TRIM8: Making the Right Decision between the Oncogene and Tumour Suppressor Role

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
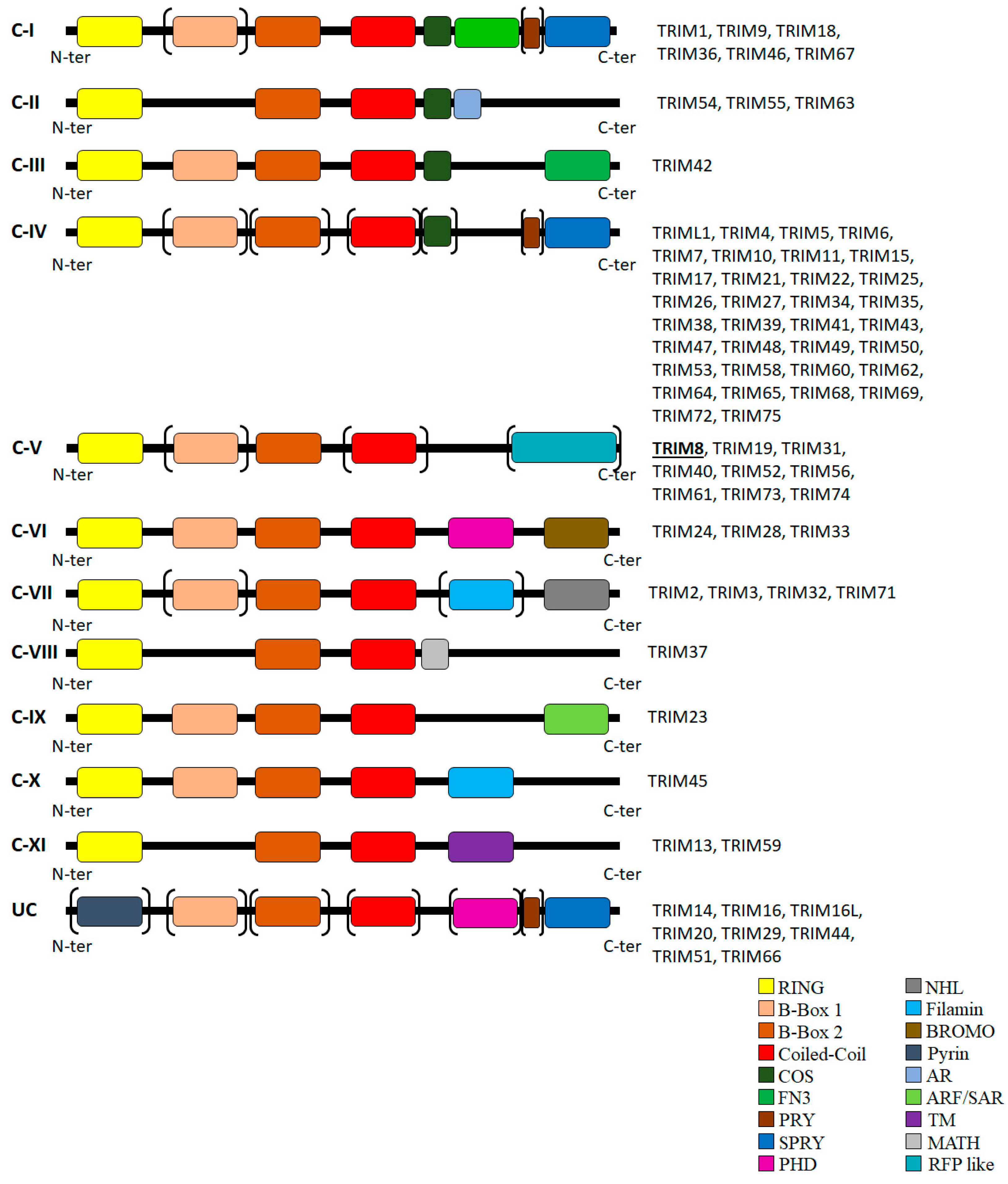
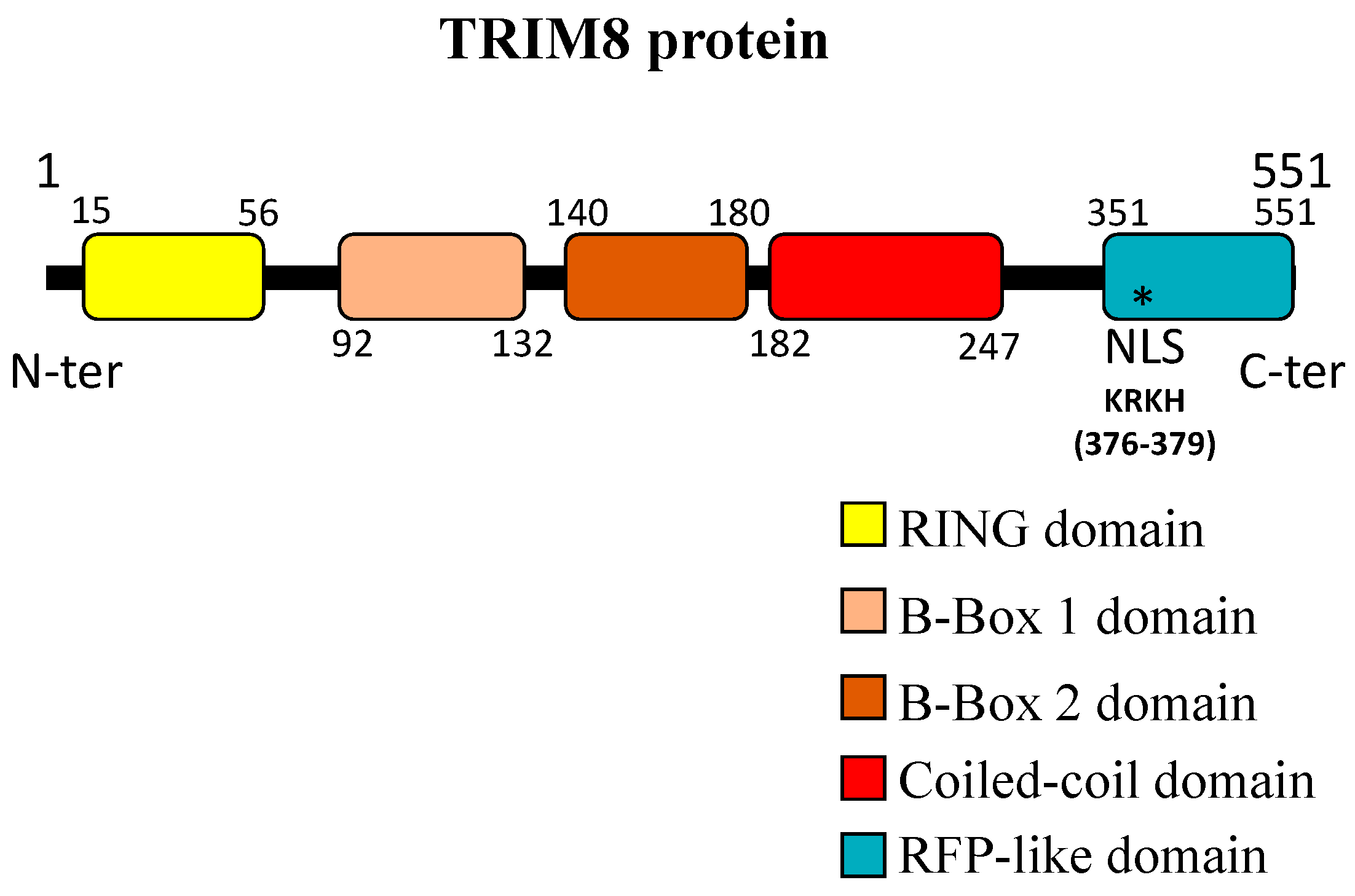
1. TRIM8: A Representative Member of the Large TRIM Family of Proteins
2. The Role of TRIM8 in Tumorigenesis: Tumour Suppressor or Oncogene?
2.1. The Tumour Suppressor Role of TRIM8
2.2. Targeting TRIM8 as A Therapeutic Approach for Human Malignancies
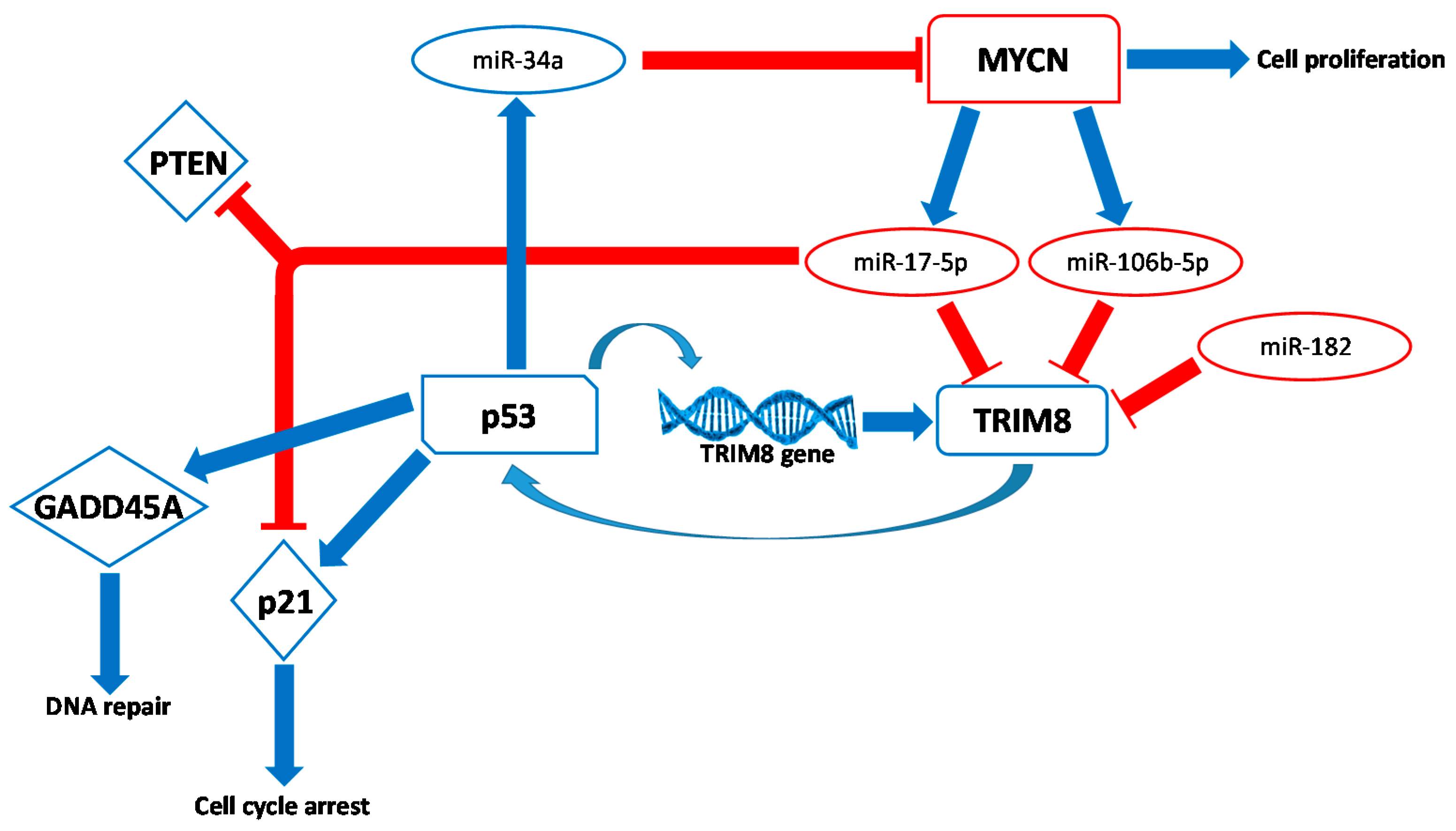
2.3. The Flipside of the Coin: TRIM8 as An Oncogene
3. TRIM8 and Stemness
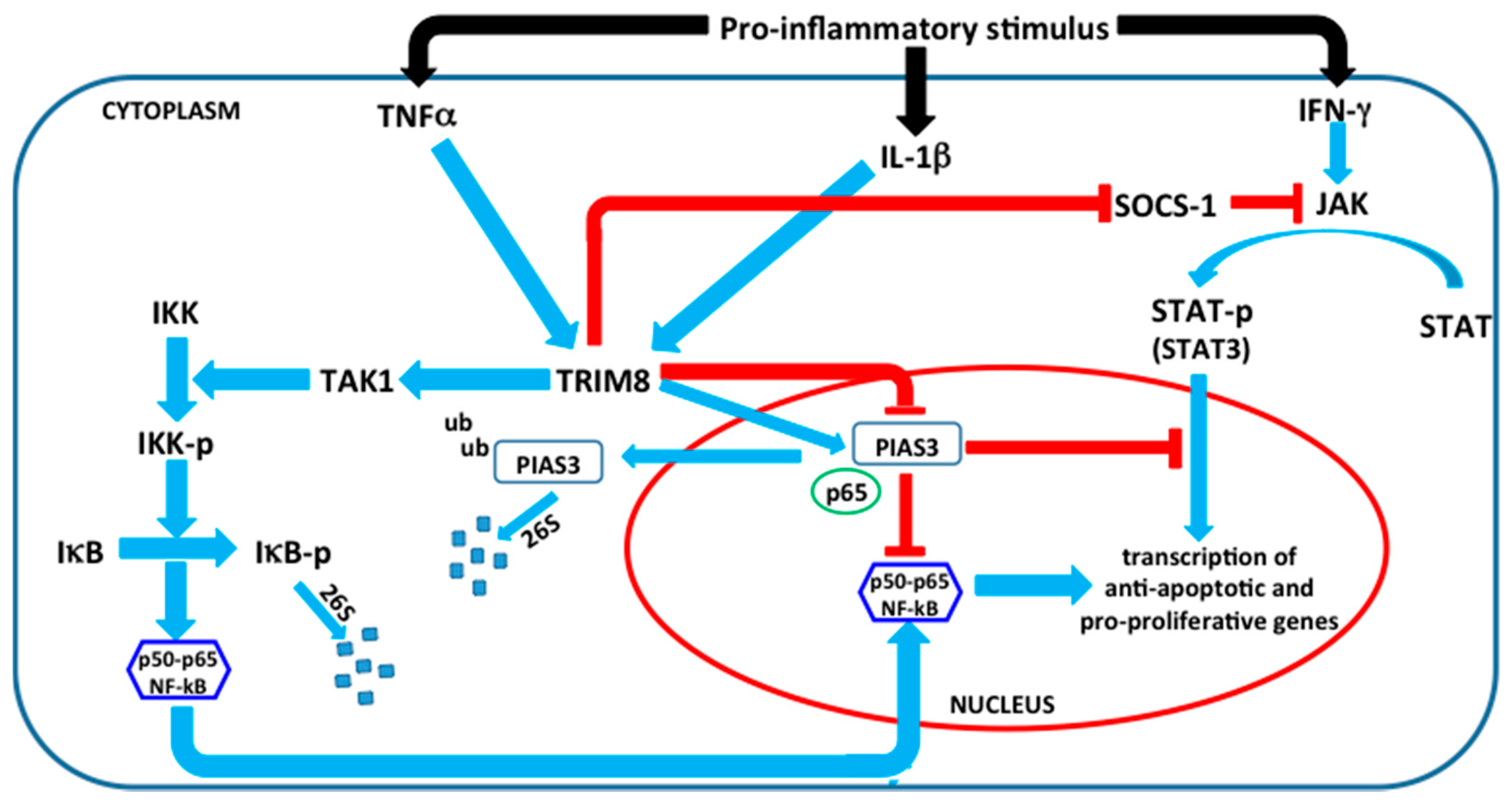
4. TRIM8 and Its Emerging Role in Innate Immunity
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Stoye, J.P.; Saïb, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med. 2011, 3, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; Benke, S.; García-Sastre, A.; Rajsbaum, R. InTRIMsic immunity: Positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev. 2014, 25, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.L. RING domains: Master builders of molecular scaffolds? J. Mol. Biol. 2000, 295, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Lovering, R.; Hanson, I.M.; Borden, K.L.; Martin, S.; O’Reilly, N.J.; Evan, G.I.; Rahman, D.; Pappin, D.J.; Trowsdale, J.; Freemont, P.S. Identification and preliminary characterization of a protein motif related to the zinc finger. Proc. Natl. Acad. Sci. USA 1993, 90, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Freemont, P.S. RING for destruction? Curr. Biol. 2000, 10, R84–R87. [Google Scholar] [CrossRef]
- Joazeiro, C.A.; Weissman, A.M. RING finger proteins: Mediators of ubiquitin ligase activity. Cell 2000, 102, 549–552. [Google Scholar] [CrossRef]
- Chu, Y.; Yang, X. SUMO E3 ligase activity of TRIM proteins. Oncogene 2011, 30, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.; Meroni, G. TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life 2012, 64, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur. J. Immunol. 2008, 38, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Cammas, F.; Khetchoumian, K.; Chambon, P.; Losson, R. TRIM involvement in transcriptional regulation. Adv. Exp. Med. Biol. 2012, 770, 59–76. [Google Scholar] [PubMed]
- Cambiaghi, V.; Giuliani, V.; Lombardi, S.; Marinelli, C.; Toffalorio, F.; Pelicci, P.G. TRIM proteins in cancer. Adv. Exp. Med. Biol. 2012, 770, 77–91. [Google Scholar] [PubMed]
- Vincent, S.R.; Kwasnicka, D.A.; Fretier, P. A novel RING finger-B boxcoiled-coil protein, GERP. Biochem. Biophys. Res. Commun. 2000, 279, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Carinci, F.; Arcelli, D.; Lo Muzio, L.; Francioso, F.; Valentini, D.; Evangelisti, R.; Volinia, S.; D’Angelo, A.; Meroni, G.; Zollo, M.; et al. Molecular classification of nodal metastasis in primary larynx squamous cell carcinoma. Transl. Res. 2007, 150, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Caratozzolo, M.F.; Valletti, A.; Gigante, M.; Aiello, I.; Mastropasqua, F.; Marzano, F.; Ditonno, P.; Carrieri, G.; Simonnet, H.; D’Erchia, A.M.; et al. TRIM8 anti-proliferative action against chemo-resistant renal cell carcinoma. Oncotarget 2014, 5, 7446–7457. [Google Scholar] [CrossRef] [PubMed]
- Caratozzolo, M.F.; Micale, L.; Turturo, M.G.; Cornacchia, S.; Fusco, C.; Marzano, F.; Augello, B.; D’Erchia, A.M.; Guerrini, L.; Pesole, G.; et al. TRIM8 modulates p53 activity to dictate cell cycle arrest. Cell Cycle 2012, 11, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Mastropasqua, F.; Marzano, F.; Valletti, A.; Aiello, I.; Di Tullio, G.; Morgano, A.; Liuni, S.; Ranieri, E.; Guerrini, L.; Gasparre, G.; et al. TRIM8 restores p53 tumour suppressor function by blunting N-MYC activity in chemo-resistant tumours. Mol. Cancer 2017, 16, 67. [Google Scholar] [CrossRef] [PubMed]
- Bomben, R.; Gobessi, S.; Dal Bo, M.; Volinia, S.; Marconi, D.; Tissino, E.; Benedetti, D.; Zucchetto, A.; Rossi, D.; Gaidano, G.; et al. The miR-17∼92 family regulates the response to Toll-like receptor 9 triggering of CLL cells with unmutated IGHV genes. Leukemia 2012, 26, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Micale, L.; Fusco, C.; Fontana, A.; Barbano, R.; Augello, B.; De Nittis, P.; Copetti, M.; Pellico, M.T.; Mandriani, B.; Cocciadiferro, D.; et al. TRIM8 downregulation in glioma affects cell proliferation and it is associated with patients survival. BMC Cancer 2015, 15, 470. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Wang, Y.; Xie, L. TRIM8 regulates the chemoresistance of colorectal cancer in a p53-dependent manner. Oncol. Lett. 2016, 12, 4807–4812. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, B.; Shi, T.; Qin, H. miR-182 promotes tumor growth and increases chemoresistance of human anaplastic thyroid cancer by targeting tripartite motif 8. Onco Targets Ther. 2017, 10, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Elabd, S.; Meroni, G.; Blattner, C. TRIMming p53’s anticancer activity. Oncogene 2016, 35, 5577–5584. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Kirsh, O.; Pearson, M.; Itahana, K.; Pelicci, P.G.; Dejean, A. Deconstructing PML-induced premature senescence. EMBO J. 2002, 21, 3358–3369. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Moller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Droge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.M.; Kim, J.Y.; Jeong, J.B.; Seong, K.M.; Nam, S.Y.; Yang, K.H.; Kim, C.S.; Kim, H.S.; Jeong, M.; An, S.; et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur. J. Cell Biol. 2011, 90, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Villagra, A.; Peng, L.; Coppola, D.; Glozak, M.; Sotomayor, E.M.; Chen, J.; Lane, W.S.; Seto, E. The ATDC (TRIM29) protein binds p53 and antagonizes p53-mediated functions. Mol. Cell. Biol. 2010, 30, 3004–3015. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xu-Monette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, N.J.; Chen, C.; Tang, W.; Kornbluth, S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc. Natl. Acad. Sci. USA 2012, 109, 20931–20936. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ji, Z.; Wang, Y.; Li, J.; Cao, H.; Zhu, H.H.; Gao, W.Q. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 2014, 147, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Y.; Yang, H.; Shi, G.; Xu, G.; Shi, J.; Yin, N.; Chen, D. TRIM66 overexpresssion contributes to osteosarcoma carcinogenesis and indicates poor survival outcome. Oncotarget 2015, 6, 23708–23719. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.F.; Mankaruos, M.; Scorilas, A.; Youssef, Y.; Girgis, A.; Mossad, S.; Metias, S.; Rofael, Y.; Honey, R.J.; Stewart, R.; et al. The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J. Urol. 2010, 183, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Arabi, L.; Gsponer, J.R.; Smida, J.; Nathrath, M.; Perrina, V.; Jundt, G.; Ruiz, C.; Quagliata, L.; Baumhoer, D. Upregulation of the miR-17-92 cluster and its two paraloga in osteosarcoma—Reasons and consequences. Genes Cancer 2014, 5, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Li, Y.; Lim, S.G.; Tan, T.M. miR-106b-25/miR-17-92 clusters: Polycistrons with oncogenic roles in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 5962–5972. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yan, J.; Mao, A.P.; Li, C.; Ran, Y.; Shu, H.B.; Wang, Y.Y. Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β-triggered NF-kappaB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 19341–19346. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Sripada, L.; Prajapati, P.; Singh, R.; Singh, A.K.; Singh, R. Nucleo-cytoplasmic trafficking of TRIM8, a novel oncogene, is involved in positive regulation of TNF induced NF-kappaB pathway. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Tergaonkar, V. Roles of NF-kappaB in health and disease: Mechanisms and therapeutic potential. Clin. Sci. 2009, 116, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baltimore, D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Z.; Hu, C.C.; Chen, Y.H.; Stern, A.; Cheng, J.T. Differentiation status modulates transcription factor NF-kappaB activity in unstimulated human hepatocellular carcinoma cell lines. Cancer Lett. 2000, 151, 49–56. [Google Scholar] [CrossRef]
- Liu, F.T.; Jia, L.; Wang, P.; Wang, H.; Farren, T.W.; Agrawal, S.G. STAT3 and NF-κB cooperatively control in vitro spontaneous apoptosis and poor chemo-responsiveness in patients with chronic lymphocytic leukemia. Oncotarget 2016, 7, 2031–3245. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A positive feedback loop involving EGFR/Akt/mTORC1 and IKK/NF-kB regulates head and neck squamous cell carcinoma proliferation. Oncotarget 2016, 7, 31892–31906. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, X.; Li, J.J. The role of NF-kappaB in the regulation of cell stress responses. Int. Immunopharmacol. 2002, 2, 1509–1520. [Google Scholar] [CrossRef]
- Wang, Y.; Paszek, P.; Horton, C.A.; Yue, H.; White, M.R.; Kell, D.B.; Muldoon, M.R.; Broomhead, D.S. A systematic survey of the response of a model NF-κB signalling pathway to TNFα stimulation. J. Theor. Biol. 2012, 297, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, J.L.; Yu, K.K.; Xu, M.; Xu, Y.Z.; Chen, L.; Lu, Y.M.; Fang, H.S.; Wang, X.Y.; Hu, Z.Q.; et al. Activation of the NF-κB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 2015, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Joyce, D.; Albanese, C.; Steer, J.; Fu, M.; Bouzahzah, B.; Pestell, R.G. NF-kappaB and cell-cycle regulation: The cyclin connection. Cytokine Growth Factor Rev. 2001, 12, 73–90. [Google Scholar] [CrossRef]
- Huang, C.; Li, J.; Ding, M.; Wang, L.; Shi, X.; Castranova, V.; Vallyathan, V.; Ju, G.; Costa, M. Arsenic-induced NFkappaB transactivation through Erks- and JNKs-dependent pathways in mouse epidermal JB6 cells. Mol. Cell. Biochem. 2001, 222, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Pettaway, C.A.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Shirakabe, K.; Shibuya, H.; Irie, K.; Oishi, I.; Ueno, N.; Taniguchi, T.; Nishida, E.; Matsumoto, K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 1995, 270, 2008–2011. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya-Tsuji, J.; Kishimoto, K.; Hiyama, A.; Inoue, J.; Cao, Z.; Matsumoto, K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999, 398, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Ninomiya-Tsuji, J.; Qian, Y.; Matsumoto, K.; Li, X. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol. Cell. Biol. 2002, 22, 7158–7167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Matsunaga, Y.; Katayama, Y.; Nakayama, K.I.; Hatakeyama, S. TRIM8 modulates STAT3 activity through negative regulation of PIAS3. J. Cell Sci. 2010, 123, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Lluva, S.; Tatham, M.H.; Jones, R.C.; Jaffray, E.G.; Edmondson, R.D.; Hay, R.T.; Malliri, A. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat. Cell Biol. 2010, 12, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.M.; Yan, M.G.; Yang, D.H.; Sun, W.W.; Zhang, J.X. PIAS3 expression in human gastric carcinoma and its adjacent non-tumor tissues. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Brantley, E.C.; Nabors, L.B.; Gillespie, G.Y.; Choi, Y.H.; Palmer, C.A.; Harrison, K.; Roarty, K.; Benveniste, E.N. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: Implications for STAT-3 activation and gene expression. Clin. Cancer Res. 2008, 14, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Toniato, E.; Chen, X.P.; Losman, J.; Flati, V.; Donahue, L.; Rothman, P. TRIM8/GERP RING finger protein interacts with SOCS-1. J. Biol. Chem. 2002, 277, 37315–37322. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.S. Suppressors of cytokine signalling (SOCS) in the immune system. Nat. Rev. Immunol. 2002, 2, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; De Groot, R.P.; Jove, R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: Clinical implications. Clin. Cancer Res. 2007, 13, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Goff, S.P. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 2007, 131, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kong, J.; Tucker-Burden, C.; Anand, M.; Rong, Y.; Rahman, F.; Moreno, C.S.; Van Meir, E.G.; Hadjipanayis, C.G.; Brat, D.J. Human Brat ortholog TRIM3 is a tumor suppressor that regulates asymmetric cell division in glioblastoma. Cancer Res. 2014, 74, 4536–4548. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Tucker-Burden, C.; Zhang, C.; Moberg, K.; Read, R.; Hadjipanayis, C.; Brat, D.J. Drosophila Brat and human ortholog TRIM3 maintain stem cell equilibrium and suppress brain tumorigenesis by attenuating Notch nuclear transport. Cancer Res. 2016, 76, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Raychaudhuri, B.; Haqqi, T.; Prayson, R.; Van Meir, E.G.; Vogelbaum, M.; Haque, S.J. Stat3 activation is required for the growth of U87 cell derived tumours in mice. Eur. J. Cancer 2009, 45, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Cherryholmes, G.; Schroeder, A.; Phallen, J.; Alizadeh, D.; Xin, H.; Wang, T.; Lee, H.; Lahtz, C.; Swiderski, P.; et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. 2014, 74, 5218–5228. [Google Scholar] [CrossRef] [PubMed]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM gene expression in response to interferons. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, Y.; Yang, Y.; Yang, M.; Zhou, L.; Huang, X.; Qin, Q. Fish TRIM8 exerts antiviral roles through regulation of the proinflammatory factors and interferon signaling. Fish Shellfish Immunol. 2016, 54, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Conklin, J.F.; Sage, J. Keeping an eye on retinoblastoma control of human embryonic stem cells. J. Cell. Biochem. 2009, 108, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Cicalese, A.; Insinga, A.; Pelicci, P.G. The emerging role of p53 in stem cells. Trends Mol. Med. 2012, 18, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Espinoza, L.A.; Kinders, R.J.; Lawrence, S.M.; Pfister, T.D.; Zhou, M.; Veenstra, T.D.; Thorgeirsson, S.S.; Jessup, J.M. NANOG modulates stemness in human colorectal cancer. Oncogene 2013, 32, 4397–4405. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhu, H.; Shan, H.; Lu, J.; Chang, X.; Li, X.; Lu, J.; Fan, X.; Zhu, S.; Wang, Y.; et al. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer Lett. 2013, 340, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mazur, S.J.; Lin, T.; Appella, E.; Xu, Y. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene 2014, 33, 2655–2664. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caratozzolo, M.F.; Marzano, F.; Mastropasqua, F.; Sbisà, E.; Tullo, A. TRIM8: Making the Right Decision between the Oncogene and Tumour Suppressor Role. Genes 2017, 8, 354. https://doi.org/10.3390/genes8120354
Caratozzolo MF, Marzano F, Mastropasqua F, Sbisà E, Tullo A. TRIM8: Making the Right Decision between the Oncogene and Tumour Suppressor Role. Genes. 2017; 8(12):354. https://doi.org/10.3390/genes8120354
Chicago/Turabian StyleCaratozzolo, Mariano Francesco, Flaviana Marzano, Francesca Mastropasqua, Elisabetta Sbisà, and Apollonia Tullo. 2017. "TRIM8: Making the Right Decision between the Oncogene and Tumour Suppressor Role" Genes 8, no. 12: 354. https://doi.org/10.3390/genes8120354
APA StyleCaratozzolo, M. F., Marzano, F., Mastropasqua, F., Sbisà, E., & Tullo, A. (2017). TRIM8: Making the Right Decision between the Oncogene and Tumour Suppressor Role. Genes, 8(12), 354. https://doi.org/10.3390/genes8120354