Genome Editing Tools in Plants



 and
and 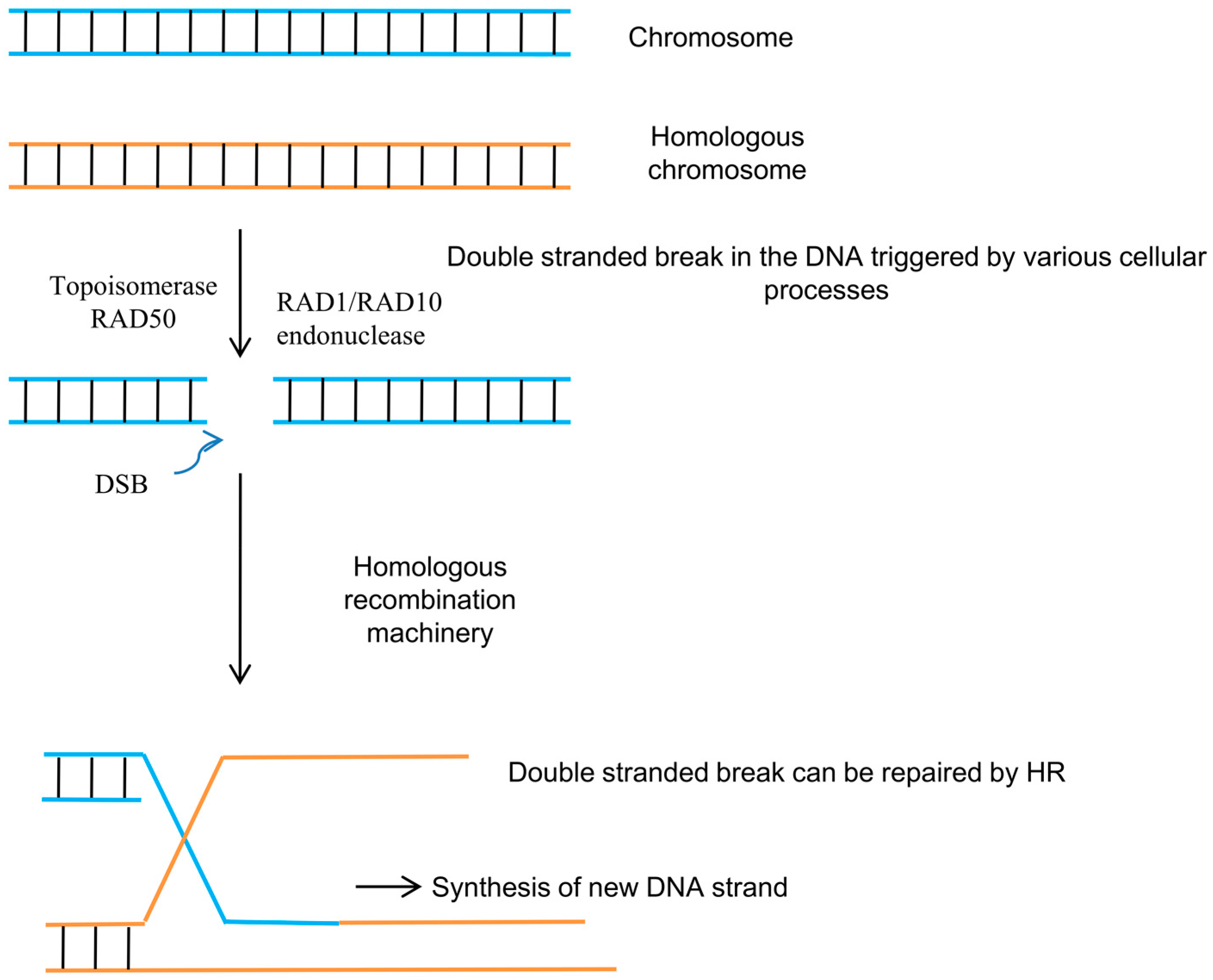{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Homologous Recombination
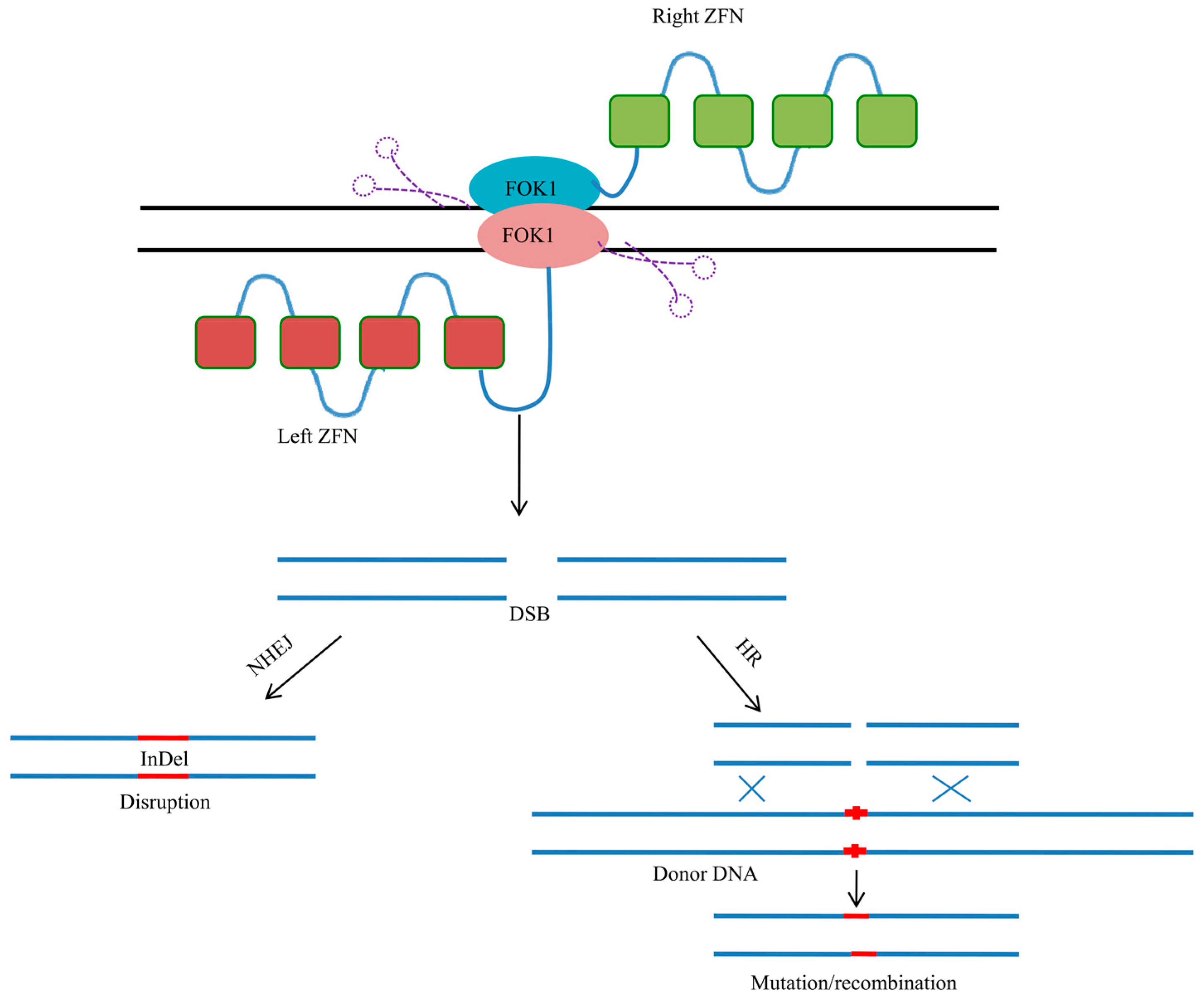
3. Zinc Finger Nucleases
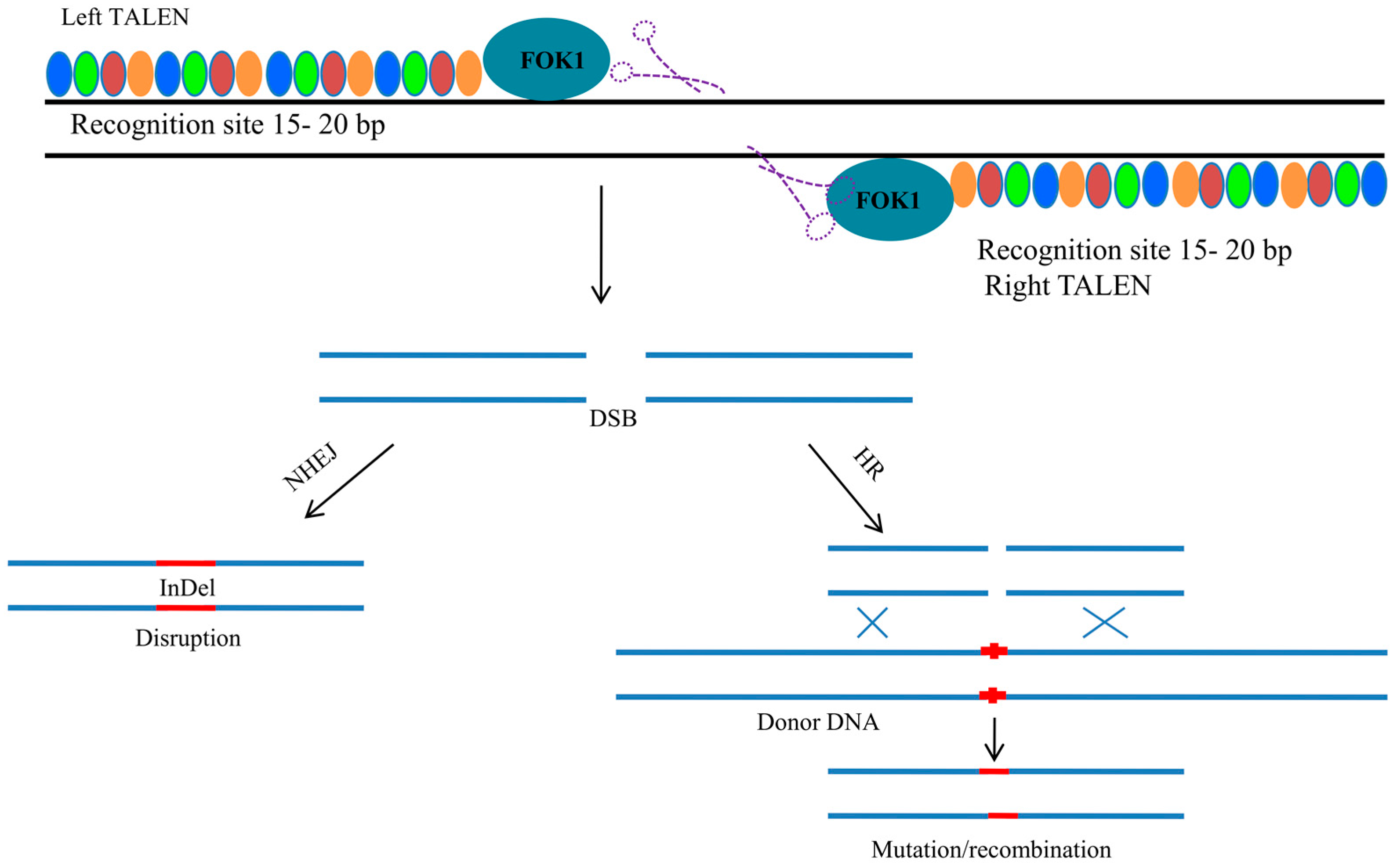
4. Transcription Activator-Like Effector Nuclease
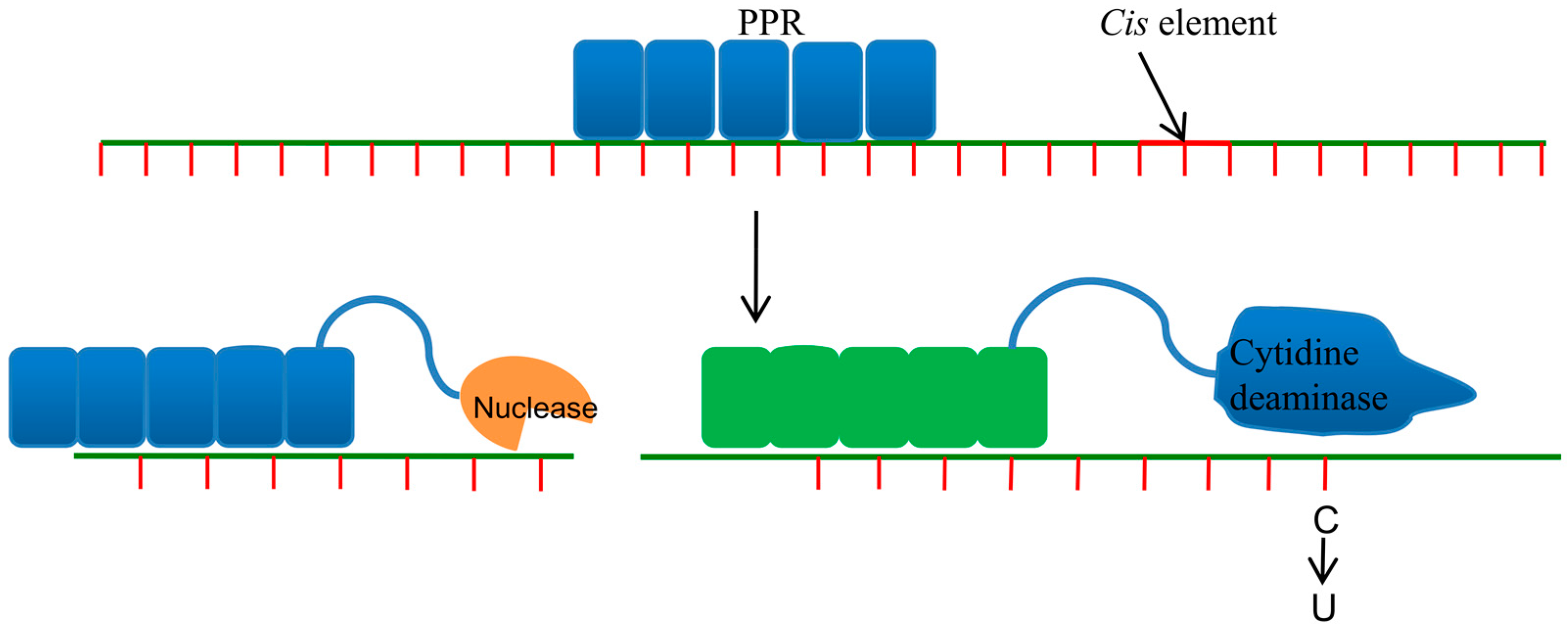
5. Pentatricopeptide Repeat Proteins
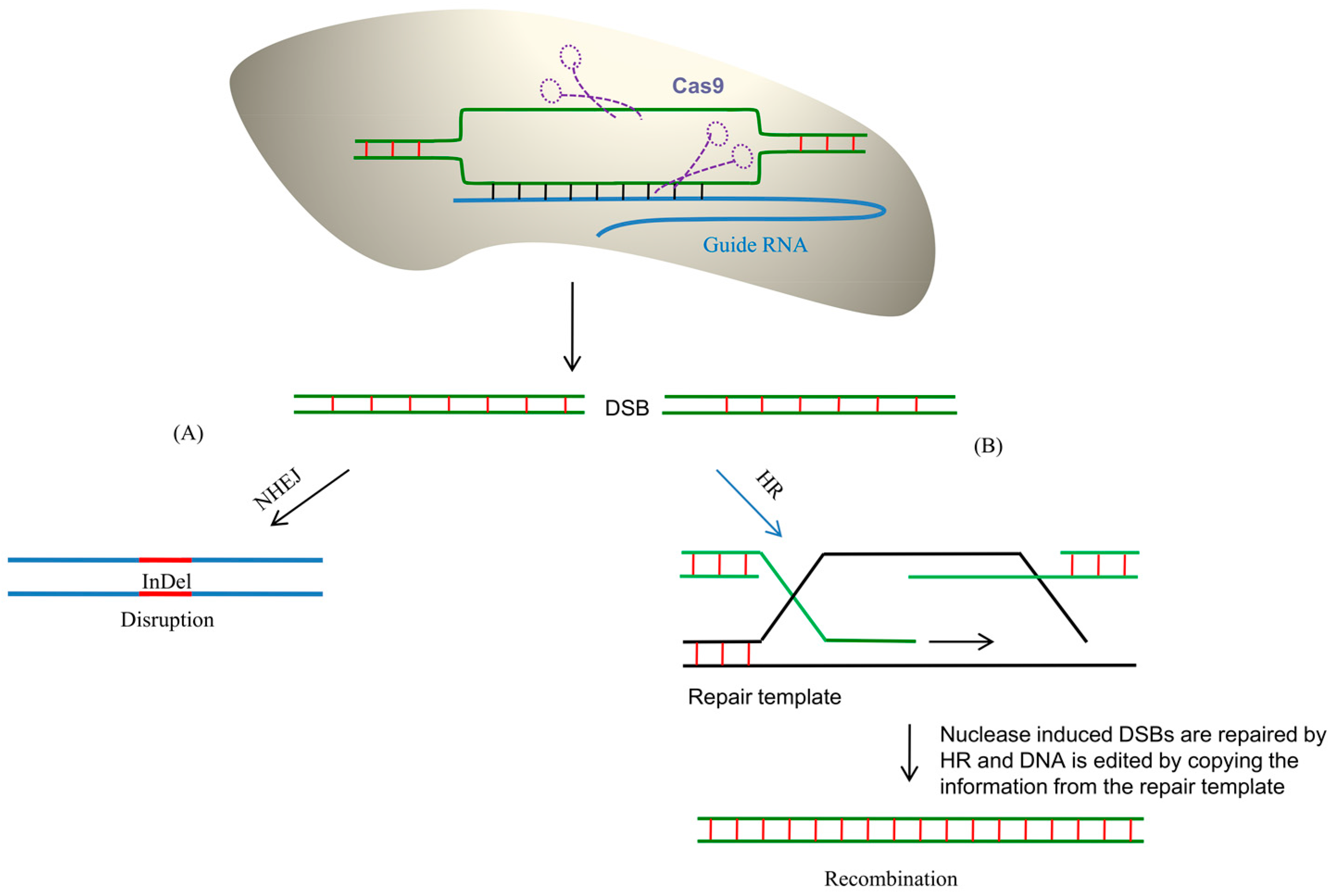
6. CRISPR/Cas9
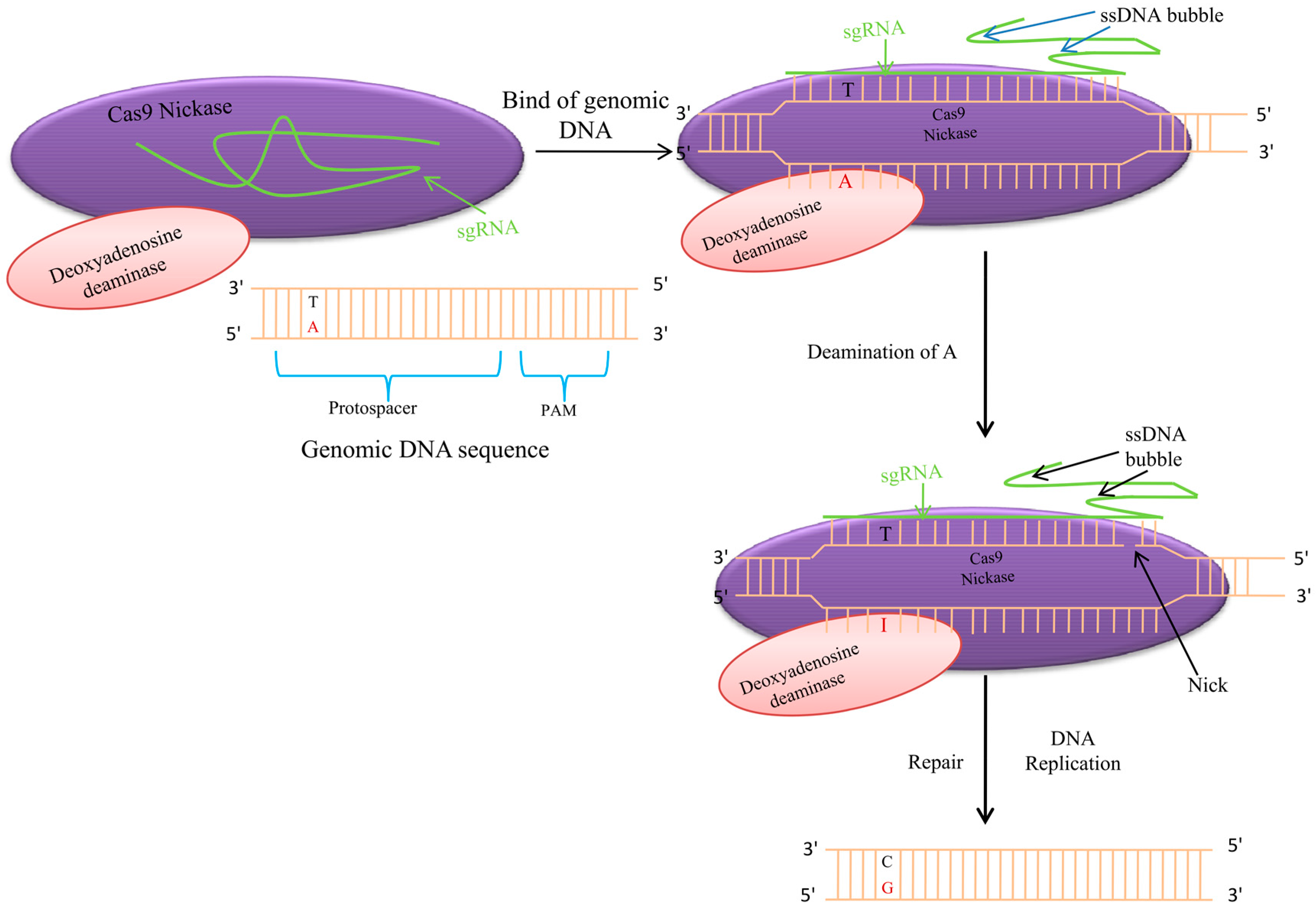
7. Adenine Base Editor
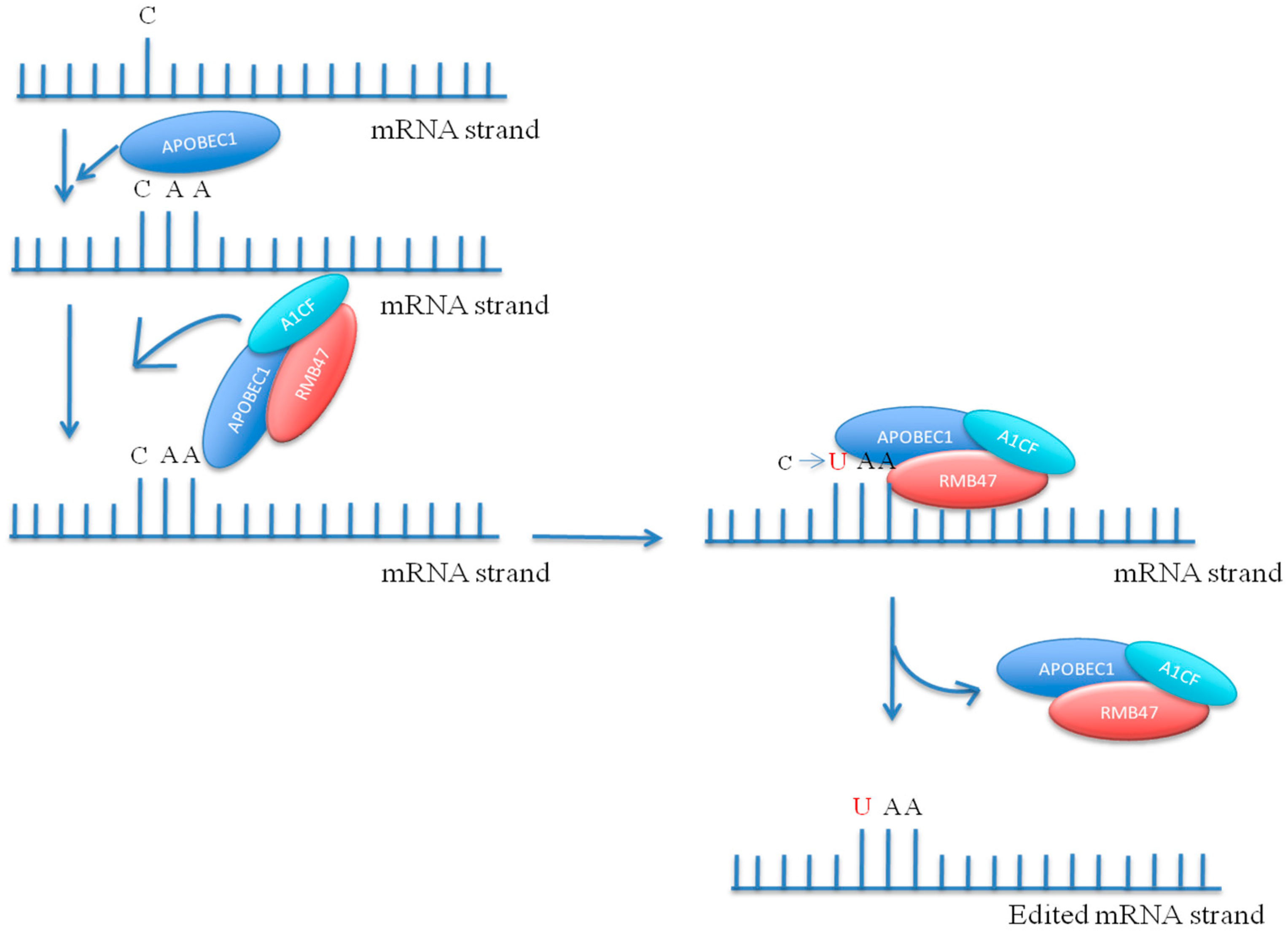
8. RNA Interference
9. Site-Directed Sequence Editing
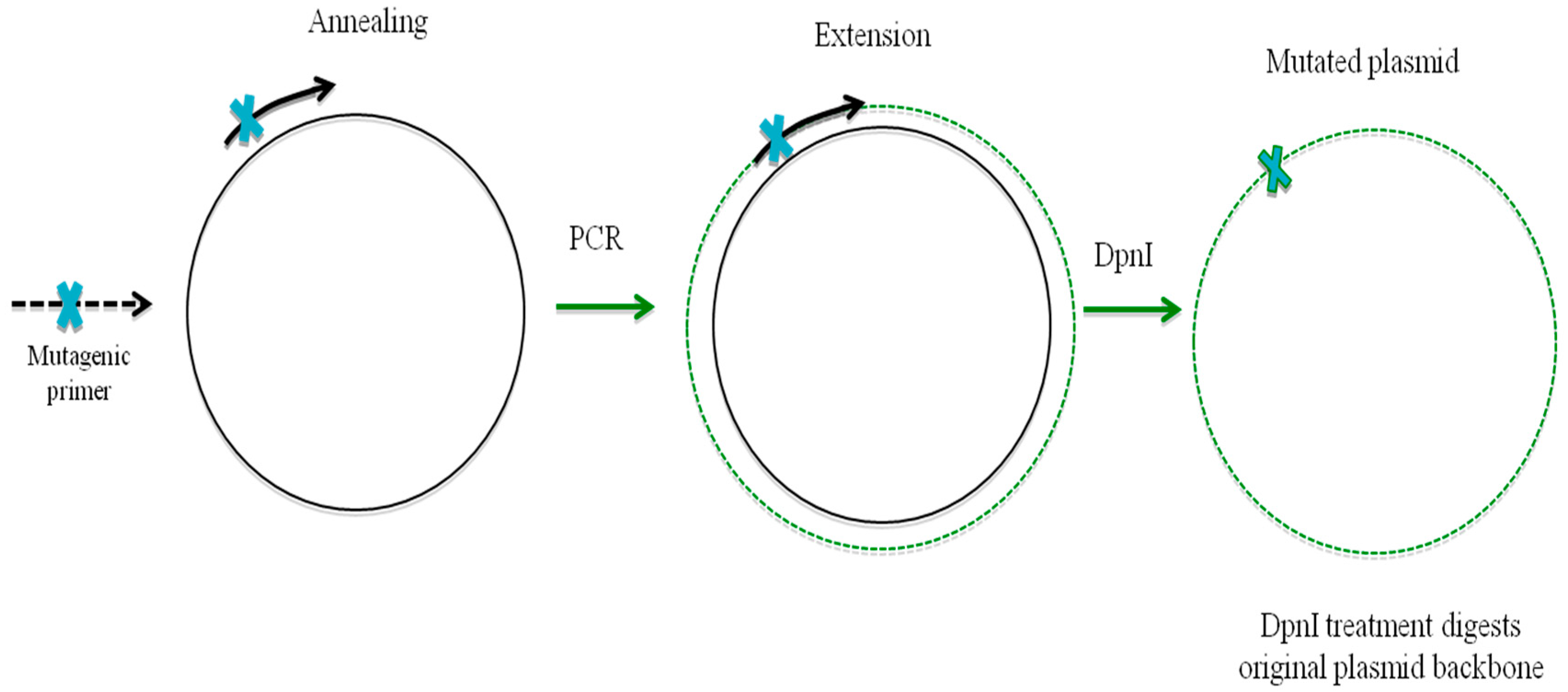
10. Oligonucleotide-Directed Mutagenesis
11. Cisgenesis and Intragenesis
12. Plastid Genome and Synthetic Genomics
13. Conclusions and Future Perspectives
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohanta, T.K.; Bashir, T.; Hashem, A.; Abd_Allah, E.F. Systems biology approach in plant abiotic stresses. Plant Physiol. Biochem. 2017, 121, 58–73. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, I.C.; Deans, T.L. Tools and applications in synthetic biology. Adv. Drug Deliv. Rev. 2016, 105 Pt A, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Paszkowski, J.; Baur, M.; Bogucki, A.; Potrykus, I. Gene targeting in plants. EMBO J. 1988, 7, 4021–4026. [Google Scholar] [PubMed]
- Templeton, N.S.; Roberts, D.D.; Safer, B. Efficient gene targeting in mouse embryonic stem cells. Gene Ther. 1997, 4, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, H.; Maandag, E.R.; Berns, A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proc. Natl. Acad. Sci. USA 1992, 89, 5128–5132. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Vaghchhipawala, Z.; Vasudevan, B.; Lee, L.-Y.; Shen, Y.; Singer, K.; Waterworth, W.M.; Zhang, Z.J.; West, C.E.; Mysore, K.S.; et al. Agrobacterium T-DNA integration into the plant genome can occur without the activity of key non-homologous end-joining proteins. Plant J. 2015, 81, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Shi, L. Screening of mutations by TILLING in plants BT—Plant genotyping: Methods and protocols. In Plant Genotyping; Batley, J., Ed.; Springer: New York, NY, USA, 2015; pp. 193–203. ISBN 978-1-4939-1966-6. [Google Scholar]
- Kurowska, M.; Daszkowska-Golec, A.; Gruszka, D.; Marzec, M.; Szurman, M.; Szarejko, I.; Maluszynski, M. TILLING - a shortcut in functional genomics. J. Appl. Genet. 2011, 52, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Till, B.J.; Comai, L. TILLING. Traditional Mutagenesis Meets Functional Genomics. Plant Physiol. 2004, 135, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Puchta, H. Gene replacement by homologous recombination in plants. Plant Mol. Biol. 2002, 48, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Puchta, H.; Dujon, B.; Hohn, B. Two different but related mechanisms are used in plants for the repair of genomic double-strand breaks by homologous recombination. Proc. Natl. Acad. Sci. USA 1996, 93, 5055–5060. [Google Scholar] [CrossRef] [PubMed]
- Roy, S. Maintenance of genome stability in plants: Repairing DNA double strand breaks and chromatin structure stability. Front. Plant Sci. 2014, 5, 487. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Puchta, H.; Fauser, F. Synthetic nucleases for genome engineering in plants: Prospects for a bright future. Plant J. 2014, 78, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607. [Google Scholar] [CrossRef] [PubMed]
- Knoll, A.; Higgins, J.D.; Seeliger, K.; Reha, S.J.; Dangel, N.J.; Bauknecht, M.; Schröpfer, S.; Franklin, F.C.H.; Puchta, H. The fanconi anemia ortholog FANCM ensures ordered homologous recombination in both somatic and meiotic cells in Arabidopsis. Plant Cell 2012, 24, 1448–1464. [Google Scholar] [CrossRef] [PubMed]
- Hartung, F.; Suer, S.; Bergmann, T.; Puchta, H. The role of AtMUS81 in DNA repair and its genetic interaction with the helicase AtRecQ4A. Nucleic Acids Res. 2006, 34, 4438–4448. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.; Wang, Z.; Friedberg, E.C.; Tomkinson, A.E. Identification of functional domains within the RAD1.RAD10 repair and recombination endonuclease of Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 20551–20558. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Charbonnel, C.; Allain, E.; Gallego, M.E.; White, C.I. Kinetic analysis of DNA double-strand break repair pathways in Arabidopsis. DNA Repair 2011, 10, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Mengiste, T.; Paszkowski, J. Prospects for the precise engineering of plant genomes by homologous recombination. Biol. Chem. 1999, 380, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Liberman-Lazarovich, M.; Levy, A. Homologous recombination in plants: An antireview. Methods Mol. Biol. 2011, 701, 51–65. [Google Scholar]
- Petolino, J.F. Genome editing in plants via designed zinc finger nucleases. Vitr. Cell. Dev. Biol. 2015, 51, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.F.; Fahrenkrug, S.C.; Hackett, P.B. Targeting DNA with fingers and TALENs. Mol. Ther. Nucleic Acids 2012, 1, e3. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Christensen, R.G.; Rayla, A.L.; Lakshmanan, A.; Stormo, G.D.; Wolfe, S.A. An optimized two-finger archive for ZFN-mediated gene targeting. Nat. Methods 2012, 9, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Pabo, C.O.; Peisach, E.; Grant, R.A. Design and selection of novel Cys2His2 zinc finger proteins. Annu. Rev. Biochem. 2001, 70, 313–340. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; Gersbach, C.A. Genome engineering for therapeutic applications. In Translating Gene Therapy to the Clinic; Laurence, J., Franklin, N., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 27–43. ISBN 978-0-12-800563-7. [Google Scholar]
- Pavletich, N.P.; Pabo, C.O. Zinc finger-DNA recognition: Crystal structure of a Zif268-DNA complex at 2.1 A. Science 1991, 252, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Elrod-Erickson, M.; Pabo, C.O. Binding Studies with mutants of Zif268: Contribution of individual side chains to binding affinity and specificity in the ZIF268 zinc finger-DNA complex. J. Biol. Chem. 1999, 274, 19281–19285. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Berg, J.M. A direct comparison of the properties of natural and designed zinc-finger proteins. Chem. Biol. 1995, 2, 83–89. [Google Scholar] [CrossRef]
- Liu, Q.; Segal, D.J.; Ghiara, J.B.; Barbas, C.F. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 5525–5530. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Segal, D.J.; Dreier, B.; Barbas, C.F. Toward controlling gene expression at will: Specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. USA 1998, 95, 14628–14633. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Pabo, C.O. Getting a handhold on DNA: Design of poly-zinc finger proteins with femtomolar dissociation constants. Proc. Natl. Acad. Sci. USA 1998, 95, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Bibikova, M.; Whitby, F.G.; Reddy, A.R.; Chandrasegaran, S.; Carroll, D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000, 28, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Mani, M.; Kandavelou, K.; Dy, F.J.; Durai, S.; Chandrasegaran, S. Design, engineering, and characterization of zinc finger nucleases. Biochem. Biophys. Res. Commun. 2005, 335, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Alwin, S.; Gere, M.B.; Guhl, E.; Effertz, K.; Barbas, C.F.; Segal, D.J.; Weitzman, M.D.; Cathomen, T. Custom zinc-finger nucleases for use in human cells. Mol. Ther. 2005, 12, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zia, Z.; Case, C. Validated zinc finger protein designs for all 16 GNN DNA triplet targets. J. Biol. Chem. 2001, 277, 3850–3856. [Google Scholar] [CrossRef] [PubMed]
- Dreier, B.; Beerli, R.R.; Segal, D.J.; Flippin, J.D.; Barbas, C.F. Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J. Biol. Chem. 2001, 276, 29466–29478. [Google Scholar] [CrossRef] [PubMed]
- Dreier, B.; Segal, D.J.; Barbas, C.F. Insights into the molecular recognition of the 5′-GNN-3′ family of DNA sequences by zinc finger domains. J. Mol. Biol. 2000, 303, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Dreier, B.; Fuller, R.P.; Segal, D.J.; Lund, C.V.; Blancafort, P.; Huber, A.; Koksch, B.; Barbas, C.F. Development of zinc finger domains for recognition of the 5’-CNN-3’ family DNA sequences and their use in the construction of artificial transcription factors. J. Biol. Chem. 2005, 280, 35588–35597. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.C.; Miller, J.C.; Pabo, C.O. Drug discovery with engineered zinc-finger proteins. Nat. Rev. Drug. Discov. 2003, 2, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Pavletich, N.P.; Pabo, C.O. Crystal structure of a five-finger GLI-DNA complex: New perspectives on zinc fingers. Science 1993, 261, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Greisman, H.A.; Pabo, C.O. A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science 1997, 275, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Durai, S.; Mani, M.; Kandavelou, K.; Wu, J.; Porteus, M.H.; Chandrasegaran, S. Zinc finger nucleases: Custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005, 33, 5978–5990. [Google Scholar] [CrossRef] [PubMed]
- Mani, M.; Smith, J.; Kandavelou, K.; Berg, J.M.; Chandrasegaran, S. Binding of two zinc finger nuclease monomers to two specific sites is required for effective double-strand DNA cleavage. Biochem. Biophys. Res. Commun. 2005, 334, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H. Mammalian gene targeting with designed zinc finger nucleases. Mol. Ther. 2006, 13, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Durai, S.; Bosley, A.; Abulencia, A.B.; Chandrasegaran, S.; Ostermeier, M. A bacterial one-hybrid selection system for interrogating zinc finger-DNA interactions. Comb. Chem. High Throughput Screen. 2006, 9, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Bonas, U.; Stall, R.E.; Staskawicz, B. Genetic and structural characterization of the avirulence gene avrBs3 from Xanthomonas campestris pv. vesicatoria. Mol. Gen. Genet. MGG 1989, 218, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10570–10575. [Google Scholar] [CrossRef] [PubMed]
- Wah, D.A.; Bitinaite, J.; Schildkraut, I.; Aggarwal, A.K. Structure of FokI has implications for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10564–10569. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, B.; Spalding, M.H.; Weeks, D.P.; Yang, B. High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 2012, 30, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Small, I.D.; Rackham, O.; Filipovska, A. Organelle transcriptomes: Products of a deconstructed genome. Curr. Opin. Microbiol. 2013, 16, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Linneweber, C.; Small, I. Pentatricopeptide repeat proteins: A socket set for organelle gene expression. Trends Plant Sci. 2008, 13, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Myouga, F.; Motohashi, R.; Shinozaki, K.; Shikanai, T. Conserved domain structure of pentatricopeptide repeat proteins involved in chloroplast RNA editing. Proc. Natl. Acad. Sci. USA 2007, 104, 8178–8183. [Google Scholar] [CrossRef] [PubMed]
- Shikanai, T. RNA editing in plant organelles: Machinery, physiological function and evolution. Cell. Mol. Life Sci. C. 2006, 63, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Lurin, C.; Andrés, C.; Aubourg, S.; Bellaoui, M.; Bitton, F.; Bruyère, C.; Caboche, M.; Debast, C.; Gualberto, J.; Hoffmann, B.; et al. Genome-Wide Analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 2004, 16, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Boussardon, C.; Avon, A.; Kindgren, P.; Bond, C.S.; Challenor, M.; Lurin, C.; Small, I. The cytidine deaminase signature HxE(x)nCxxC of DYW1 binds zinc and is necessary for RNA editing of ndhD-1. New Phytol. 2014, 203, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Rojas, M.; Fujii, S.; Yap, A.; Chong, Y.S.; Bond, C.S.; Small, I. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 2012, 8, e1002910. [Google Scholar] [CrossRef] [PubMed]
- Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 2015, 113, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Li, Q.; Yan, C.; Liu, Y.; Liu, J.; Yu, F.; Wang, Z.; Long, J.; He, J.; Wang, H.-W.; et al. Structural basis for the modular recognition of single-stranded RNA by PPR proteins. Nature 2013, 504, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Yagi, Y.; Hayashi, S.; Kobayashi, K.; Hirayama, T.; Nakamura, T. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS ONE 2013, 8, e57286. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Bond, C.S.; Small, I.D. Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc. Natl. Acad. Sci. USA 2011, 108, 1723–1728. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, N.; Hattori, M.; Andres, C.; Iida, K.; Lurin, C.; Schmitz-Linneweber, C.; Sugita, M.; Small, I. On the expansion of the pentatricopeptide repeat gene family in plants. Mol. Biol. Evol. 2008, 25, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- De Longevialle, A.F.; Hendrickson, L.; Taylor, N.L.; Delannoy, E.; Lurin, C.; Badger, M.; Millar, A.H.; Small, I. The pentatricopeptide repeat gene OTP51 with two LAGLIDADG motifs is required for the cis-splicing of plastid ycf3 intron 2 in Arabidopsis thaliana. Plant J. 2008, 56, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, R.; Kroeger, T.; Belcher, S.; Schöttler, M.A.; Barkan, A.; Schmitz-Linneweber, C. The pentatricopeptide repeat-SMR protein ATP4 promotes translation of the chloroplast atpB/E mRNA. Plant J. 2012, 72, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, R.; Qu, Y.; Zubo, Y.O.; Börner, T.; Schmitz-Linneweber, C. Mutation of the pentatricopeptide repeat-SMR protein SVR7 impairs accumulation and translation of chloroplast ATP synthase subunits in Arabidopsis thaliana. J. Plant Res. 2013, 126, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Melonek, J.; Boykin, L.M.; Small, I.; Howell, K.A. PPR-SMRs: Ancient proteins with enigmatic functions. RNA Biol. 2013, 10, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, S.; Wang, X.; Chang, J.; Gao, J.; Shi, R.; Zhang, J.; Lu, W.; Liu, Y.; Zhao, P.; et al. Highly efficient multiplex targeted mutagenesis and genomic structure variation in Bombyx mori cells using CRISPR/Cas9. Insect Biochem. Mol. Biol. 2014, 49, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Yang, Y. RNA-Guided Genome editing in plants using a CRISPR–Cas system. Mol. Plant 2013, 6, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Mao, Y.; Xu, N.; Zhang, B.; Wei, P.; Yang, D.; Wang, Z. Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 4632–4637. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Wang, N. Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS ONE 2014, 9, e93806. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-F.; Aach, J.; Norville, J.E.; McCormack, M.; Zhang, D.; Bush, J.; Church, G.M.; Sheen, J. Multiplex and homologous recombination-mediated plant genome editing via guide RNA/Cas9. Nat. Biotechnol. 2013, 31, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, V.; Staskawicz, B.; Weigel, D.; Jones, J.D.G.; Kamoun, S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.-L.; et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Kumar, J.; Alok, A.; Tuli, R. RNA-guided genome editing for target gene mutations in wheat. G3 2013, 3, 2233–2238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, B.; Weeks, D.P.; Spalding, M.H.; Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014, 42, 10903–10914. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Guo, D.; Zhang, J.; Huang, Q.; Qin, G.; Zhang, X.; Wan, J.; Gu, H.; Qu, L.-J. Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res 2013, 23, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, G.; Ma, S.; Xie, X.; Wu, X.; Zhang, X.; Wu, Y.; Zhao, P.; Xia, Q. CRISPR/Cas9-mediated targeted mutagenesis in Nicotiana tabacum. Plant Mol. Biol. 2015, 87, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, C.R.; Roth, D.B. An Unbiased method for detection of genome-wide off-target effects in cell lines treated with zinc finger nucleases. In Gene Correction; Storici, F., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 353–369. ISBN 978-1-62703-761-7. [Google Scholar]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.W.; Li, Z.; Peterson, R.T.; Yeh, J.-R. J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 variants with undetectable genome-wide off-targets. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat Meth 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Marraffini, L.A. Control of gene expression by CRISPR-Cas systems. F1000Prime Rep. 2013, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-guided transcriptional regulation in planta via synthetic dCas9-based transcription factors. Plant Biotechnol. J. 2015, 13, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Esvelt, K.M.; Mali, P.; Braff, J.L.; Moosburner, M.; Yaung, S.J.; Church, G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 2013, 10, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Anton, T.; Bultmann, S.; Leonhardt, H.; Markaki, Y. Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus 2014, 5, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and endogenous gene activation using programmable TALE-TET1 fusions. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhu, J.-K. Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system. Mol. Plant 2017, 10, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Wang, Y.; Li, C.; Zhang, R.; Chen, K.; Ran, Y.; Qiu, J.-L.; Wang, D.; Gao, C. Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat. Biotechnol. 2017, 35, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Small, I. RNAi for revealing and engineering plant gene functions. Curr. Opin. Biotechnol. 2007, 18, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.M.; Helliwell, C.A. Exploring plant genomes by RNA-induced gene silencing. Nat. Rev. Genet. 2003, 4, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Jan, C.; Rajagopalan, R.; Bartel, D.P. A two-hit trigger for siRNA biogenesis in plants. Cell 2006, 127, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Dasaradhi, P.V.N.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. RNA Interference: Biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 2003, 67, 657–685. [Google Scholar] [CrossRef] [PubMed]
- Carrington, J.C.; Ambros, V. Role of microRNAs in plant and animal development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.G.; Kingston, R.E.; Lau, N.C. The coming of age for Piwi proteins. Mol. Cell 2007, 26, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Siomi, M.C.; Sato, K.; Pezic, D.; Aravin, A.A. PIWI-interacting small RNAs: The vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 2011, 12, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Klattenhoff, C.; Theurkauf, W. Biogenesis and germline functions of piRNAs. Development 2008, 135, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widspread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, A.; Anderson, E.; Sullivan, K.; Reynolds, A.; Boese, Q.; Leake, D.; Karpilow, J.; Khvorova, A. A protocol for designing siRNAs with high functionality and specificity. Nat. Protoc. 2007, 2, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef] [PubMed]
- Redfern, A.D.; Colley, S.M.; Beveridge, D.J.; Ikeda, N.; Epis, M.R.; Li, X.; Foulds, C.E.; Stuart, L.M.; Barker, A.; Russell, V.J.; et al. RNA-induced silencing complex (RISC) Proteins PACT, TRBP, and Dicer are SRA binding nuclear receptor coregulators. Proc. Natl. Acad. Sci. USA 2013, 110, 6536–6541. [Google Scholar] [CrossRef] [PubMed]
- Iki, T.; Ishikawa, M.; Yoshikawa, M. In vitro formation of plant rna-induced silencing complexes using an extract of evacuolated tobacco protoplasts. In Plant Argonaute Proteins; Carbonell, A., Ed.; Springer: New York, NY, USA, 2017; pp. 39–53. ISBN 978-1-4939-7165-7. [Google Scholar]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-L.; Luo, G.; Wise, J.A.; Lou, H. Regulation of alternative splicing by local histone modifications: Potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014, 42, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Yu, B. siRNA-directed DNA methylation in plants. Curr. Genom. 2015, 16, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Taira, K. Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Nature 2004, 431, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhou, Y.; Castillo-González, C.; Lu, A.; Ge, C.; Zhao, Y.-T.; Duan, L.; Li, Z.; Axtell, M.J.; Wang, X.-J.; Zhang, X. Bi-directional processing of pri-miRNAs with branched terminal loops by Arabidopsis Dicer-like1. Nat. Struct. Mol. Biol. 2013, 20, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An Abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Kurzynska-Kokorniak, A.; Koralewska, N.; Pokornowska, M.; Urbanowicz, A.; Tworak, A.; Mickiewicz, A.; Figlerowicz, M. The many faces of Dicer: The complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res. 2015, 43, 4365–4380. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Martin-Hernandez, A.M.; Peart, J.R.; Malcuit, I.; Baulcombe, D.C. Virus-induced gene silencing in plants. Methods 2003, 30, 296–303. [Google Scholar] [CrossRef]
- Lange, M.; Yellina, A.L.; Orashakova, S.; Becker, A. virus-induced gene silencing (VIGS) in plants: An Overview of target species and the virus-derived vector systems. In Virus-Induced Gene Silencing; Becker, A., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 1–14. ISBN 978-1-62703-278-0. [Google Scholar]
- Burch-Smith, T.M.; Schiff, M.; Liu, Y.; Dinesh-Kumar, S.P. Efficient virus-induced gene silencing in Arabidopsis. Plant Physiol. 2006, 142, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Warnefors, M.; Liechti, A.; Halbert, J.; Valloton, D.; Kaessmann, H. Conserved microRNA editing in mammalian evolution, development and disease. Genome Biol. 2014, 15, R83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Mao, L. Evolution of plant microRNA gene families. Cell Res. 2007, 17, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Sharma, D.; Trivedi, P.K. Artificial microRNA mediated gene silencing in plants: Progress and perspectives. Plant Mol. Biol. 2014, 86, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schwab, R.; Weigel, D. Gene silencing in plants using artificial microRNAs and other small RNAs. Plant J. 2008, 53, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Gasparis, S.; Kała, M.; Przyborowski, M.; Orczyk, W.; Nadolska-Orczyk, A. Artificial MicroRNA-based specific gene silencing of grain hardness genes in polyploid cereals appeared to be not stable over transgenic plant generations. Front. Plant Sci. 2017, 7, 2017. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Small, I. The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 2011, 191, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Zhang, D.; Rogozin, I.B.; Aravind, L. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res. 2011, 39, 9473–9497. [Google Scholar] [CrossRef] [PubMed]
- Koito, A.; Ikeda, T. Apolipoprotein B mRNA-editing, catalytic polypeptide cytidine deaminases and retroviral restriction. Wiley Interdiscip. Rev. RNA 2012, 3, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Teng, B.-B.; Ochsner, S.; Zhang, Q.; Soman, K.V.; Lau, P.P.; Chan, L. Mutational analysis of apolipoprotein B mRNA editing enzyme (APOBEC1): Structure–function relationships of RNA editing and dimerization. J. Lipid Res. 1999, 40, 623–635. [Google Scholar] [PubMed]
- Yoshinaga, K.; Iinuma, H.; Masuzawa, T.; Uedal, K. Extensive RNA editing of U to C in addition to C to U substitution in the rbcL transcripts of hornwort chloroplasts and the origin of RNA editing in green plants. Nucleic Acids Res. 1996, 24, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Simons, J.P.; Owen, J.S. Oligonucleotide-directed gene-editing technology: Mechanisms and future prospects. Expert Opin. Biol. Ther. 2012, 12, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Evans, R.R.; Dahmer, M.L.; Singh, B.K.; Shaner, D.L. Imidazolinone-tolerant crops: History, current status and future. Pest 2005, 61, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Hartung, F.; Schiemann, J.; Quedlinburg, D. Precise plant breeding using new genome editing techniques: Opportunities, safety and regulation in the EU. Plant J. 2014, 78, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Atlihan, N.; Lu, Z.-X. New biotechnology enhances the application of cisgenesis in plant breeding. Front. Plant Sci. 2014, 5, 389. [Google Scholar] [CrossRef] [PubMed]
- Schouten, H.J.; Krens, F.A.; Jacobsen, E. Do cisgenic plants warrant less stringent oversight? Nat. Biotechnol. 2006, 24, 753. [Google Scholar] [CrossRef] [PubMed]
- Holme, I.B.; Wendt, T.; Holm, P.B. Intragenesis and cisgenesis as alternatives to transgenic crop development. Plant Biotechnol. J. 2013, 11, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, A.; Giancaspro, A.; Blechl, A.E.; Blanco, A. A transgenic durum wheat line that is free of marker genes and expresses 1Dy10. J. Cereal Sci. 2008, 48, 439–445. [Google Scholar] [CrossRef]
- Han, K.M.; Dharmawardhana, P.; Arias, R.S.; Ma, C.; Busov, V.; Strauss, S.H. Gibberellin-associated cisgenes modify growth, stature and wood properties in Populus. Plant Biotechnol. J. 2011, 9, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Rujan, T.; Richly, E.; Hansen, A.; Cornelsen, S.; Lins, T.; Leister, D.; Stoebe, B.; Hasegawa, M.; Penny, D. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. USA 2002, 99, 12246–12251. [Google Scholar] [CrossRef] [PubMed]
- Lilly, J.W.; Havey, M.J.; Jackson, S.A.; Jiang, J. Cytogenomic Analyses Reveal the Structural Plasticity of the Chloroplast Genome in Higher Plants. Plant Cell 2001, 13, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Scharff, L.; Koop, H. Linear molecules of tobacco ptDNA end at known replication origins and additional loci. Plant Mol. Biol. 2006, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Day, A.; Madesis, P. DNA replication, recombination, and repair in plastids BT - cell and molecular biology of plastids. In Cell and Molecular Biology of Plastids; Bock, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 65–119. ISBN 978-3-540-75376-6. [Google Scholar]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, S.; Xia, E.-H.; Jiang, J.-J.; Zeng, F.-C.; Gao, L.-Z. Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci. Rep. 2016, 6, 30135. [Google Scholar] [CrossRef] [PubMed]
- Scharff, L.B.; Bock, R. Synthetic biology in plastids. Plant J. 2014, 78, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A. Expression of plastid genes: Organelle-specific elaborations on a prokaryotic scaffold. Plant Physiol. 2011, 155, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.R. Regulation of plastid gene expression in the chloroplast-to-chromoplast transition. Plant Cell 2008, 20, 823. [Google Scholar] [CrossRef]
- Babiychuk, E.; Vandepoele, K.; Wissing, J.; Garcia-Diaz, M.; De Rycke, R.; Akbari, H.; Joubès, J.; Beeckman, T.; Jänsch, L.; Frentzen, M.; Van Montagu, M.C.E.; Kushnir, S. Plastid gene expression and plant development require a plastidic protein of the mitochondrial transcription termination factor family. Proc. Natl. Acad. Sci. USA 2011, 108, 6674–6679. [Google Scholar] [CrossRef] [PubMed]
- Bräutigam, K.; Dietzel, L.; Pfannschmidt, T. Plastid-nucleus communication: Anterograde and retrograde signalling inthe development and function of plastids. In Cell and Molecular Biology of Plastids; Bock, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 409–455. ISBN 978-3-540-75376-6. [Google Scholar]
- Verhounig, A.; Karcher, D.; Bock, R. Inducible gene expression from the plastid genome by a synthetic riboswitch. Proc. Natl. Acad. Sci. USA 2010, 107, 6204–6209. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Rijzaani, H.; Karcher, D.; Ruf, S.; Bock, R. Efficient metabolic pathway engineering in transgenic tobacco and tomato plastids with synthetic multigene operons. Proc. Natl. Acad. Sci. USA 2013, 110. [Google Scholar] [CrossRef] [PubMed]
- Elghabi, Z.; Ruf, S.; Bock, R. Biolistic co-transformation of the nuclear and plastid genomes. Plant J. 2011, 67, 941–948. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanta, T.K.; Bashir, T.; Hashem, A.; Abd_Allah, E.F.; Bae, H. Genome Editing Tools in Plants. Genes 2017, 8, 399. https://doi.org/10.3390/genes8120399
Mohanta TK, Bashir T, Hashem A, Abd_Allah EF, Bae H. Genome Editing Tools in Plants. Genes. 2017; 8(12):399. https://doi.org/10.3390/genes8120399
Chicago/Turabian StyleMohanta, Tapan Kumar, Tufail Bashir, Abeer Hashem, Elsayed Fathi Abd_Allah, and Hanhong Bae. 2017. "Genome Editing Tools in Plants" Genes 8, no. 12: 399. https://doi.org/10.3390/genes8120399
APA StyleMohanta, T. K., Bashir, T., Hashem, A., Abd_Allah, E. F., & Bae, H. (2017). Genome Editing Tools in Plants. Genes, 8(12), 399. https://doi.org/10.3390/genes8120399