempi, G.; et al. Temporal profiling of cytokine-induced genes in pancreatic beta-cells by meta-analysis and network inference. Genomics 2014, 103, 264–275. [Google Scholar] [CrossRef]
- Huang, K.; Yanai, A.; Kang, R.; Arstikaitis, P.; Singaraja, R.R.; Metzler, M.; Mullard, A.; Haigh, B.; Gauthier-Campbell, C.; Gutekunst, C.A.; et al. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron 2004, 44, 977–986. [Google Scholar] [CrossRef]
- Lage, K.; Karlberg, E.O.; Storling, Z.M.; Olason, P.I.; Pedersen, A.G.; Rigina, O.; Hinsby, A.M.; Tumer, Z.; Pociot, F.; Tommerup, N.; et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007, 25, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, L.A.; Storling, Z.M.; Ortis, F.; Lage, K.; Bang-Berthelsen, C.; Bergholdt, R.; Hald, J.; Brorsson, C.A.; Eizirik, D.L.; Pociot, F.; et al. Huntingtin-interacting protein 14 is a type 1 diabetes candidate protein regulating insulin secretion and beta-cell apoptosis. Proc. Natl. Acad. Sci. USA 2011, 108, E681–E688. [Google Scholar] [CrossRef] [PubMed]
- Skotte, N.H.; Sanders, S.S.; Singaraja, R.R.; Ehrnhoefer, D.E.; Vaid, K.; Qiu, X.; Kannan, S.; Verma, C.; Hayden, M.R. Palmitoylation of caspase-6 by HIP14 regulates its activation. Cell Death Differ. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lyu, H.; Huang, J.; Liu, B. Targeting of ErbB3 receptor to overcome resistance in cancer treatment. Mol. Cancer 2014, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Hakonarson, H.; Qu, H.Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A novel susceptibility locus for type 1 diabetes on chr12q13 identified by a genome-wide association study. Diabetes 2008, 57, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Keene, K.L.; Quinlan, A.R.; Hou, X.; Hall, I.M.; Mychaleckyj, J.C.; Onengut-Gumuscu, S.; Concannon, P. Evidence for two independent associations with type 1 diabetes at the 12q13 locus. Genes Immun. 2012, 13, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F.; et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007, 39, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium; The Australo-Anglo-American Spondylitis Consortium; Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007, 39, 1329–1337. [Google Scholar]
- Kaur, S.; Mirza, A.H.; Brorsson, C.A.; Floyel, T.; Storling, J.; Mortensen, H.B.; Pociot, F.; Hvidoere International Study Group. The genetic and regulatory architecture of ErbB3-type 1 diabetes susceptibility locus. Mol. Cell. Endocrinol. 2016, 419, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ErbB2 and discovering ErbB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Nejentsev, S.; Walker, N.; Riches, D.; Egholm, M.; Todd, J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009, 324, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Downes, K.; Pekalski, M.; Angus, K.L.; Hardy, M.; Nutland, S.; Smyth, D.J.; Walker, N.M.; Wallace, C.; Todd, J.A. Reduced expression of IFIH1 is protective for type 1 diabetes. PLoS ONE 2010, 5, e12646. [Google Scholar] [CrossRef] [PubMed]
- Colli, M.L.; Moore, F.; Gurzov, E.N.; Ortis, F.; Eizirik, D.L. MDA5 and PTPN2, two candidate genes for type 1 diabetes, modify pancreatic beta-cell responses to the viral by-product double-stranded RNA. Hum. Mol. Genet. 2010, 19, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Skog, O.; Korsgren, O.; Frisk, G. Modulation of innate immunity in human pancreatic islets infected with enterovirus in vitro. J. Med. Virol. 2011, 83, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Santin, I.; Eizirik, D.L. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and beta-cell apoptosis. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 71–81. [Google Scholar] [CrossRef] [PubMed]
- Lincez, P.J.; Shanina, I.; Horwitz, M.S. Reduced expression of the MDA5 gene ifih1 prevents autoimmune diabetes. Diabetes 2015, 64, 2184–2193. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.Y.; Smyth, D.J.; Howson, J.M.; Cooper, J.D.; Walker, N.M.; Stevens, H.; Wicker, L.S.; Todd, J.A. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/tnfaip3 as a susceptibility locus. Genes Immun. 2009, 10, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Liuwantara, D.; Elliot, M.; Smith, M.W.; Yam, A.O.; Walters, S.N.; Marino, E.; McShea, A.; Grey, S.T. Nuclear factor-kappab regulates beta-cell death: A critical role for a20 in beta-cell protection. Diabetes 2006, 55, 2491–2501. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, M.; Brorsson, C.A.; Meyerovich, K.; Catrysse, L.; Delaroche, D.; Vanzela, E.C.; Ortis, F.; Beyaert, R.; Nielsen, L.B.; Andersen, M.L.; et al. A20 inhibits beta-cell apoptosis by multiple mechanisms and predicts residual beta-cell function in type 1 diabetes. Mol. Endocrinol. 2016, 30, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Grey, S.T.; Arvelo, M.B.; Hasenkamp, W.; Bach, F.H.; Ferran, C. A20 inhibits cytokine-induced apoptosis and nuclear factor kappab-dependent gene activation in islets. J. Exp. Med. 1999, 190, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Grey, S.T.; Longo, C.; Shukri, T.; Patel, V.I.; Csizmadia, E.; Daniel, S.; Arvelo, M.B.; Tchipashvili, V.; Ferran, C. Genetic engineering of a suboptimal islet graft with a20 preserves beta cell mass and function. J. Immunol. 2003, 170, 6250–6256. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.Y.; Lin, B.; Zhang, Z.L.; Guo, L.H. Direct transfer of a20 gene into pancreas protected mice from streptozotocin-induced diabetes. Acta Pharmacol. Sin. 2004, 25, 721–726. [Google Scholar] [PubMed]
- Cho, Y.S.; Chen, C.H.; Hu, C.; Long, J.; Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east asians. Nat. Genet. 2011, 44, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Senee, V.; Chelala, C.; Duchatelet, S.; Feng, D.; Blanc, H.; Cossec, J.C.; Charon, C.; Nicolino, M.; Boileau, P.; Cavener, D.R.; et al. Mutations in glis3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat. Genet. 2006, 38, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Kim, Y.S.; ZeRuth, G.; Beak, J.Y.; Gerrish, K.; Kilic, G.; Sosa-Pineda, B.; Jensen, J.; Pierreux, C.E.; Lemaigre, F.P.; et al. Transcription factor glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol. Cell. Biol. 2009, 29, 6366–6379. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chang, B.H.; Chan, L. Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol. Med. 2013, 5, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chang, B.H.; Samson, S.L.; Li, M.V.; Chan, L. The kruppel-like zinc finger protein GLIS3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. 2009, 37, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, T.C.; Paula, F.M.; Villate, O.; Colli, M.L.; Moura, R.F.; Cunha, D.A.; Marselli, L.; Marchetti, P.; Cnop, M.; Julier, C.; et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein bim. PLoS Genet. 2013, 9, e1003532. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Barker, A.; Sharp, S.J.; Timpson, N.J.; Bouatia-Naji, N.; Warrington, N.M.; Kanoni, S.; Beilin, L.J.; Brage, S.; Deloukas, P.; Evans, D.M.; et al. Association of genetic loci with glucose levels in childhood and adolescence: A meta-analysis of over 6000 children. Diabetes 2011, 60, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.; Colli, M.L.; Cnop, M.; Esteve, M.I.; Cardozo, A.K.; Cunha, D.A.; Bugliani, M.; Marchetti, P.; Eizirik, D.L. PTPN2, a candidate gene for type 1 diabetes, modulates interferon-gamma-induced pancreatic beta-cell apoptosis. Diabetes 2009, 58, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Santin, I.; Moore, F.; Colli, M.L.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic beta-cell apoptosis via regulation of the BH3-only protein bim. Diabetes 2011, 60, 3279–3288. [Google Scholar] [CrossRef] [PubMed]
- Floyel, T.; Brorsson, C.; Nielsen, L.B.; Miani, M.; Bang-Berthelsen, C.H.; Friedrichsen, M.; Overgaard, A.J.; Berchtold, L.A.; Wiberg, A.; Poulsen, P.; et al. Ctsh regulates beta-cell function and disease progression in newly diagnosed type 1 diabetes patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10305–10310. [Google Scholar] [CrossRef] [PubMed]
- GTEx Portal. Available online: https://gtexportal.org (accessed on 1st November 2016).
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, J.A.; Clayton, D.G. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.; Mine, K.; Inoue, Y.; Teshima, M.; Ogawa, S.; Kai, Y.; Kurafuji, T.; Hirakawa, K.; Miyakawa, D.; Ikeda, H.; et al. Reduced TYK2 gene expression in beta-cells due to natural mutation determines susceptibility to virus-induced diabetes. Nat. Commun. 2015, 6, 6748. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Floyel, T.; Grieco, F.A.; Santin, I.; Op de Beeck, A.; Marselli, L.; Marchetti, P.; Pociot, F.; Eizirik, D.L. Tyk2, a candidate gene for type 1 diabetes, modulates apoptosis and the innate immune response in human pancreatic beta-cells. Diabetes 2015, 64, 3808–3817. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, H.; Zhao, M.; Chang, C.; Lu, Q. The bach family of transcription factors: A comprehensive review. Clin. Rev. Allergy Immunol. 2016, 50, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Santin, I.; Dos Santos, R.S.; Marselli, L.; Marchetti, P.; Eizirik, D.L. BACH2, a candidate risk gene for type 1 diabetes, regulates apoptosis in pancreatic beta-cells via jnk1 modulation and crosstalk with the candidate gene ptpn2. Diabetes 2014, 63, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Brorsson, C.A.; Nielsen, L.B.; Andersen, M.L.; Kaur, S.; Bergholdt, R.; Hansen, L.; Mortensen, H.B.; Pociot, F.; Storling, J.; Hvidoere Study Group on Childhood Diabetes. Genetic risk score modelling for disease progression in new-onset type 1 diabetes patients: Increased genetic load of islet-expressed and cytokine-regulated candidate genes predicts poorer glycemic control. J. Diabetes Res. 2016, 2016, 9570424. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jorgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Farquharson, M.A.; Hardman, R. Aberrant expression of class II major histocompatibility complex molecules by b cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1987, 30, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, T.; Miyazaki, A.; Miyagawa, J.; Tamura, S.; Inada, M.; Yamada, K.; Shinji, Y.; Katsura, H.; Yamagata, K.; Itoh, N.; et al. Examination of islets in the pancreas biopsy specimens from newly diagnosed type 1 (insulin-dependent) diabetic patients. Diabetologia 1990, 33, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Storling, J.; Overgaard, A.J.; Brorsson, C.A.; Piva, F.; Bang-Berthelsen, C.H.; Haase, C.; Nerup, J.; Pociot, F. Do post-translational beta cell protein modifications trigger type 1 diabetes? Diabetologia 2013, 56, 2347–2354. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Kalis, M.; Bolmeson, C.; Esguerra, J.L.; Gupta, S.; Edlund, A.; Tormo-Badia, N.; Speidel, D.; Holmberg, D.; Mayans, S.; Khoo, N.K.; et al. Beta-cell specific deletion of DICER1 leads to defective insulin secretion and diabetes mellitus. PLoS ONE 2011, 6, e29166. [Google Scholar] [CrossRef] [PubMed]
- Melkman-Zehavi, T.; Oren, R.; Kredo-Russo, S.; Shapira, T.; Mandelbaum, A.D.; Rivkin, N.; Nir, T.; Lennox, K.A.; Behlke, M.A.; Dor, Y.; et al. Mirnas control insulin content in pancreatic beta-cells via downregulation of transcriptional repressors. EMBO J. 2011, 30, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, S. Minireview: Microrna function in pancreatic beta cells. Mol. Endocrinol. 2014, 28, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- De Jong, V.M.; Zaldumbide, A.; van der Slik, A.R.; Persengiev, S.P.; Roep, B.O.; Koeleman, B.P. Post-transcriptional control of candidate risk genes for type 1 diabetes by rare genetic variants. Genes Immun. 2013, 14, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Xiao, D.; Ming, G.; Yin, J.; Zhou, H.; Liu, Z. Type 2 diabetes mellitus-related genetic polymorphisms in microRNAs and microRNA target sites. J. Diabetes 2014, 6, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Menoud, V.; Rome, S.; Regazzi, R. Horizontal transfer of exosomal micrornas transduce apoptotic signals between pancreatic beta-cells. Cell Commun. Signal. 2015, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Samandari, N.; Mirza, A.H.; Nielsen, L.B.; Kaur, S.; Hougaard, P.; Fredheim, S.; Mortensen, H.B.; Pociot, F. Circulating microrna levels predict residual beta cell function and glycaemic control in children with type 1 diabetes mellitus. Diabetologia 2017, 60, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Grieco, F.A.; Sebastiani, G.; Juan-Mateu, J.; Villate, O.; Marroqui, L.; Ladriere, L.; Tugay, K.; Regazzi, R.; Bugliani, M.; Marchetti, P.; et al. MicroRNAs mir-23a-3p, mir-23b-3p, and mir-149–5p regulate the expression of proapoptotic BH3-only proteins DP5 and puma in human pancreatic beta-cells. Diabetes 2017, 66, 100–112. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Størling, J.; Pociot, F. Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis. Genes 2017, 8, 72. https://doi.org/10.3390/genes8020072
Størling J, Pociot F. Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis. Genes. 2017; 8(2):72. https://doi.org/10.3390/genes8020072
Chicago/Turabian StyleStørling, Joachim, and Flemming Pociot. 2017. "Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis" Genes 8, no. 2: 72. https://doi.org/10.3390/genes8020072
APA StyleStørling, J., & Pociot, F. (2017). Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis. Genes, 8(2), 72. https://doi.org/10.3390/genes8020072