
High-Throughput Sequencing of Small RNA Transcriptomes in Maize Kernel Identifies miRNAs Involved in Embryo and Endosperm Development

 ,
, 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. sRNA Isolation, Library Preparation and Sequencing
2.2. sRNA Bioinformatic Analysis
2.3. miRNA Gene Expression Analysis and Calculation of the Fold-Changes and p-Values
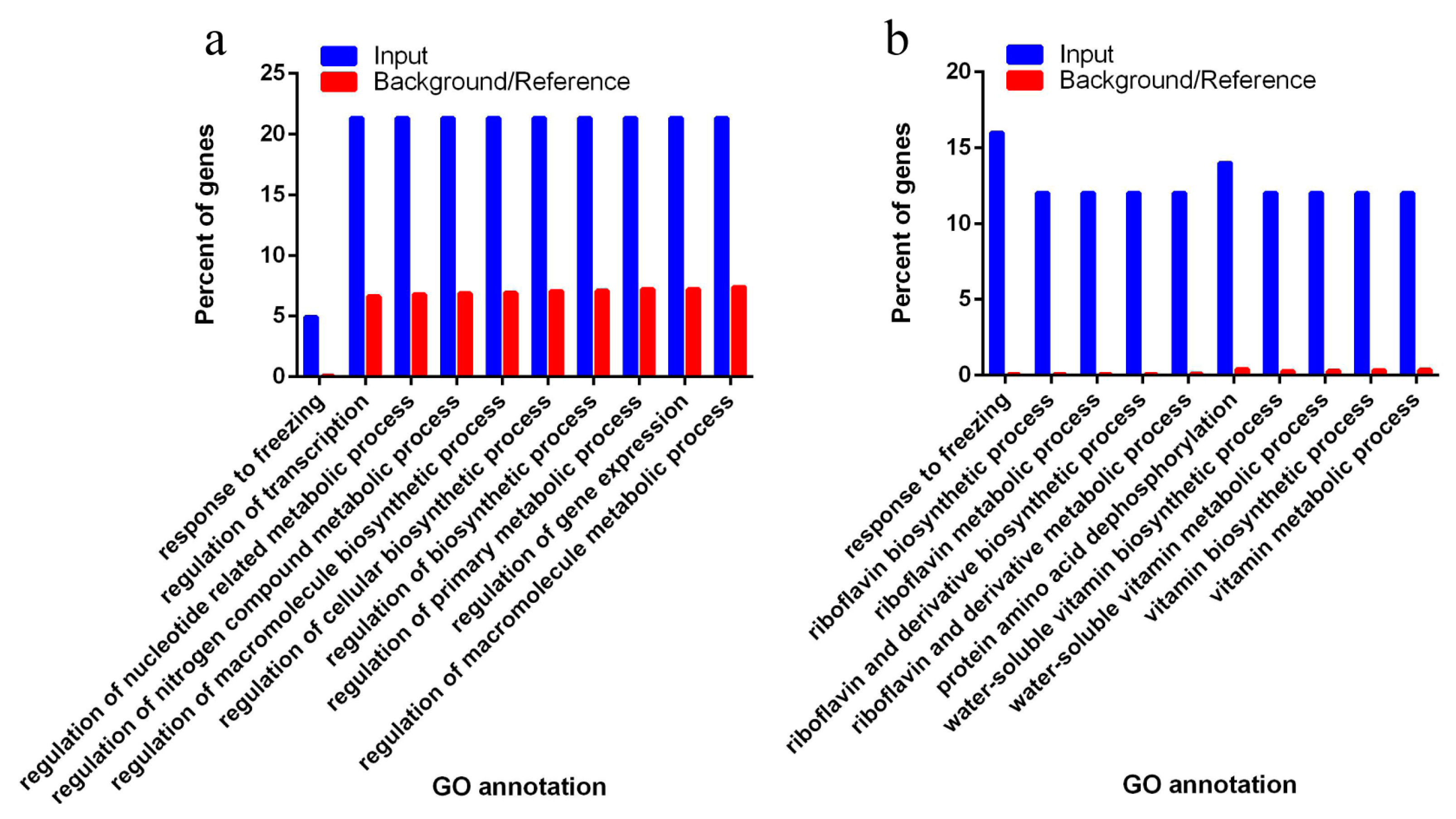
2.4. Prediction of miRNA Targets and Functional Annotation
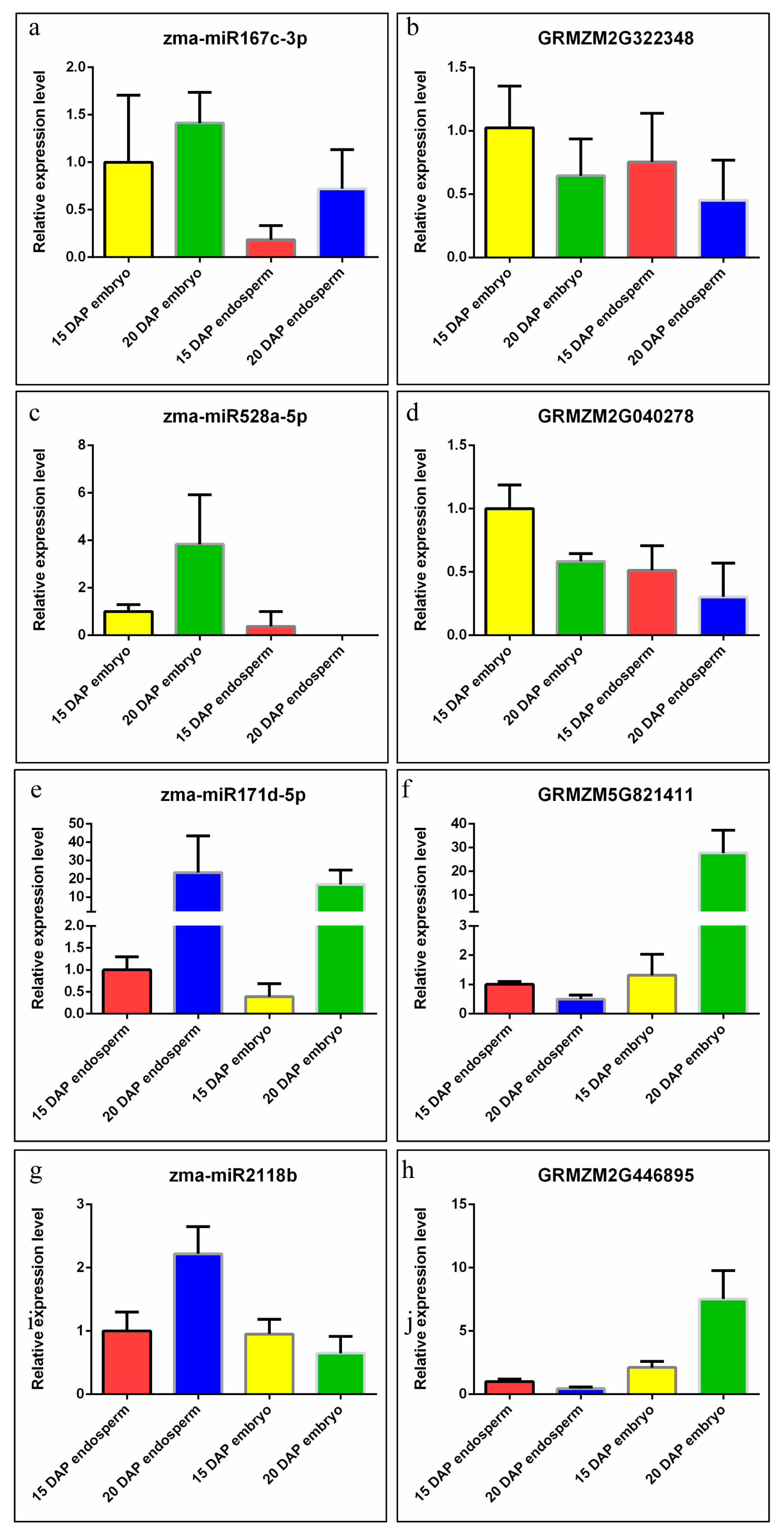
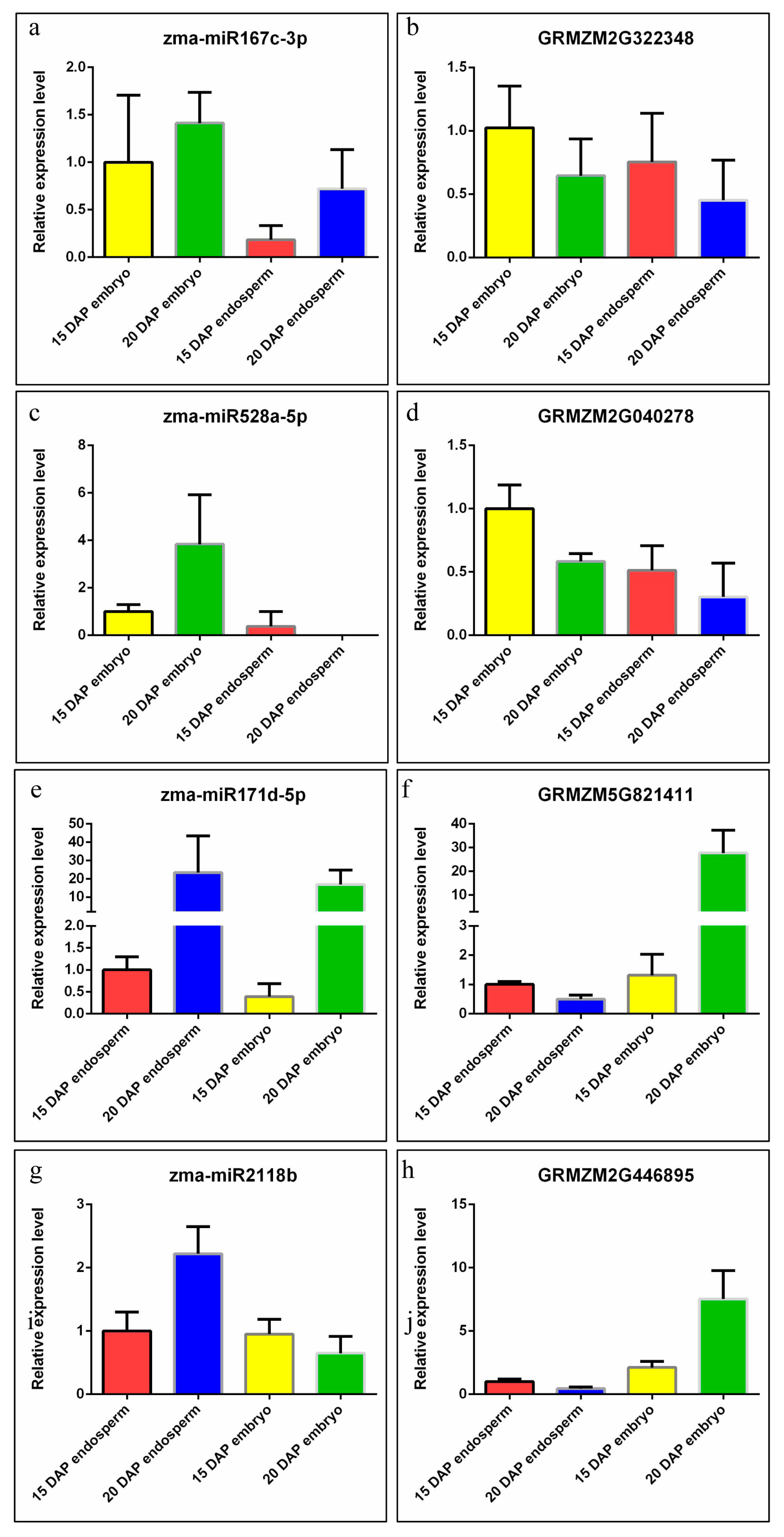
2.5. Expression Validation of miRNA and Their Targets
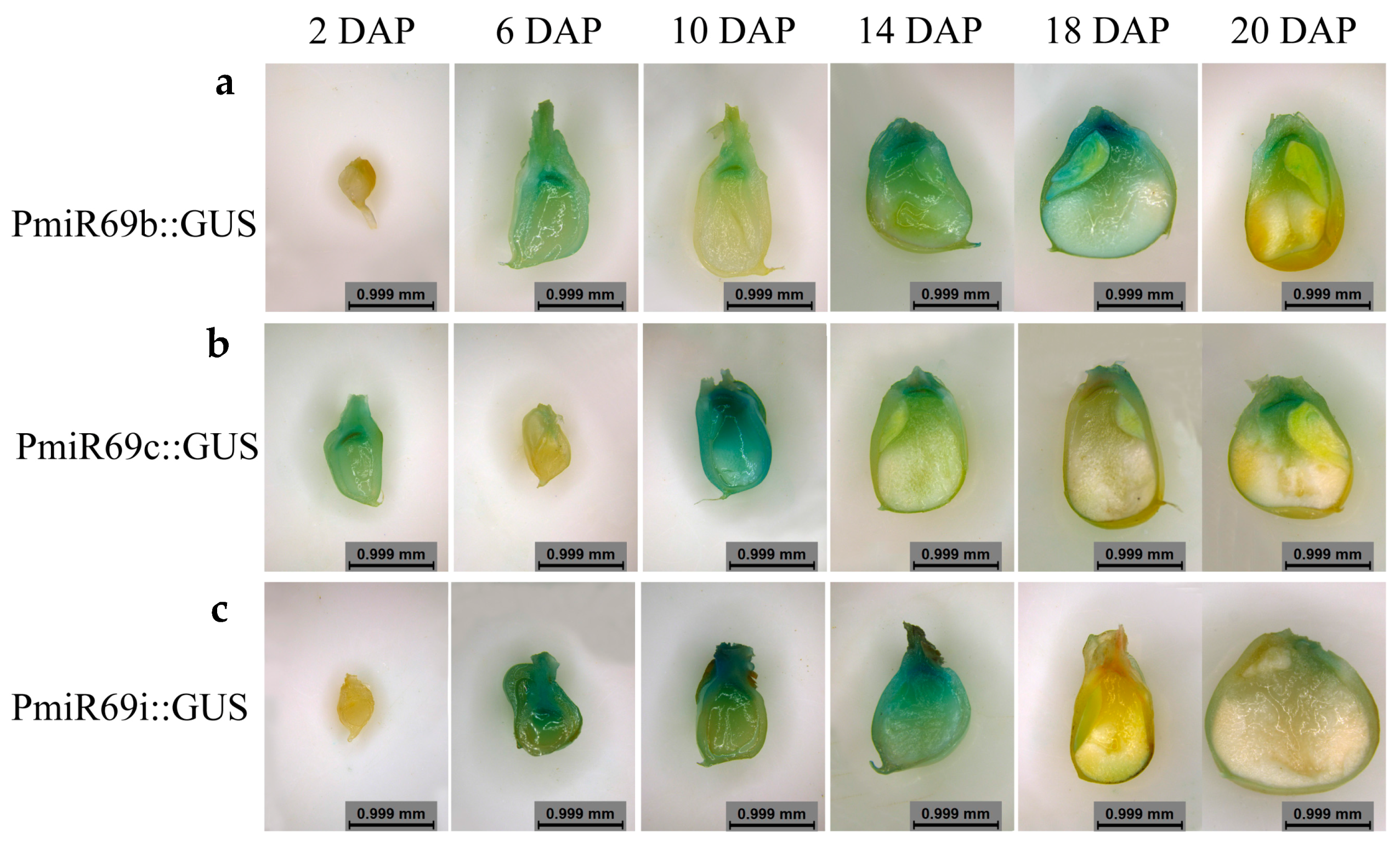
2.6. Histological Observation of Transgenic Kernel
3. Results
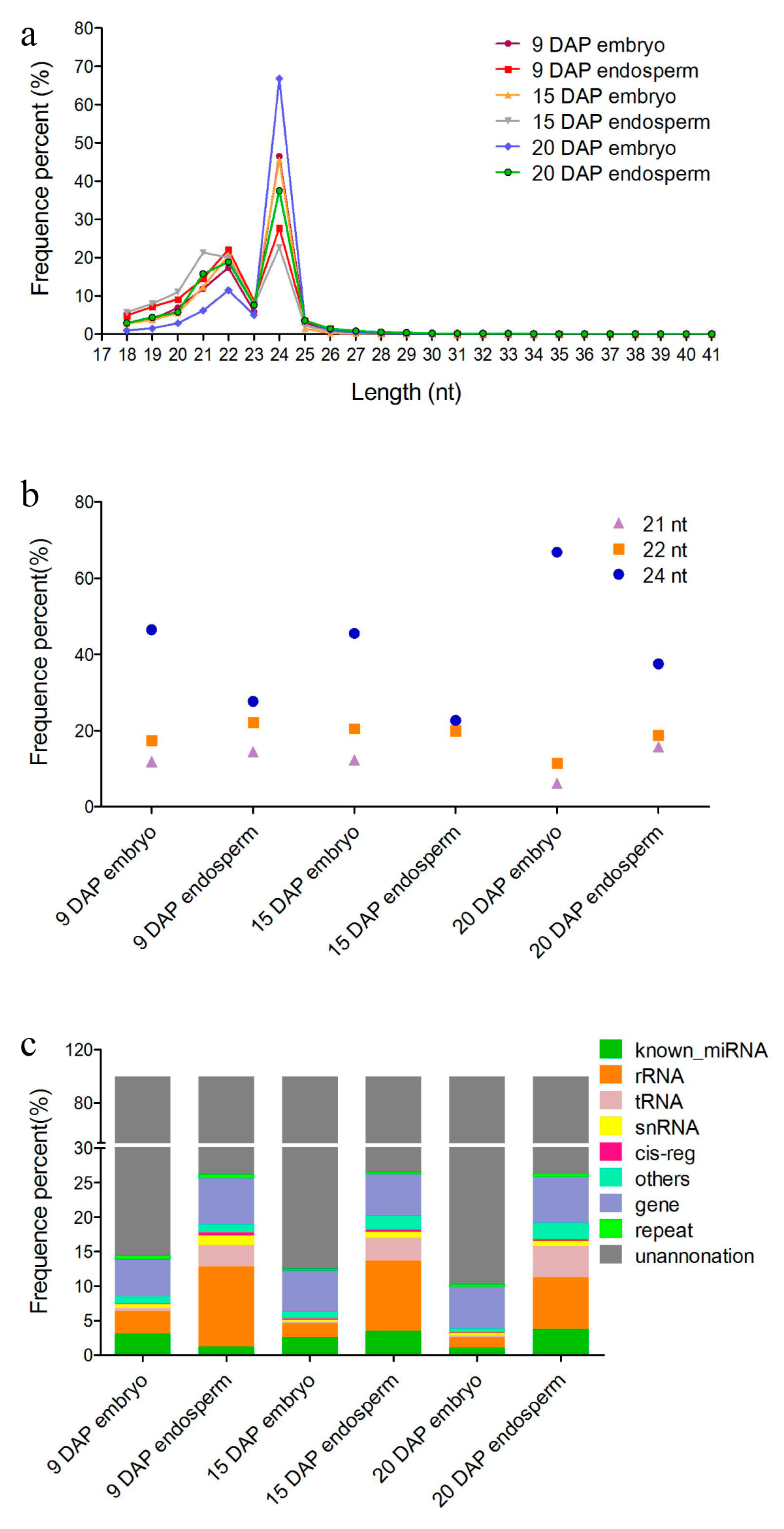
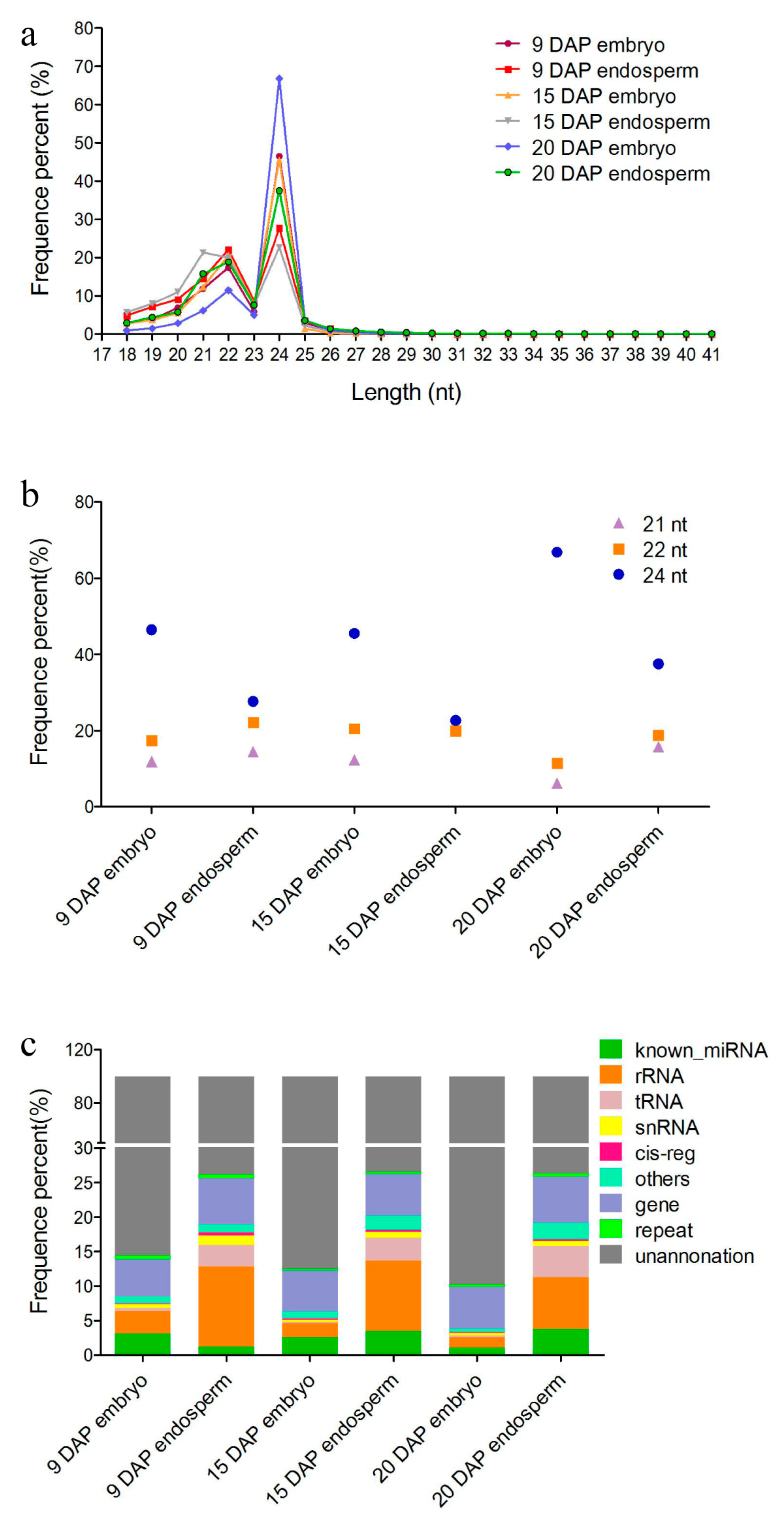
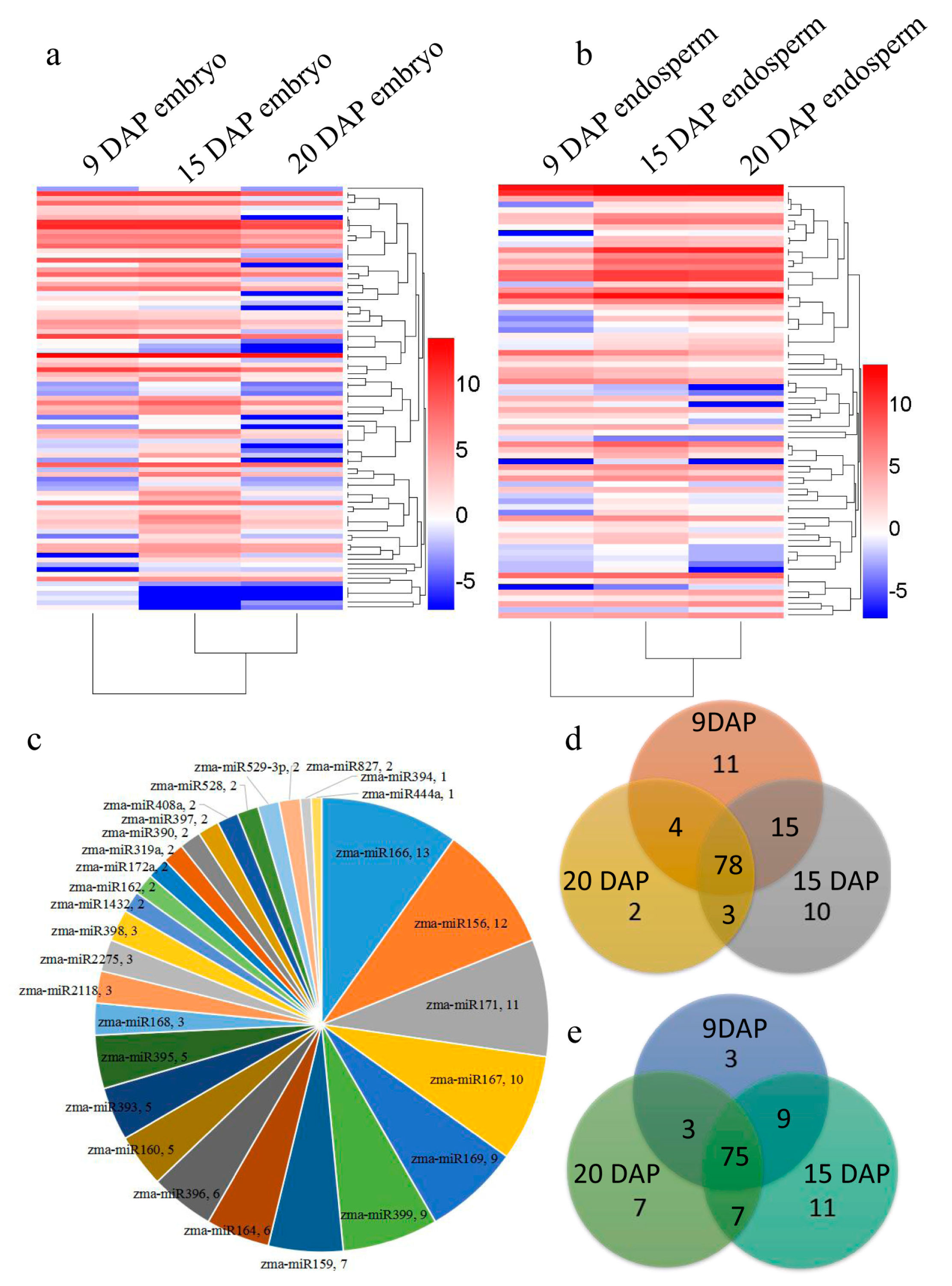
3.1. Overview of sRNAs Expressed in Maize Embryo and Endosperm during Kernel Development
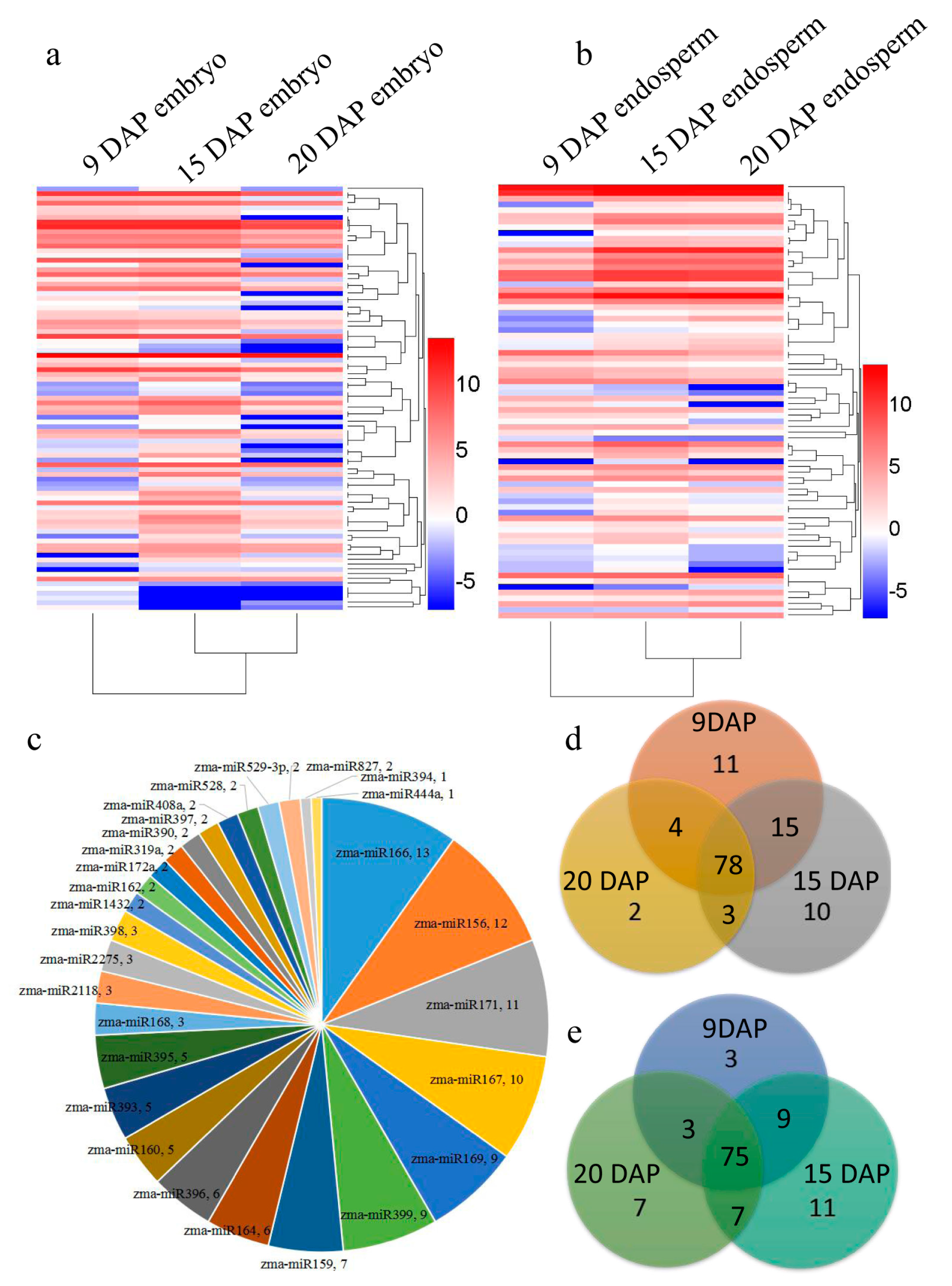
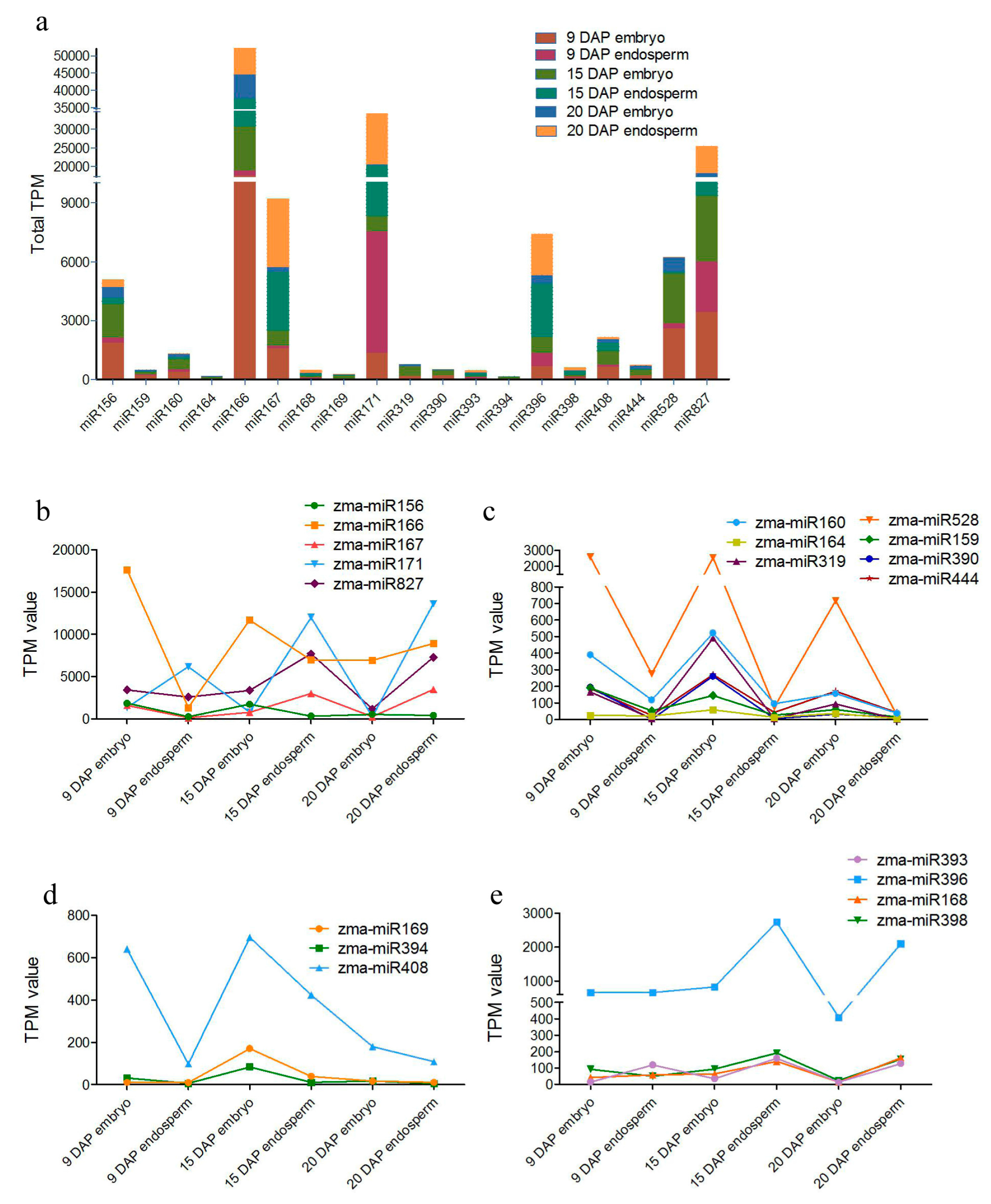
3.2. Identification of Known and Novel miRNAs Expressed in Maize Embryo and Endosperm
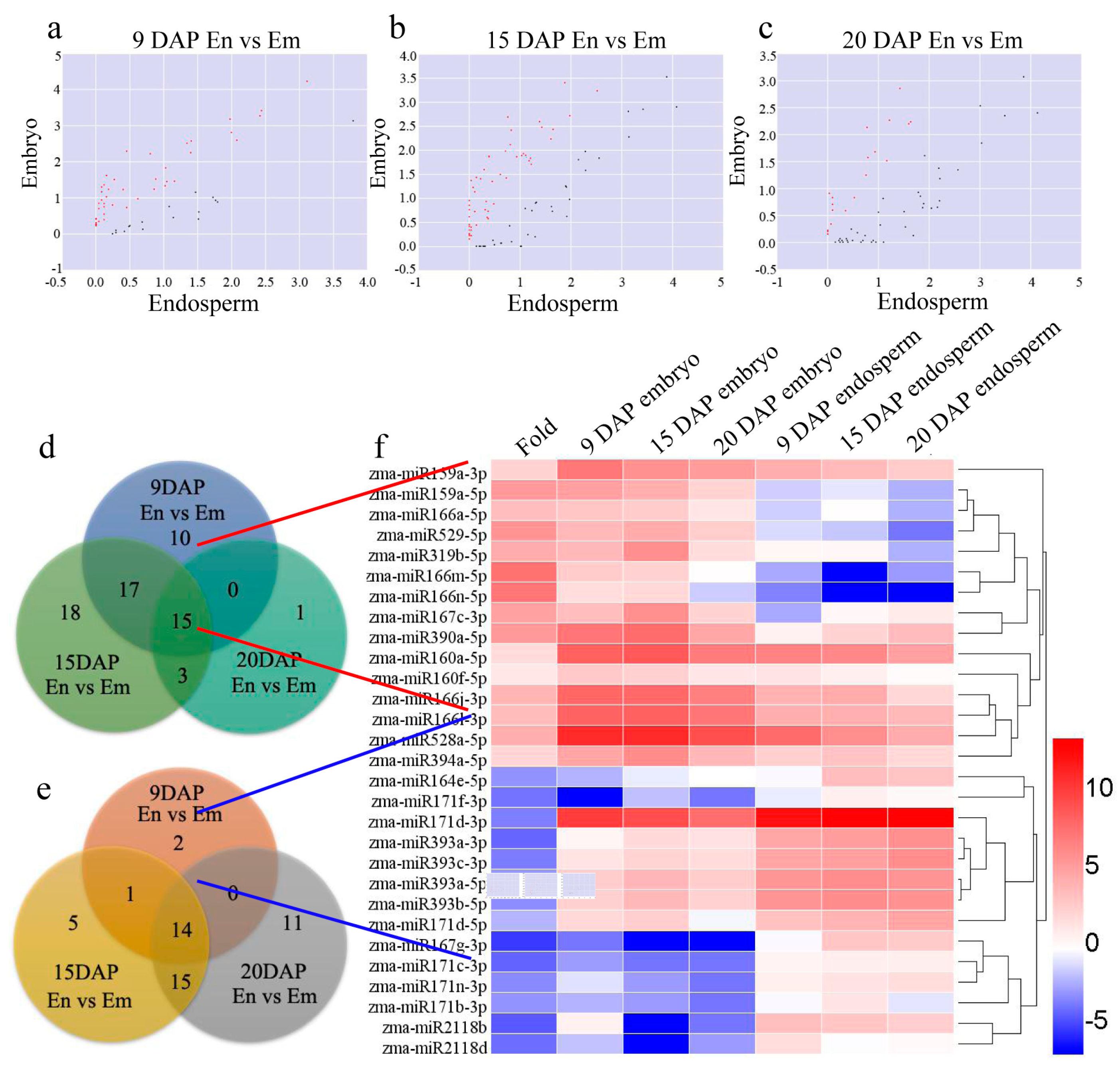
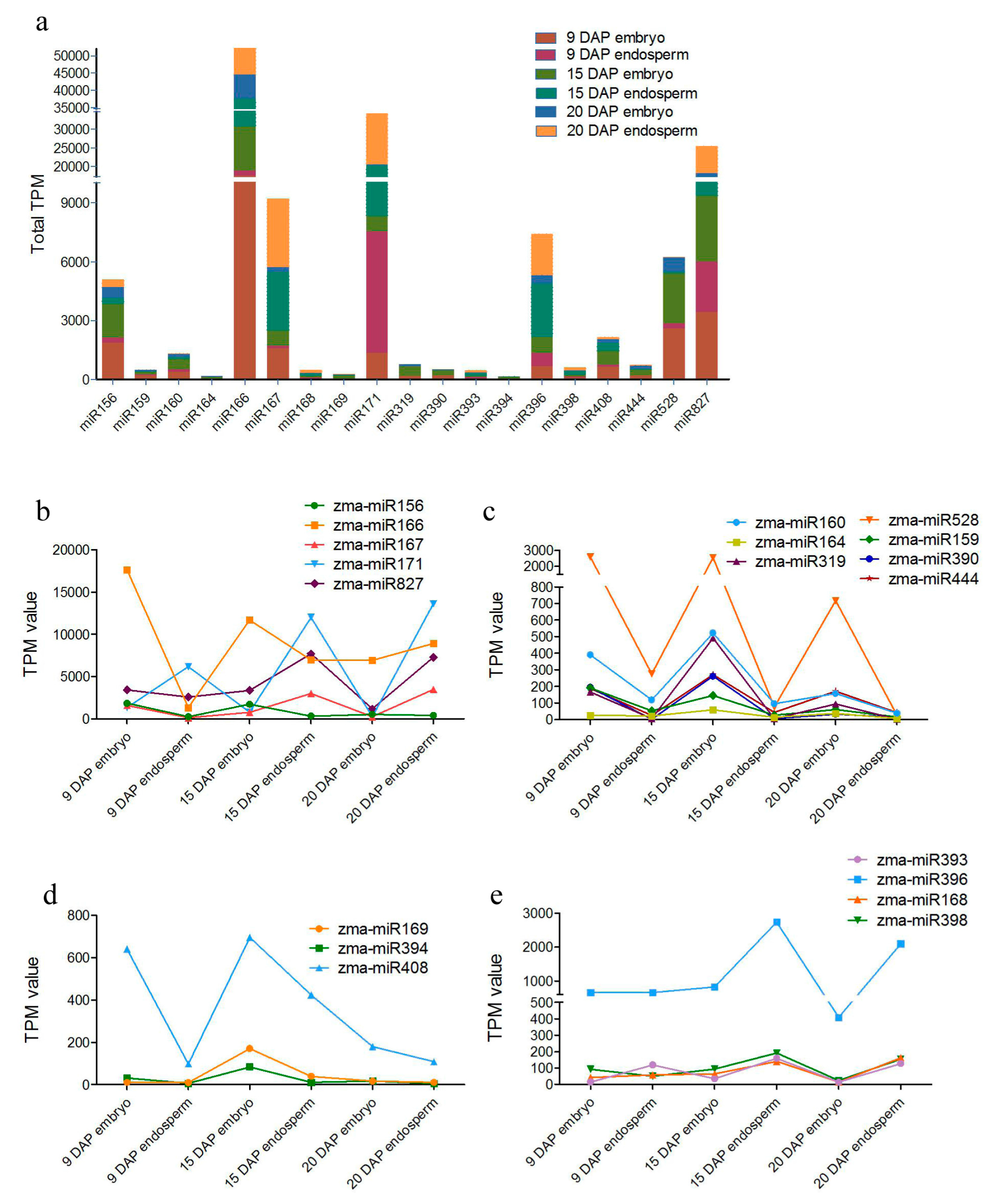
3.3. Differential Expression of miRNAs during Kernel Development
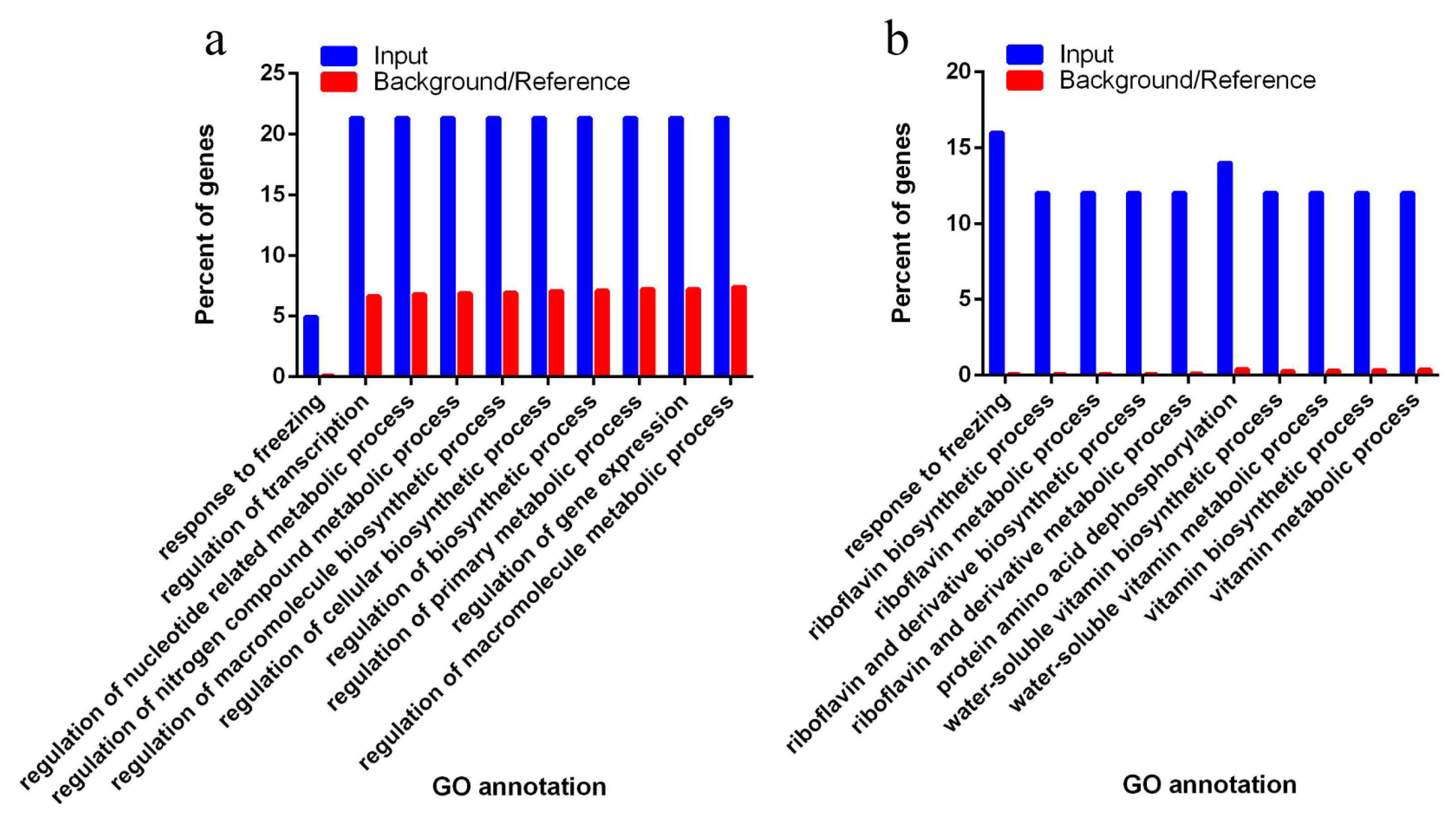
3.4. Target of Differentially Expressed Known miRNAs Related to Biosynthetic and Metabolic Process
3.5. Zma-miR169 Family Involved in Kernel Development of Maize
4. Discussion
4.1. Differential and Specific Expression Patterns of miRNAs May Determine the Differential Kernel Development Patterns of the Embryo and Endosperm
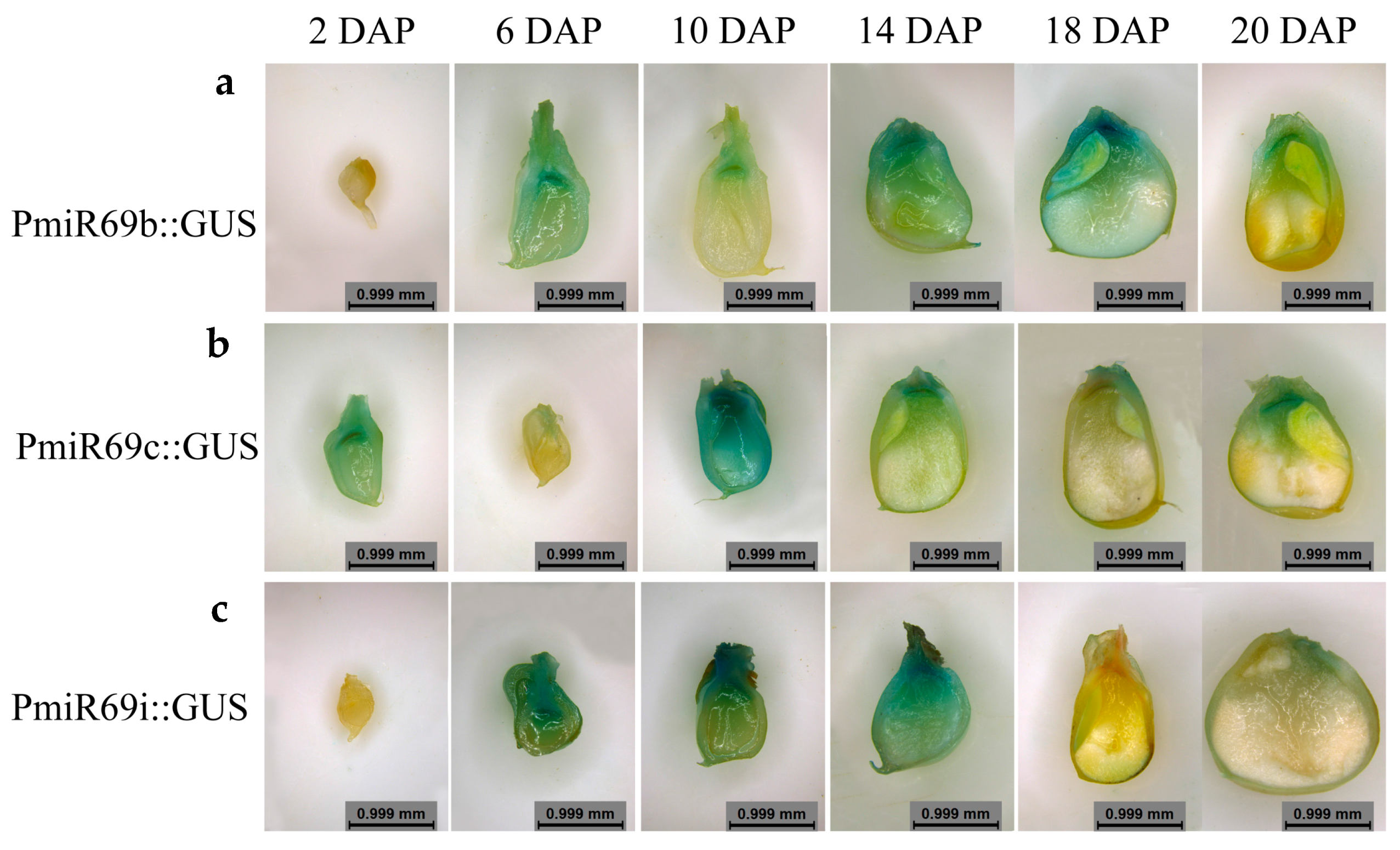
4.2. miR169 Family Members Specifically Were Expressed in Maize Kernel
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Consonni, G.; Gavazzi, G.; Dolfini, S. Genetic analysis as a tool to investigate the molecular mechanisms underlying seed development in maize. Ann. Bot. 2005, 96, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.A. Endosperm development: Cellularization and cell fate specification. Annu. Rev. Plant Physiol. Mol. 2001, 52, 233–267. [Google Scholar] [CrossRef] [PubMed]
- Sabelli, P.A.; Larkins, B.A. The development of endosperm in grasses. Plant Physiol. 2009, 149, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Doll, N.M.; Depege-Fargeix, N.; Rogowsky, P.M.; Widiez, T. Signaling in early maize kernel development. Mol. Plant 2017, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, D.; Shu, D.; Zhang, Z.; Wang, W.; Klukas, C.; Chen, L.L.; Fan, Y.; Chen, M.; Zhang, C. The differential transcription network between embryo and endosperm in the early developing maize seed. Plant Physiol. 2013, 162, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- MiRbase. Available online: http://www.mirbase.org/ (accessed on 3 July 2014).
- Tang, J.; Chu, C. MicroRNAs in crop improvement: Fine-tuners for complex traits. Nat. Plants 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.J.; Zhang, J.J.; Xue, H.W. Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res. 2009, 37, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Sun, H.; Qiao, M.; Zhao, Y.; Du, Y.; Zhang, J.; Li, J.; Tang, G.; Zhao, Q. Differentially expressed microRNA cohorts in seed development may contribute to poor grain filling of inferior spikelets in rice. BMC Plant Biol. 2014, 14, 196. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ikeda, M.; Matsubara, A.; Song, X.J.; Ito, M.; Asano, K.; Matsuoka, M.; Kitano, H.; Ashikari, M. OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 2010, 42, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Ni, S.; Wang, J.; Zhang, B.; Xu, R.; Wang, Y.; Chen, H.; Zhu, X.; Li, Y. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat. Plants 2015, 2, 15203. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, K.; Liu, Y.; Chen, Y.; Chen, P.; Shi, Z.; Luo, J.; Jiang, D.; Fan, F.; Zhu, Y.; et al. Blocking miR396 increases rice yield by shaping inflorescence architecture. Nat. Plants 2015, 2, 15196. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, S.; Xu, Y.; Li, C.; Zhang, Z.; Zhang, D.; Xu, S.; Zhang, C.; Chong, K. OsmiR396d-regulated osGRFs function in floral organogenesis in rice through binding to their targets osJMJ706 and osCR4. Plant Physiol. 2014, 165, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, Y.; Fang, Y.; Zeng, L.; Xu, J.; Yu, H.; Shi, Z.; Pan, J.; Zhang, D.; Kang, S.; et al. A rare allele of GS2 enhances grain size and grain yield in rice. Mol. Plant 2015, 8, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Yu, Y.; Wang, C.Y.; Li, Z.Y.; Liu, Q.; Xu, J.; Liao, J.Y.; Wang, X.J.; Qu, L.H.; Chen, F.; et al. Overexpression of microRNA osmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat. Biotechnol. 2013, 31, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Houston, K.; McKim, S.M.; Comadran, J.; Bonar, N.; Druka, I.; Uzrek, N.; Cirillo, E.; Guzy-Wrobelska, J.; Collins, N.C.; Halpin, C.; et al. Variation in the interaction between alleles of HvAPETALA2 and microRNA172 determines the density of grains on the barley inflorescence. Proc. Natl. Acad. Sci. USA 2013, 110, 16675–16680. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Kong, J.; Lai, T.; Manning, K.; Wu, C.; Ying, W.; Cheng, Q.; Li, B.; Yu, Z.; Xian, Z. Tuning LeSPL-CNR expression by SlymiR157 affects tomato fruit ripening. Sci. Rep. 2015, 5, 7852. [Google Scholar] [CrossRef] [PubMed]
- Ferreira e Silva, G.F.; Silva, E.M.; Azevedo Mda, S.; Guivin, M.A.; Ramiro, D.A.; Figueiredo, C.R.; Carrer, H.; Peres, L.E.; Nogueira, F.T. microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 2014, 78, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Z.; Zhang, J.; Zhang, Y.; Han, Q.; Hu, T.; Xu, X.; Liu, H.; Li, H.; Ye, Z. Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant. FEBS Lett. 2011, 585, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Aravind, J.; Rinku, S.; Pooja, B.; Shikha, M.; Kaliyugam, S.; Mallikarjuna, M.G.; Kumar, A.; Rao, A.R.; Nepolean, T. Identification, characterization, and functional validation of drought-responsive microRNAs in subtropical maize inbreds. Front. Plant Sci. 2017, 8, 941. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Zhang, M.; Zhao, Y.; He, X.; Ding, C.; Wang, S.; Feng, Y.; Song, X.; Li, P.; Wang, B. Identification of salt tolerance-related microRNAs and their targets in maize (Zea mays L.) using high-throughput sequencing and degradome analysis. Front. Plant Sci. 2017, 8, 864. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kumari, M.; Kumar, H.; Varadwaj, P.K. Genome-wide analysis of miRNAs and Tasi-RNAs in Zea mays in response to phosphate deficiency. Funct. Integr. Genomics 2017, 17, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Long, J.; Zheng, L.; Li, Y.; Hu, Y.; Yu, G.; Liu, H.; Liu, Y.; Huang, Z.; Zhang, J.; et al. Identification and characterization of microRNAs in maize endosperm response to exogenous sucrose using small RNA sequencing. Genomics 2016, 108, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhang, M.; Xing, L.; Li, W.; Jiang, H.; Wang, L.; Xu, M. Transcriptomic analysis of long non-coding RNAs and coding genes uncovers a complex regulatory network that is involved in maize seed development. Genes 2017, 8, 274. [Google Scholar] [CrossRef] [PubMed]
- NCBI SRA. Available online: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP118425 (accessed on 22 September 2017).
- Cutadapt 1.14. Available online: http://cutadapt.readthedocs.io/en/stable/ (accessed on 16 May 2017).
- FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 2 February 2010).
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Bowtie. Available online: http://bowtie-bio.sourceforge.net/index.shtml (accessed on 13 June 2017).
- Plant SnoRNAbase v1.2. Available online: http://bioinf.scri.sari.ac.uk/cgi-bin/plant_snorna/home (accessed on 2 November 2017).
- Genomic tRNA Database. Available online: http://gtrnadb.ucsc.edu/download.html (accessed on 1 February 2017).
- Rfam 13.0. Available online: http://rfam.xfam.org/ (accessed on 1 September 2017).
- Blast. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 15 November 2017).
- Bowtie 2 Software. Available online: http://bowtie-bio.sourceforge.net/bowtie2/index.shtml (accessed on 5 October 2017).
- RepBase. Available online: http://www.girinst.org/server/RepBase/ (accessed on 27 January 2017).
- RepeatMasker. Available online: http://www.repeatmasker.org/ (accessed on 29 August 2016).
- RNAfold Software. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi (accessed on 14 November 2017).
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- PsRNATarget. Available online: http://plantgrn.noble.org/psRNATarget/ (accessed on 5 June 2009).
- MaizeGDB. Available online: http://www.maizegdb.org/ (accessed on 7 November 2017).
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. AgriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Shi, B.; Hou, N.; Cao, Y.; Meng, Y.; Bian, H.; Zhu, M.; Han, N. MicroRNAs participate in gene expression regulation and phytohormone cross-talk in barley embryo during seed development and germination. BMC Plant Biol. 2017, 17, 150. [Google Scholar] [CrossRef] [PubMed]
- MiRdeep2. Available online: https://www.mdc-berlin.de/8551903/en/ (accessed on 1 May 2016).
- Dai, X.; Zhao, P.X. PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- AgriGO. Available online: http://systemsbiology.cau.edu.cn/agriGOv2/index.php (accessed on 24 November 2017).
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, X.; Zhu, M.; Xu, M.; Wang, L. Transcription factors NF-YA2 and NF-YA10 regulate leaf growth via auxin signaling in Arabidopsis. Sci. Rep. 2017, 7, 1395. [Google Scholar] [CrossRef] [PubMed]
- Luan, M.; Xu, M.; Lu, Y.; Zhang, Q.; Zhang, L.; Zhang, C.; Fan, Y.; Lang, Z.; Wang, L. Family-wide survey of miR169s and NF-YAs and their expression profiles response to abiotic stress in maize roots. PLoS ONE 2014, 9, e91369. [Google Scholar] [CrossRef] [PubMed]
- Luan, M.; Xu, M.; Lu, Y.; Zhang, L.; Fan, Y.; Wang, L. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 2015, 555, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.S.; Li, J.; Stahle, M.I.; Dubroue, A.; Gubler, F.; Millar, A.A. Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc. Natl. Acad. Sci. USA 2007, 104, 16371–16376. [Google Scholar] [CrossRef] [PubMed]
- Juarez, M.T.; Kui, J.S.; Thomas, J.; Heller, B.A.; Timmermans, M.C. MicroRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 2004, 428, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Carlsbecker, A.; Lee, J.Y.; Roberts, C.J.; Dettmer, J.; Lehesranta, S.; Zhou, J.; Lindgren, O.; Moreno-Risueno, M.A.; Vaten, A.; Thitamadee, S.; et al. Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 2010, 465, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Tagiri, A. MicroRNA-targeted transcription factor gene RDD1 promotes nutrient ion uptake and accumulation in rice. Plant J. 2016, 85, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xia, K.; Liang, Z.; Chen, K.; Gao, C.; Zhang, M. MicroRNA393 is involved in nitrogen-promoted rice tillering through regulation of auxin signal transduction in axillary buds. Sci. Rep. 2016, 6, 32158. [Google Scholar] [CrossRef] [PubMed]
- Khraiwesh, B.; Zhu, J.K.; Zhu, J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 2012, 1819, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yoo, S.J.; Lee, J.H.; Kim, W.; Yoo, S.K.; Fitzgerald, H.; Carrington, J.C.; Ahn, J.H. Genetic framework for flowering-time regulation by ambient temperature-responsive miRNAs in Arabidopsis. Nucleic Acids Res. 2010, 38, 3081–3093. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Oono, Y.; Zhu, J.; He, X.J.; Wu, J.M.; Iida, K.; Lu, X.Y.; Cui, X.; Jin, H.; Zhu, J.K. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 2008, 20, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, G.; Sutoh, K.; Zhu, J.K.; Zhang, W. Identification of cold-inducible microRNAs in plants by transcriptome analysis. Biochim. Biophys. Acta 2008, 1779, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Liu, Y.; Li, W.; Song, L.; Zhang, J.; Guo, C. Genome-wide investigation of microRNAs and their targets in response to freezing stress in Medicago sativa L., based on high-throughput sequencing. G3 Genes Genomes Genet. 2016, 6, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Candar-Cakir, B.; Arican, E.; Zhang, B. Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [PubMed]
- Rathore, P.; Geeta, R.; Das, S. Microsynteny and phylogenetic analysis of tandemly organised miRNA families across five members of Brassicaceae reveals complex retention and loss history. Plant Sci. 2016, 247, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, N.; Li, H.; Liu, J.; Fu, C.; Xiao, Z.; Wei, C.; Lu, X.; Feng, J.; Zhou, Y. Identification of drought-responsive microRNAs and their targets in Ammopiptanthus Mongolicus by using high-throughput sequencing. Sci. Rep. 2016, 6, 34601. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Chen, J.; Jin, L.; Duns, G.J.; Ouyang, P. Differential expression of miRNAs under salt stress in Spartina alterniflora leaf tissues. J. Nanosci. Nanotechnol. 2015, 15, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, S.L.; Li, J.L.; Hu, X.H.; Wang, H.; Cao, X.L.; Xu, Y.J.; Zhao, Z.X.; Xiao, Z.Y.; Yang, N.; et al. Osa-miR169 negatively regulates rice immunity against the blast fungus Magnaporthe Oryzae. Front. Plant Sci. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Wang, J.; Wang, T.; Wei, L.; Li, J.; Liu, L. Identification of rapeseed microRNAs involved in early stage seed germination under salt and drought stresses. Front. Plant Sci. 2016, 7, 658. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, L.; Zhu, M.; Zhang, M.; Li, W.; Jiang, H.; Zou, J.; Wang, L.; Xu, M. High-Throughput Sequencing of Small RNA Transcriptomes in Maize Kernel Identifies miRNAs Involved in Embryo and Endosperm Development. Genes 2017, 8, 385. https://doi.org/10.3390/genes8120385
Xing L, Zhu M, Zhang M, Li W, Jiang H, Zou J, Wang L, Xu M. High-Throughput Sequencing of Small RNA Transcriptomes in Maize Kernel Identifies miRNAs Involved in Embryo and Endosperm Development. Genes. 2017; 8(12):385. https://doi.org/10.3390/genes8120385
Chicago/Turabian StyleXing, Lijuan, Ming Zhu, Min Zhang, Wenzong Li, Haiyang Jiang, Junjie Zou, Lei Wang, and Miaoyun Xu. 2017. "High-Throughput Sequencing of Small RNA Transcriptomes in Maize Kernel Identifies miRNAs Involved in Embryo and Endosperm Development" Genes 8, no. 12: 385. https://doi.org/10.3390/genes8120385
APA StyleXing, L., Zhu, M., Zhang, M., Li, W., Jiang, H., Zou, J., Wang, L., & Xu, M. (2017). High-Throughput Sequencing of Small RNA Transcriptomes in Maize Kernel Identifies miRNAs Involved in Embryo and Endosperm Development. Genes, 8(12), 385. https://doi.org/10.3390/genes8120385