
Proliferation Cycle Causes Age Dependent Mitochondrial Deficiencies and Contributes to the Aging of Stem Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fly Maintenance, Fecundity and Egg Hatching Assay
2.2. Mitochondrial Activity Staining on Ovaries
2.3. Selection for mtDNA Mutations
2.4. Quantitative Polymerase Chain Reaction Analysis of Telomere Length
2.5. Immunofluorescence Staining, EdU Staining and In Situ Hybridization
2.6. Rolling Cycle Amplification and Restriction Fragment Length Polymorphism analysis of mtDNA
3. Results
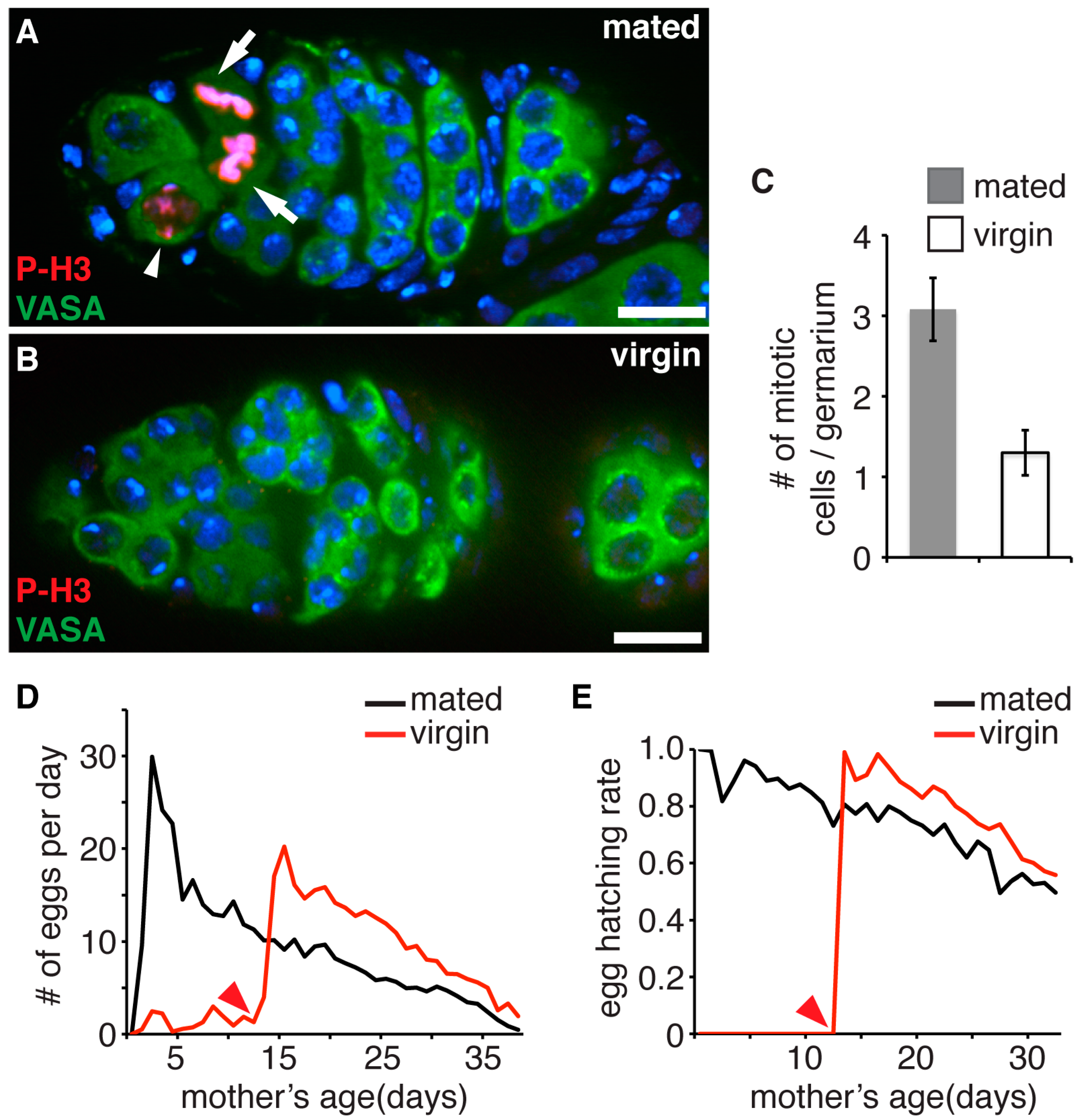
3.1. Accumulation of Proliferation Cycles in Germline Stem Cells Causes Age Dependent Decline of Female Fecundity and Reduced Fitness of Their Progeny
3.2. Aging Does Not Lead to Telomere Shortening in Germline Stem Cells
3.3. Aged GSCs Have Increased mtDNA Mutations Loads and Defective Electron Chain Complexes
3.4. Aged Germline Stem Cells Have Rearrangements on mtDNA AT-Rich Region and Impaired mtDNA Replication
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wallace, D.C. Mitochondria as chi. Genetics 2008, 179, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Pesole, G.; Gissi, C.; De Chirico, A.; Saccone, C. Nucleotide substitution rate of mammalian mitochondrial genomes. J. Mol. Evol. 1999, 48, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Greaves, L.C.; Reeve, A.K.; Turnbull, D. The ageing mitochondrial genome. Nucleic Acids Res. 2007, 35, 7399–7405. [Google Scholar] [CrossRef] [PubMed]
- Linnane, A.W.; Marzuki, S.; Ozawa, T.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 1, 642–645. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Gidlöf, S.; Oldfors, A.; Wibom, R.; Törnell, J.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Jacobs, H.T.; Kajander, O.A. Mitochondrial DNA—All things bad? Trends Genet. 1999, 15, 91–93. [Google Scholar] [CrossRef]
- Khrapko, K.; Vijg, J. Mitochondrial DNA mutations and aging: A case closed? Nat. Genet. 2007, 39, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.-G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Menzies, R.A.; Gold, P.H. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. 1971, 246, 2425–2429. [Google Scholar] [PubMed]
- Baines, H.L.; Turnbull, D.M.; Greaves, L.C. Human stem cell aging: Do mitochondrial DNA mutations have a causal role? Aging Cell 2014, 13, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C.; et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Investig. 2003, 112, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.A.C.; Greaves, L.C.; Gutierrez-Gonzalez, L.; Rodriguez-Justo, M.; Deheragoda, M.; Leedham, S.J.; Taylor, R.W.; Lee, C.Y.; Preston, S.L.; Lovell, M.; et al. Mechanisms of field cancerization in the human stomach: The expansion and spread of mutated gastric stem cells. Gastroenterology 2008, 134, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Fellous, T.G.; Islam, S.; Tadrous, P.J.; Elia, G.; Kocher, H.M.; Bhattacharya, S.; Mears, L.; Turnbull, D.M.; Taylor, R.W.; Greaves, L.C.; et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology 2009, 49, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hämäläinen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Götz, A.; Forsström, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.D.; Wagner, W.; Mahlknecht, U. Stem cells and ageing. EMBO Rep. 2005, 6, S35–S38. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.L.; Nelson, T.J.; Dzeja, P.P.; Terzic, A. Energy metabolism plasticity enables stemness programs. Ann. N.Y. Acad. Sci. 2012, 1254, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Duan, S.; Yi, F.; Ocampo, A.; Liu, G.-H.; Izpisua Belmonte, J.C. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Günes, C.; Rudolph, K.L. The role of telomeres in stem cells and cancer. Cell 2013, 152, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Drummond-Barbosa, D. Stem cells, their niches and the systemic environment: An aging network. Genetics 2008, 180, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Chen, S.; Weng, C.; Call, G.; Zhu, D.; Tang, H.; Zhang, N.; Xie, T. Stem cell aging is controlled both intrinsically and extrinsically in the Drosophila ovary. Cell Stem Cell 2007, 1, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Waskar, M.; Li, Y.; Tower, J. Stem cell aging in the Drosophila ovary. Age 2005, 27, 201–212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, R.; Xuan, Y.; Li, X.; Xi, R. Age-related changes of germline stem cell activity, niche signaling activity and egg production in Drosophila. Aging Cell 2008, 7, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; DeLuca, S.Z.; O’Farrell, P.H. Manipulating the metazoan mitochondrial genome with targeted restriction enzymes. Science 2008, 321, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.F.; Biessmann, M.R.; Benitez, C.; Török, T.; Mason, J.M.; Biessmann, H. Effects of telomere length in Drosophila melanogaster on life span, fecundity and fertility. Chromosoma 2007, 116, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.H.; Chen, Z.; Xu, H. Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat. Genet. 2014, 46, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Lécuyer, E. High resolution fluorescent in situ hybridization in Drosophila. Methods Mol. Biol. 2011, 714, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Haag-Liautard, C.; Coffey, N.; Houle, D.; Lynch, M.; Charlesworth, B.; Keightley, P.D. Direct estimation of the mitochondrial dna mutation rate in Drosophila melanogaster. PLoS Biol. 2008, 6, e204. [Google Scholar] [CrossRef] [PubMed]
- Wolfner, M.F. The gifts that keep on giving: Physiological functions and evolutionary dynamics of male seminal proteins in Drosophila. Heredity 2002, 88, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Bindoff, L.A.; Turnbull, D.M. Cytochrome c oxidase activity in single muscle fibers: Assay techniques and diagnostic applications. Ann. Neurol. 1993, 33, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.; Wei, G.; Shen, T.; Han, M.; Lian, Y.; Fu, H.; Luo, Y.; Yang, Y.; Liu, J.; Wakabayashi, Y.; et al. MitoRCA-seq reveals unbalanced cytocine to thymine transition in Polg mutant mice. Sci. Rep. 2015, 5, 12049. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.L.; Farr, C.L.; Kaguni, L.S. Drosophila melanogaster mitochondrial DNA: Completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Biol. 1995, 4, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M. Population genetics of the cytoplasm and the units of selection on mitochondrial DNA in Drosophila melanogaster. Genetica 2011, 139, 685–697. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nekhaeva, E.; Bodyak, N.D.; Kraytsberg, Y.; McGrath, S.B.; Orsouw, N.J.V.; Pluzhnikov, A.; Wei, J.Y.; Vijg, J.; Khrapko, K. Clonally expanded mtDNA point mutations are abundant in individual cells of human tissues. PNAS 2002, 99, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- May-Panloup, P.; Boucret, L.; Chao de la Barca, J.M.; Desquiret-Dumas, V.; Ferré-L’Hotellier, V.; Morinière, C.; Descamps, P.; Procaccio, V.; Reynier, P. Ovarian ageing: the role of mitochondria in oocytes and follicles. Hum. Reprod. Update 2016, 22, 725–743. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Yin, P.-H.; Lin, J.-C.; Wu, C.-C.; Chen, C.-Y.; Wu, C.-W.; Chi, C.-W.; Tam, T.-N.; Wei, Y.-H. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann. N. Y. Acad. Sci. 2005, 1042, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; ISBN 978-0-8153-3218-3. [Google Scholar]
- Prowse, K.R.; Greider, C.W. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc. Natl. Acad. Sci. USA 1995, 92, 4818–4822. [Google Scholar] [CrossRef] [PubMed]
- Nagley, P.; Wei, Y.-H. Ageing and mammalian mitochondrial genetics. Trends Genet. 1998, 14, 513–517. [Google Scholar] [CrossRef]
- Tolkovsky, A.M. Mitophagy. Biochim. Biophys. Acta 2009, 1793, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Kai, T.; Williams, D.; Spradling, A.C. The expression profile of purified Drosophila germline stem cells. Dev. Biol. 2005, 283, 486–502. [Google Scholar] [CrossRef] [PubMed]
- Margineantu, D.H.; Hockenbery, D.M. Mitochondrial functions in stem cells. Curr. Opin. Genet. Dev. 2016, 38, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Iv, C.A.E.; Ramalho-Santos, J.; Houten, B.V.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Q.; Zhang, F.; Xu, H. Proliferation Cycle Causes Age Dependent Mitochondrial Deficiencies and Contributes to the Aging of Stem Cells. Genes 2017, 8, 397. https://doi.org/10.3390/genes8120397
Ren Q, Zhang F, Xu H. Proliferation Cycle Causes Age Dependent Mitochondrial Deficiencies and Contributes to the Aging of Stem Cells. Genes. 2017; 8(12):397. https://doi.org/10.3390/genes8120397
Chicago/Turabian StyleRen, Qiuting, Fan Zhang, and Hong Xu. 2017. "Proliferation Cycle Causes Age Dependent Mitochondrial Deficiencies and Contributes to the Aging of Stem Cells" Genes 8, no. 12: 397. https://doi.org/10.3390/genes8120397
APA StyleRen, Q., Zhang, F., & Xu, H. (2017). Proliferation Cycle Causes Age Dependent Mitochondrial Deficiencies and Contributes to the Aging of Stem Cells. Genes, 8(12), 397. https://doi.org/10.3390/genes8120397