Using Human iPSC-Derived Neurons to Uncover Activity-Dependent Non-Coding RNAs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
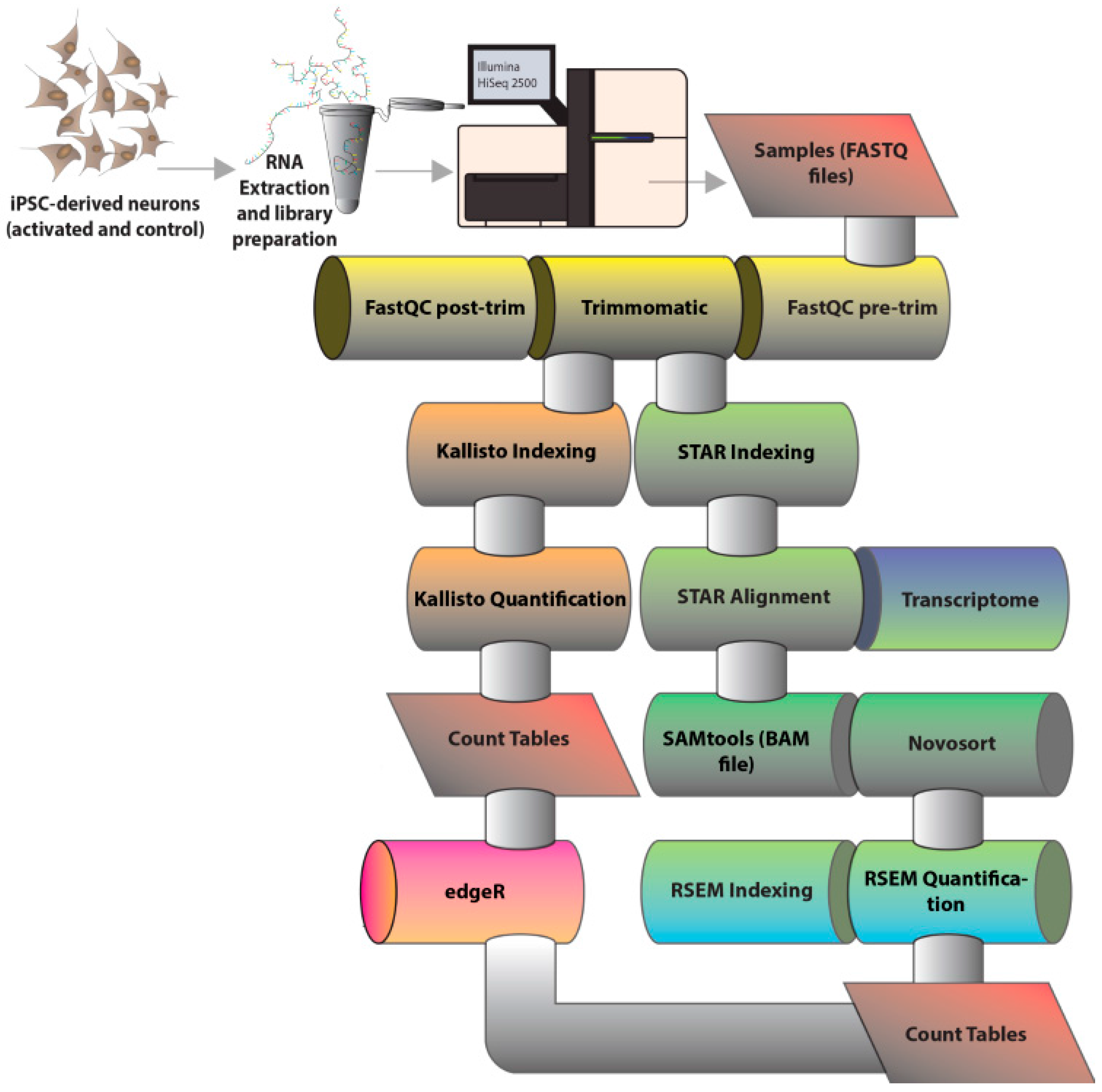
2. Materials and Methods
2.1. Generation and Differentiation of Induced Pluripotent Stem Cells
2.2. Neuronal Activation and RNA Sequencing
2.3. Bioinformatics Analysis of RNA Sequencing Results
2.4. Functional Analyses of Differentially Expressed Genes
3. Results and Discussion
3.1. Overall Quality of RNA-Sequencing Data
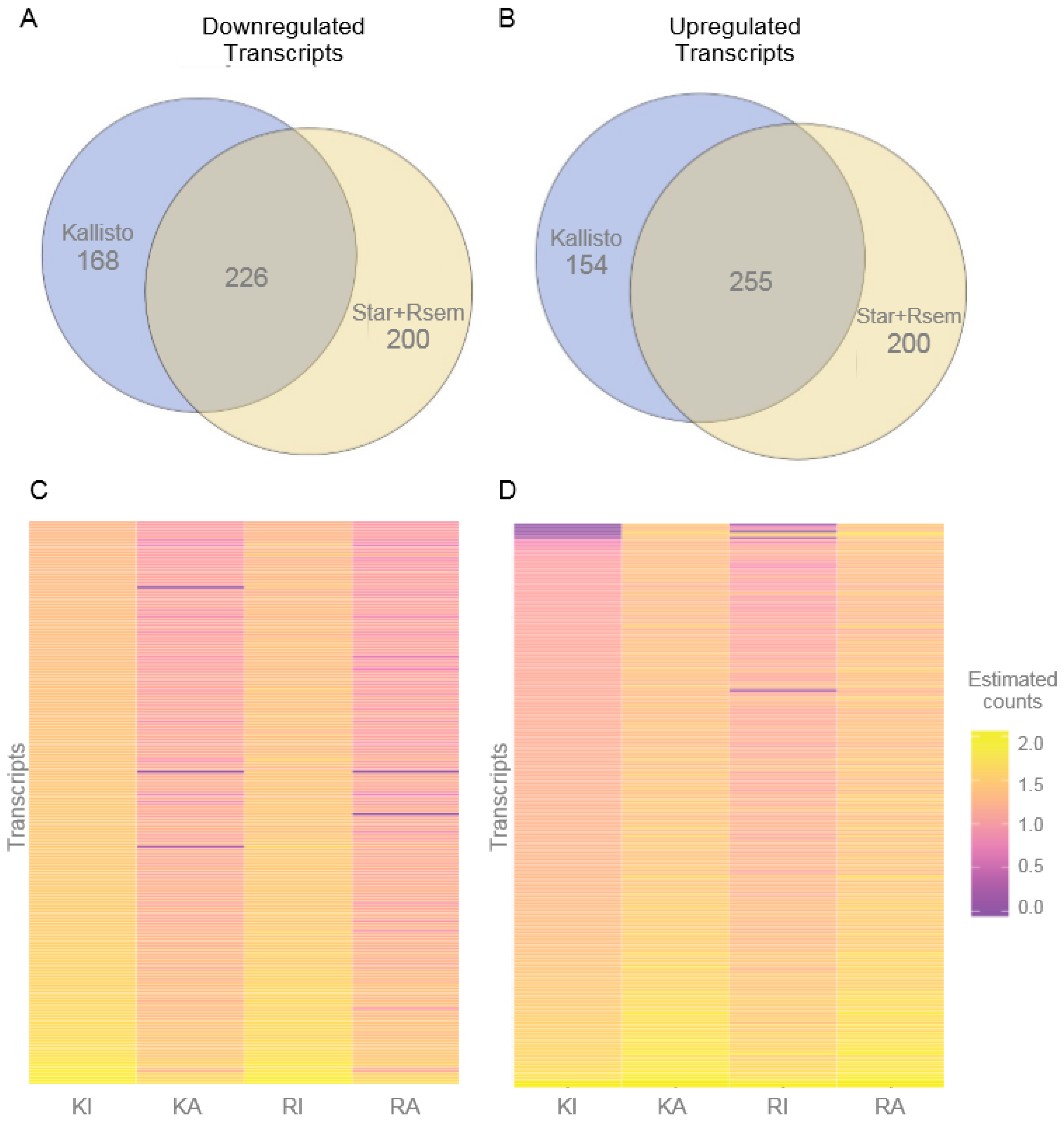
3.2. Differentially Expressed Transcripts
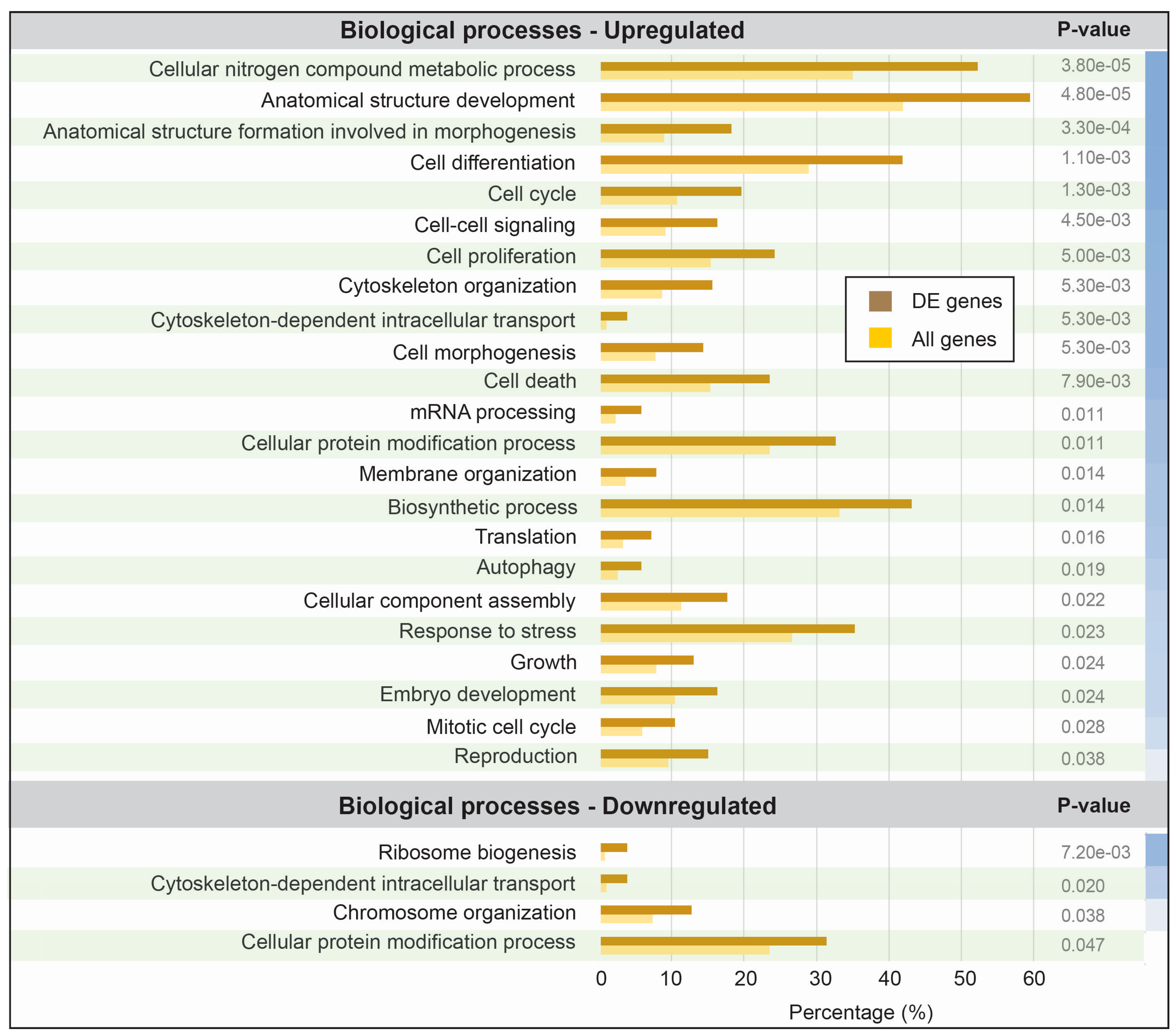
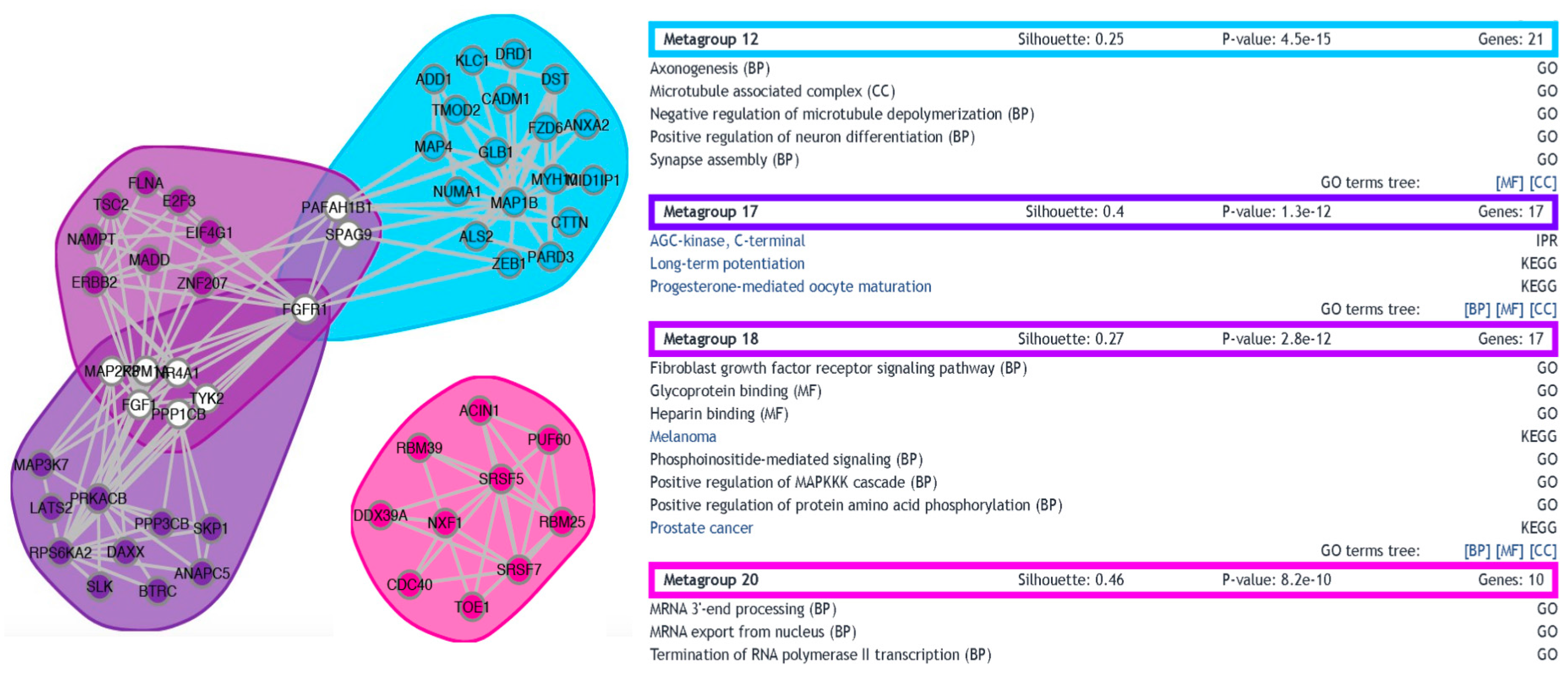
3.3. Functional Analysis of Differentially Expressed Transcripts
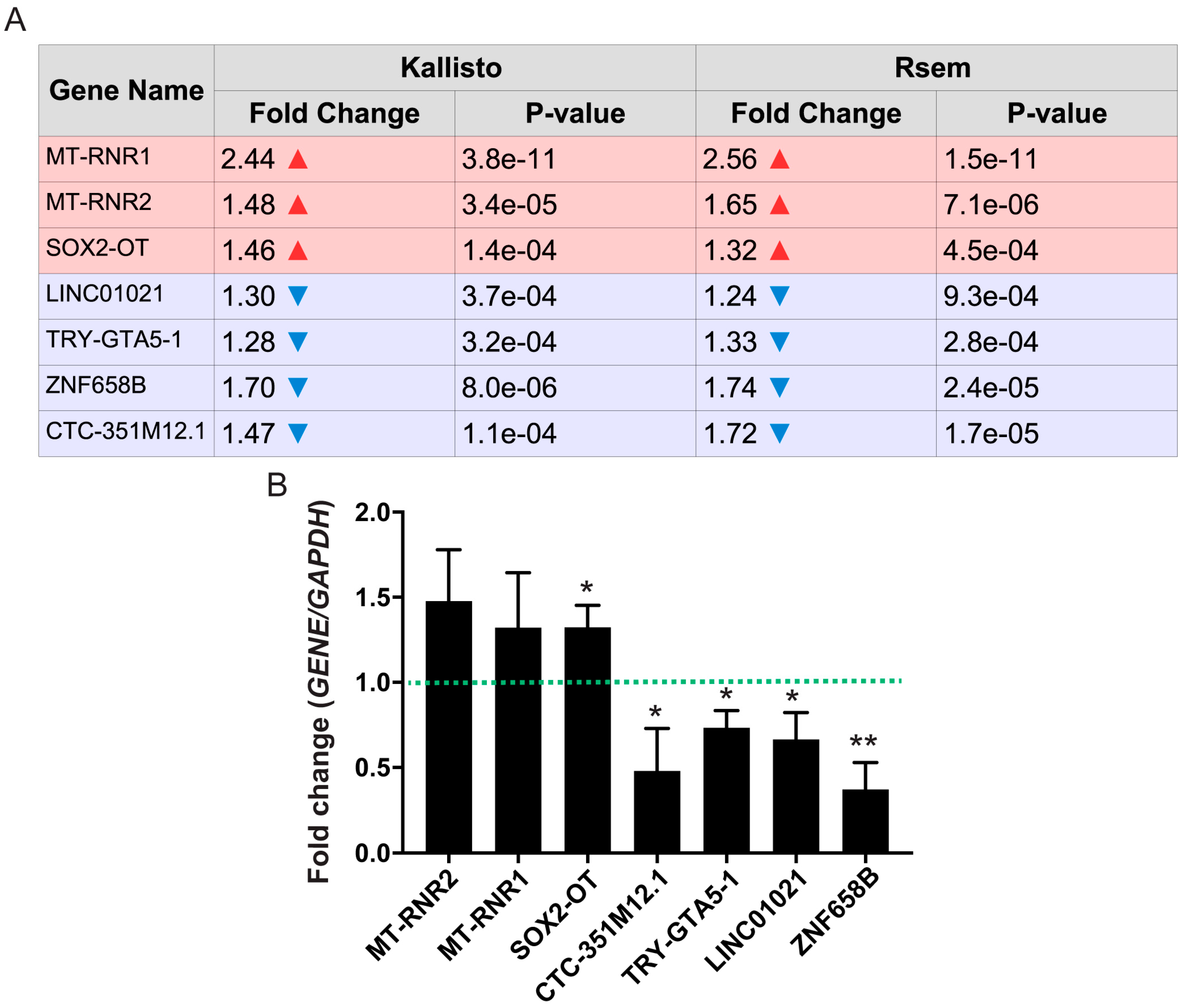
3.4. Differentially Expressed Non-Coding RNAs
3.5. Human-Specific Differentially Expressed Genes
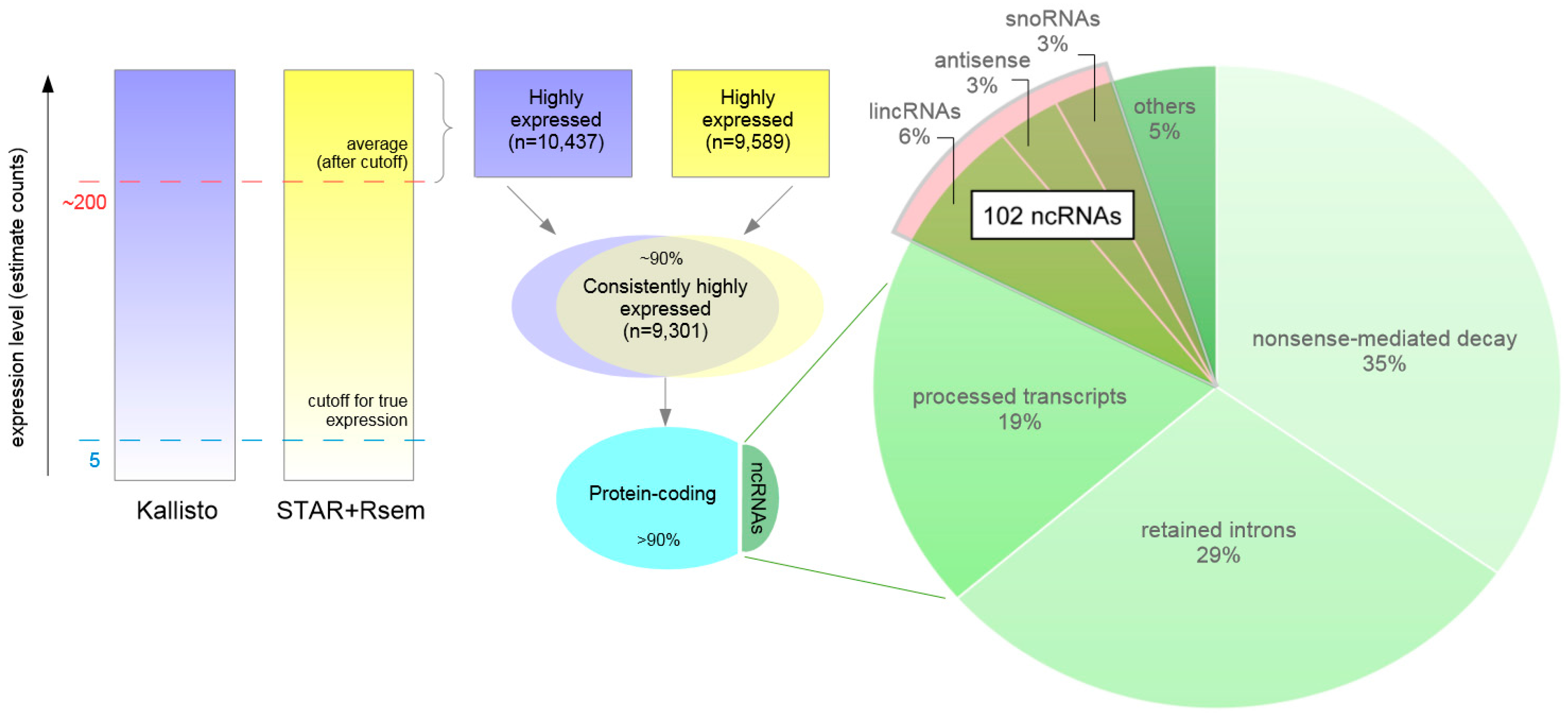
3.6. Highly Expressed Transcripts
3.7. Highly Expressed Protein-Coding Transcripts Involved in Brain Function, Development, and Evolution
3.8. Highly Expressed ncRNAs
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Calcagno, J.M.; Fuentes, A. What makes us human? Answers from evolutionary anthropology. Evol. Anthropol. 2012, 21, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Preuss, T.M. The human brain: Rewired and running hot. Ann. N. Y. Acad. Sci. 2011, 1225 (Suppl. 1), E182–E191. [Google Scholar] [CrossRef] [PubMed]
- Pillay, P.; Manger, P.R. Order-specific quantitative patterns of cortical gyrification. Eur. J. Neurosci. 2007, 25, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Chimpanzee, S.; Analysis, C. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437, 69–87. [Google Scholar]
- Xie, C.; Yuan, J.; Li, H.; Li, M.; Zhao, G.; Bu, D.; Zhu, W.; Wu, W.; Chen, R.; Zhao, Y. NONCODEv4: Exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014, 42, D98–D103. [Google Scholar] [CrossRef] [PubMed]
- Awan, H.M.; Shah, A.; Rashid, F.; Shan, G. Primate-specific long non-coding RNAs and microRNAs. Genomics Proteom. Bioinform. 2017, 15, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grutzner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E. Evolution of microRNA diversity and regulation in animals. Nat. Rev. Genet. 2011, 12, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Mattick, J.S.; Taft, R.J. A meta-analysis of the genomic and transcriptomic composition of complex life. Cell Cycle 2013, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Bammann, H.; Han, D.; Xie, G.; Khaitovich, P. Conserved expression of lincRNA during human and macaque prefrontal cortex development and maturation. RNA 2014, 20, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.G.; He, L.; Li, Z.; Xu, Y.; Li, M.; Fu, X.; Yan, Z.; Yuan, Y.; Menzel, C.; Li, N.; et al. Intergenic and repeat transcription in human, chimpanzee and macaque brains measured by RNA-Seq. PLoS Comput. Biol. 2010, 6, e1000843. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Lehner, B.; Williams, G.; Campbell, R.D.; Sanderson, C.M. Antisense transcripts in the human genome. Trends Genet. 2002, 18, 63–65. [Google Scholar] [CrossRef]
- Hu, X.; Sood, A.K.; Dang, C.V.; Zhang, L. The role of long noncoding RNAs in cancer: The dark matter matters. Curr. Opin. Genet. Dev. 2017, 48, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, R.S.; Rajagopalan, S.; Natarajan, R.; Deiuliis, J.A. Non-coding RNAs in cardiovascular disease: Pathological relevance and emerging role as biomarkers and therapeutics. Am. J. Hypertens. 2017. [Google Scholar] [CrossRef] [PubMed]
- Leti, F.; DiStefano, J.K. Long non-coding RNAas as diagnostic and therapeutic targets in type 2 diabetes and related complications. Genes 2017, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Morris, K.V.; Wood, M.J. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos. Trans. R. Soc. Lond. Ser. B 2014, 369, 20130507. [Google Scholar] [CrossRef] [PubMed]
- Barry, G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol. Psychiatry 2014, 19, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, D.; Schrewe, H.; Orom, U.A. Molecular mechanisms of long ncRNAs in neurological disorders. Front. Genet. 2014, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- D’Haene, E.; Jacobs, E.Z.; Volders, P.J.; De Meyer, T.; Menten, B.; Vergult, S. Identification of long non-coding RNAs involved in neuronal development and intellectual disability. Sci. Rep. 2016, 6, 28396. [Google Scholar] [CrossRef] [PubMed]
- Maag, J.L.; Panja, D.; Sporild, I.; Patil, S.; Kaczorowski, D.C.; Bramham, C.R.; Dinger, M.E.; Wibrand, K. Dynamic expression of long non-coding RNAs and repeat elements in synaptic plasticity. Front. Neurosci. 2015, 9, 351. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; O’Sullivan, M.L.; Sanchez, C.A.; Hwang, M.; Israel, M.A.; Brennand, K.; Deerinck, T.J.; Goldstein, L.S.; Gage, F.H.; Ellisman, M.H.; et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Pereda, A.E. Electrical synapses and their functional interactions with chemical synapses. Nat. Rev. Neurosci. 2014, 15, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Roussos, P.; Guennewig, B.; Kaczorowski, D.C.; Barry, G.; Brennand, K.J. Activity-dependent changes in gene expression in schizophrenia human-induced pluripotent stem cell neurons. JAMA Psychiatry 2016, 73, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Hook, V.; Brennand, K.J.; Kim, Y.; Toneff, T.; Funkelstein, L.; Lee, K.C.; Ziegler, M.; Gage, F.H. Human iPSC neurons display activity-dependent neurotransmitter secretion: Aberrant catecholamine levels in schizophrenia neurons. Stem Cell Rep. 2014, 3, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—FastQC A Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 February 2017).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-Seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- SAMtools. Available online: http://samtools.sourceforge.net (accessed on 1 February 2017).
- NovoSort | Novocraft. Available online: http://www.novocraft.com/documentation/novosort-2/ (accessed on 1 February 2017).
- McCarthy, F.M.; Wang, N.; Magee, G.B.; Nanduri, B.; Lawrence, M.L.; Camon, E.B.; Barrell, D.G.; Hill, D.P.; Dolan, M.E.; Williams, W.P.; et al. AgBase: A functional genomics resource for agriculture. BMC Genomics 2006, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; Fontanillo, C.; Droste, C.; De Las Rivas, J. Functional gene networks: R/Bioc package to generate and analyse gene networks derived from functional enrichment and clustering. Bioinformatics 2015, 31, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.L.; White, A.; Black, D.M.; Wallace, R.H.; Sah, P.; Bartlett, P.F. Latent stem and progenitor cells in the hippocampus are activated by neural excitation. J. Neurosci. 2008, 28, 5240–5247. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, J.; Park, D.S.; Greene, L.A.; Shelanski, M.L. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J. Neurosci. 1999, 19, 8747–8756. [Google Scholar] [PubMed]
- Herberth, B.; Pataki, A.; Jelitai, M.; Schlett, K.; Deak, F.; Spat, A.; Madarasz, E. Changes of KCl sensitivity of proliferating neural progenitors during in vitro neurogenesis. J. Neurosci. Res. 2002, 67, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ito, Y.; Niikura, T.; Shao, Z.; Hata, M.; Oyama, F.; Nishimoto, I. Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem. Biophys. Res. Commun. 2001, 283, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.S.; Duan, D.X.; Ma, R.H.; Shen, J.Y.; Li, H.L.; Ma, Z.W.; Luo, Y.; Wang, L.; Qi, X.H.; Wang, Q.; et al. Humanin attenuates Alzheimer-like cognitive deficits and pathological changes induced by amyloid β-peptide in rats. Neurosci. Bull. 2014, 30, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin peptide suppresses apoptosis by interfering with bax activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, X.J.; Han, W.N.; Li, Q.S.; Wang, Z.J.; Wu, M.N.; Yang, W.; Qi, J.S. [Gly14]-Humanin protects against amyloid β peptide-induced impairment of spatial learning and memory in rats. Neurosci. Bull. 2016, 32, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell. Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Neyt, C.; Wilkins, S.J.; Askarian-Amiri, M.E.; Sunkin, S.M.; Perkins, A.C.; Mattick, J.S. Complex architecture and regulated expression of the Sox2ot locus during vertebrate development. RNA 2009, 15, 2013–2027. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, R.; Lian, M.; Ma, H.; He, N.; Liu, H.; Wang, H.; Fang, J. Integrated analysis of long noncoding RNAs and mRNA expression profile in advanced laryngeal squamous cell carcinoma. PLoS ONE 2016, 11, e0169232. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Wang, Y.; Zhao, Z.; Zhang, J.; Lu, J.; Xu, J.; Li, X. Identification and characterization of lncRNA mediated transcriptional dysregulation dictates lncRNA roles in glioblastoma. Oncotarget 2016, 7, 45027–45041. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, M.; Chen, G.; Li, Z.; Lu, X. LncRNA SOX2-OT is a novel prognostic biomarker for osteosarcoma patients and regulates osteosarcoma cells proliferation and motility through modulating SOX2. IUBMB Life 2017, 69, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Zhao, W.; Zhou, J.; Shen, L.; Zhan, P.; Xu, C.; Chang, C.; Bi, H.; Zou, J.; Yao, X.; et al. A long noncoding RNA Sox2ot regulates lung cancer cell proliferation and is a prognostic indicator of poor survival. Int. J. Biochem. Cell. Biol. 2014, 53, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Winham, S.J.; Cuellar-Barboza, A.B.; McElroy, S.L.; Oliveros, A.; Crow, S.; Colby, C.L.; Choi, D.S.; Chauhan, M.; Frye, M.A.; Biernacka, J.M. Bipolar disorder with comorbid binge eating history: A genome-wide association study implicates APOB. J. Affect. Disord. 2014, 165, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Subramanian, M.; Jones, M.F.; Chaudhary, R.; Singh, D.K.; Zong, X.; Gryder, B.; Sindri, S.; Mo, M.; Schetter, A.; et al. Long noncoding RNA PURPL suppresses basal p53 levels and promotes tumorigenicity in colorectal cancer. Cell Rep. 2017, 20, 2408–2423. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.; Grepo, N.; Thompson, B.L.; Kim, J.; Wang, K.; Evgrafov, O.V.; Lu, W.; Knowles, J.A.; Campbell, D.B. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl. Psychiatry 2015, 5, e568. [Google Scholar] [CrossRef] [PubMed]
- Ogo, O.A.; Tyson, J.; Cockell, S.J.; Howard, A.; Valentine, R.A.; Ford, D. The zinc finger protein ZNF658 regulates the transcription of genes involved in zinc homeostasis and affects ribosome biogenesis through the zinc transcriptional regulatory element. Mol. Cell. Biol. 2015, 35, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.; Weirick, T.; John, D.; Militello, G.; Chen, W.; Dimmeler, S.; Uchida, S. Angiogenes: Knowledge database for protein-coding and noncoding RNA genes in endothelial cells. Sci. Rep. 2016, 6, 32475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E.; Landback, P.; Vibranovski, M.D.; Long, M. Accelerated recruitment of new brain development genes into the human genome. PLoS Biol. 2011, 9, e1001179. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Liu, Q.; Yu, L.; Yang, Y.; Lu, M.; Wang, H.; Luo, D.; Rong, X.; Tang, F.; Guo, J. Downregulations of CD36 and Calpain-1, inflammation, and atherosclerosis by simvastatin in apolipoprotein E knockout mice. J. Vasc. Res. 2017, 54, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gourguechon, S.; Wang, C.C. Tousled-like kinase in a microbial eukaryote regulates spindle assembly and S-phase progression by interacting with Aurora kinase and chromatin assembly factors. J. Cell Sci. 2007, 120, 3883–3894. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Sawada, A.; Chaturvedi, A.; Mishra, P.; Schuster-Gossler, K.; Galla, M.; Schambach, A.; Gossler, A.; Forster, R.; Heuser, M.; et al. Genetic deletion of SEPT7 reveals a cell type-specific role of septins in microtubule destabilization for the completion of cytokinesis. PLoS Genet. 2014, 10, e1004558. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.M.; Oliver, P.L.; Finelli, M.J.; Lee, S.; Lickiss, T.; Molnar, Z.; Davies, K.E. Laf4/Aff3, a gene involved in intellectual disability, is required for cellular migration in the mouse cerebral cortex. PLoS ONE 2014, 9, e105933. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.W.; Tsang, W.H.; Chan, S.O.; Li, H.M.; Ng, H.K.; Waye, M.M. Dyslexia-associated Kiaa0319-like protein interacts with axon guidance receptor Nogo receptor 1. Cell. Mol. Neurobiol. 2011, 31, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Qu, X.; Li, P.; Ma, Q.; Cao, C.; Liu, X. Global regulation of alternative RNA splicing by the SR-rich protein RBM39. Biochim. Biophys. Acta 2016, 1859, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Kong, R.; Zou, L.; Zhong, R.; Lou, J.; Zhou, J.; Guo, S.; Wang, J.; Zhang, X.; Zhang, J.; et al. The roles of genes in the neuronal migration and neurite outgrowth network in developmental dyslexia: Single- and multiple-risk genetic variants. Mol. Neurobiol. 2016, 53, 3967–3975. [Google Scholar] [CrossRef] [PubMed]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nature 2016, 530, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R.; Cole, S.W. Reciprocal regulation of the neural and innate immune systems. Nat. Rev. Immunol. 2011, 11, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Approaching the next revolution? Evolutionary integration of neural and immune pathogen sensing and response. Cold Spring Harb. Perspect. Biol. 2014, 7, a016360. [Google Scholar] [CrossRef] [PubMed]
- Segura-Bayona, S.; Knobel, P.A.; Gonzalez-Buron, H.; Youssef, S.A.; Pena-Blanco, A.; Coyaud, E.; Lopez-Rovira, T.; Rein, K.; Palenzuela, L.; Colombelli, J.; et al. Differential requirements for Tousled-like kinases 1 and 2 in mammalian development. Cell Death Differ. 2017, 24, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, W.; van den Berg, J.; Aprelia, M.; Medema, R.H. Tousled-like kinase 2 regulates recovery from a DNA damage-induced G2 arrest. EMBO Rep. 2016, 17, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Evolution of the human ASPM gene, a major determinant of brain size. Genetics 2003, 165, 2063–2070. [Google Scholar] [PubMed]
- Jimenez, S.A.; Piera-Velazquez, S. Potential role of human-specific genes, human-specific microRNAs and human-specific non-coding regulatory RNAs in the pathogenesis of systemic sclerosis and Sjögren’s syndrome. Autoimmun. Rev. 2013, 12, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.I.; Jayaraman, D.; Walsh, C.A. Genetic changes shaping the human brain. Dev. Cell 2015, 32, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.Y.; Nuttle, X.; Sudmant, P.H.; Antonacci, F.; Graves, T.A.; Nefedov, M.; Rosenfeld, J.A.; Sajjadian, S.; Malig, M.; Kotkiewicz, H.; et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 2012, 149, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Franchini, L.F.; Pollard, K.S. Genomic approaches to studying human-specific developmental traits. Development 2015, 142, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.L.; Cavallaro, M.; Braida, D.; Di Cristofano, A.; Canta, A.; Vezzani, A.; Ottolenghi, S.; Pandolfi, P.P.; Sala, M.; DeBiasi, S.; et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 2004, 131, 3805–3819. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feng, P.; Zhu, X.; He, S.; Duan, J.; Zhou, D. Long non-coding RNA Malat1 promotes neurite outgrowth through activation of ERK/MAPK signalling pathway in N2a cells. J. Cell. Mol. Med. 2016, 20, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Briggs, J.A.; Vanichkina, D.P.; Poth, E.M.; Beveridge, N.J.; Ratnu, V.S.; Nayler, S.P.; Nones, K.; Hu, J.; Bredy, T.W.; et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 2014, 19, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Bogu, G.K.; Soh, B.S.; Stanton, L.W. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Briggs, J.A.; Hwang, D.W.; Nayler, S.P.; Fortuna, P.R.; Jonkhout, N.; Dachet, F.; Maag, J.L.; Mestdagh, P.; Singh, E.M.; et al. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci. Rep. 2017, 7, 40127. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Rao, S.; Du, T.; Hu, H.; Liu, Z.; Shen, Y.; Xu, Q. Intergenic variants may predispose to major depression disorder through regulation of long non-coding RNA expression. Gene 2017, 601, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Donner, J.; Pirkola, S.; Silander, K.; Kananen, L.; Terwilliger, J.D.; Lonnqvist, J.; Peltonen, L.; Hovatta, I. An association analysis of murine anxiety genes in humans implicates novel candidate genes for anxiety disorders. Biol. Psychiatry 2008, 64, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Jung, S.E.; Hong, S.R.; Lee, E.H.; Lee, J.H.; Lee, S.D.; Lee, H.Y. Independent validation of DNA-based approaches for age prediction in blood. Forensic Sci. Int. Genet. 2017, 29, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Spolnicka, M.; Pospiech, E.; Peplonska, B.; Zbiec-Piekarska, R.; Makowska, Z.; Pieta, A.; Karlowska-Pik, J.; Ziemkiewicz, B.; Wezyk, M.; Gasperowicz, P.; et al. DNA methylation in ELOVL2 and C1orf132 correctly predicted chronological age of individuals from three disease groups. Int. J. Leg. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ribases, M.; Bosch, R.; Hervas, A.; Ramos-Quiroga, J.A.; Sanchez-Mora, C.; Bielsa, A.; Gastaminza, X.; Guijarro-Domingo, S.; Nogueira, M.; Gomez-Barros, N.; et al. Case-control study of six genes asymmetrically expressed in the two cerebral hemispheres: Association of BAIAP2 with attention-deficit/hyperactivity disorder. Biol. Psychiatry 2009, 66, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Luksys, G.; Ackermann, S.; Coynel, D.; Fastenrath, M.; Gschwind, L.; Heck, A.; Rasch, B.; Spalek, K.; Vogler, C.; Papassotiropoulos, A.; et al. BAIAP2 is related to emotional modulation of human memory strength. PLoS ONE 2014, 9, e83707. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Shao, M.; Sun, H.; Shen, Y.; Meng, D.; Huo, W. Long non-coding RNA SNHG14 promotes microglia activation by regulating miR-145-5p/PLA2G4A in cerebral infarction. Neuroscience 2017, 348, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Abdelmohsen, K.; Yang, X.; De, S.; Grammatikakis, I.; Noh, J.H.; Gorospe, M. LncRNA OIP5-AS1/cyrano sponges RNA-binding protein HuR. Nucleic Acids Res. 2016, 44, 2378–2392. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.N.; Starmer, J.; Miller, S.C.; Sethupathy, P.; Magnuson, T. Long noncoding RNA moderates microrna activity to maintain self-renewal in embryonic stem cells. Stem Cell Rep. 2017, 9, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Pandey, R.R.; Nagano, T.; Chakalova, L.; Mondal, T.; Fraser, P.; Kanduri, C. Kcnq1ot1/Lit1 noncoding RNA mediates transcriptional silencing by targeting to the perinucleolar region. Mol. Cell. Biol. 2008, 28, 3713–3728. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Mondal, T.; Guseva, N.; Pandey, G.K.; Kanduri, C. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 2010, 137, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Dolmetsch, R.; Geschwind, D.H. The human brain in a dish: The promise of iPSC-derived neurons. Cell 2011, 145, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.J.; Simone, A.; Tran, N.; Gage, F.H. Modeling psychiatric disorders at the cellular and network levels. Mol. Psychiatry 2012, 17, 1239–1253. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bitar, M.; Kuiper, S.; O’Brien, E.; Barry, G. Using Human iPSC-Derived Neurons to Uncover Activity-Dependent Non-Coding RNAs. Genes 2017, 8, 401. https://doi.org/10.3390/genes8120401
Bitar M, Kuiper S, O’Brien E, Barry G. Using Human iPSC-Derived Neurons to Uncover Activity-Dependent Non-Coding RNAs. Genes. 2017; 8(12):401. https://doi.org/10.3390/genes8120401
Chicago/Turabian StyleBitar, Mainá, Stefanie Kuiper, Elizabeth O’Brien, and Guy Barry. 2017. "Using Human iPSC-Derived Neurons to Uncover Activity-Dependent Non-Coding RNAs" Genes 8, no. 12: 401. https://doi.org/10.3390/genes8120401
APA StyleBitar, M., Kuiper, S., O’Brien, E., & Barry, G. (2017). Using Human iPSC-Derived Neurons to Uncover Activity-Dependent Non-Coding RNAs. Genes, 8(12), 401. https://doi.org/10.3390/genes8120401