
Functional Characterization of Rare RAB12 Variants and Their Role in Musician’s and Other Dystonias


Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Mutation Screening
2.3. cDNA Analysis
2.4. Plasmid and Viral Construction
2.5. Cell Culture and Stable Transfection
2.6. GTPase Assay
2.7. RAB12 Protein Structure Modeling and Molecular Dynamics Simulations
2.8. Immunostaining
2.9. Autophagy Inhibition and TFRC Degradation
2.10. Endogenous TFRC Levels in Patients’ Blood
2.11. Statistical Analysis
3. Results
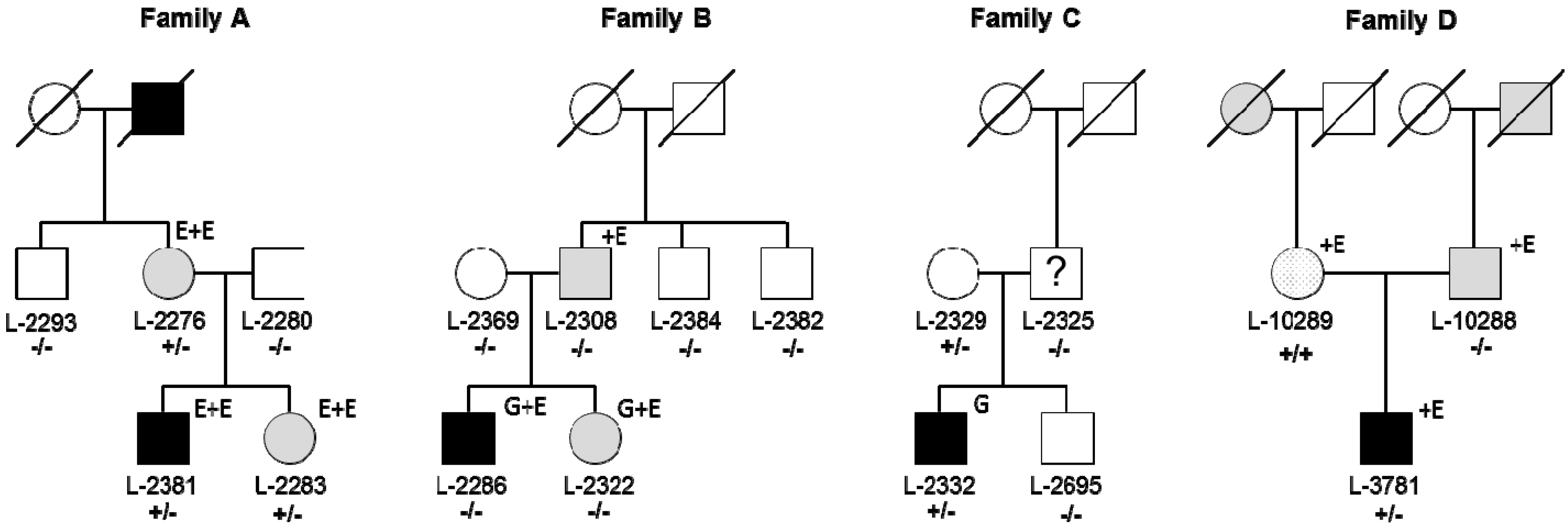
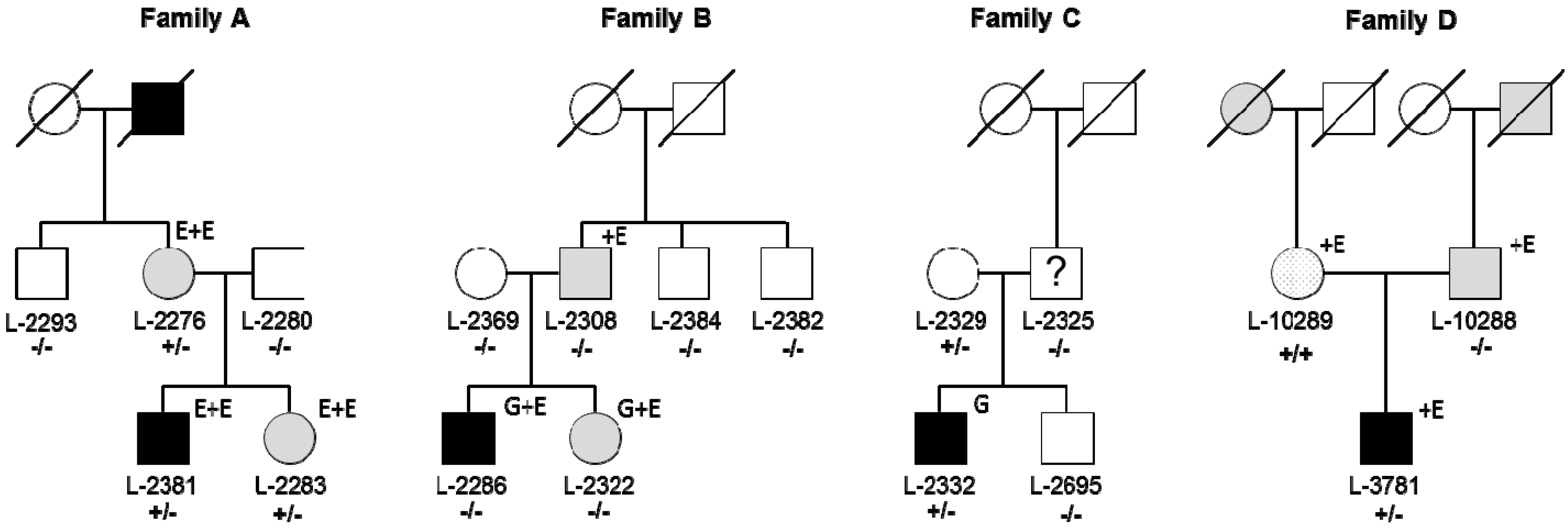
3.1. Identification of RAB12 Mutations in Patients with Musician’S Dystonia
3.2. RAB12 Variants Are More Frequent in Dystonia Patients
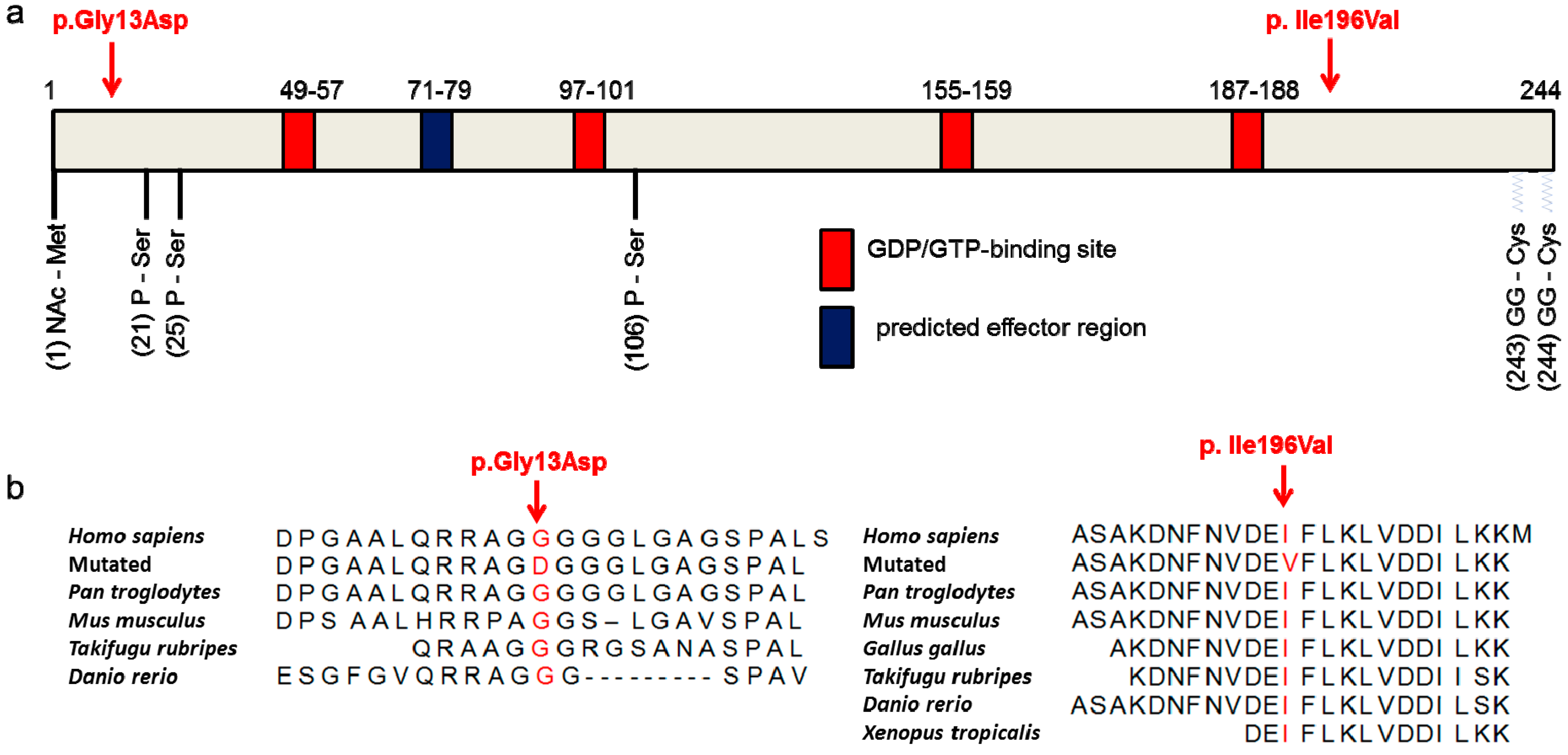
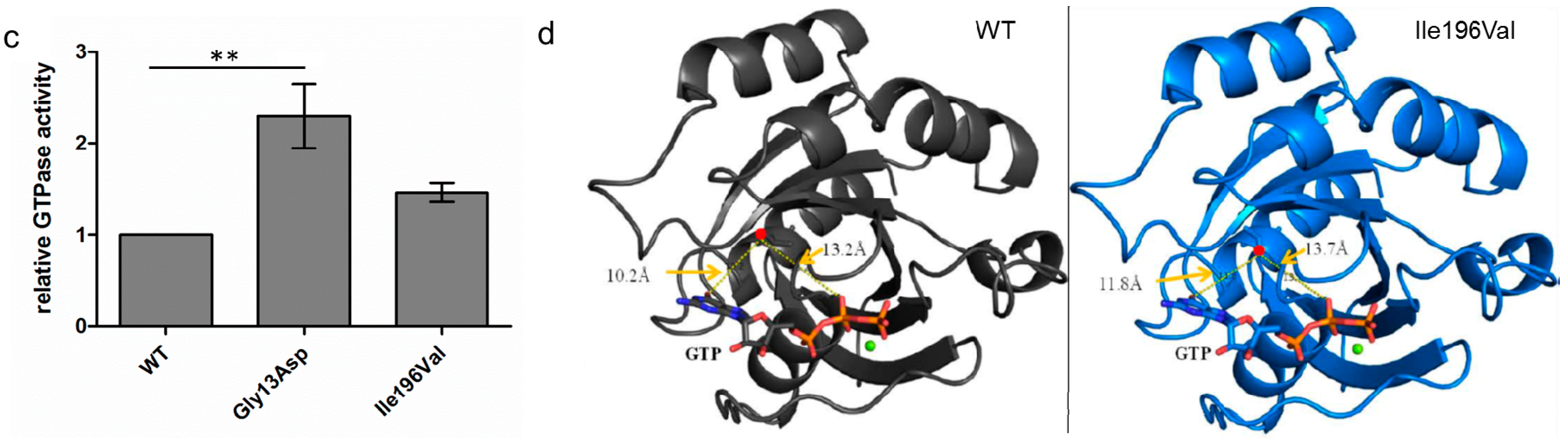
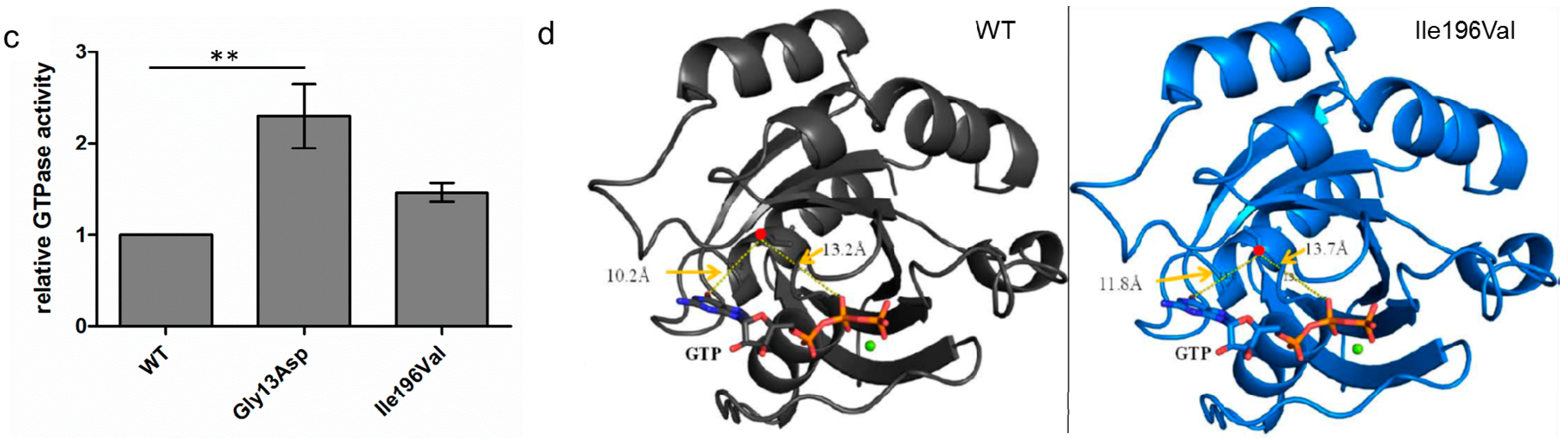
3.3. GTPase Activity Seems to Be Elevated in RAB12 Mutants
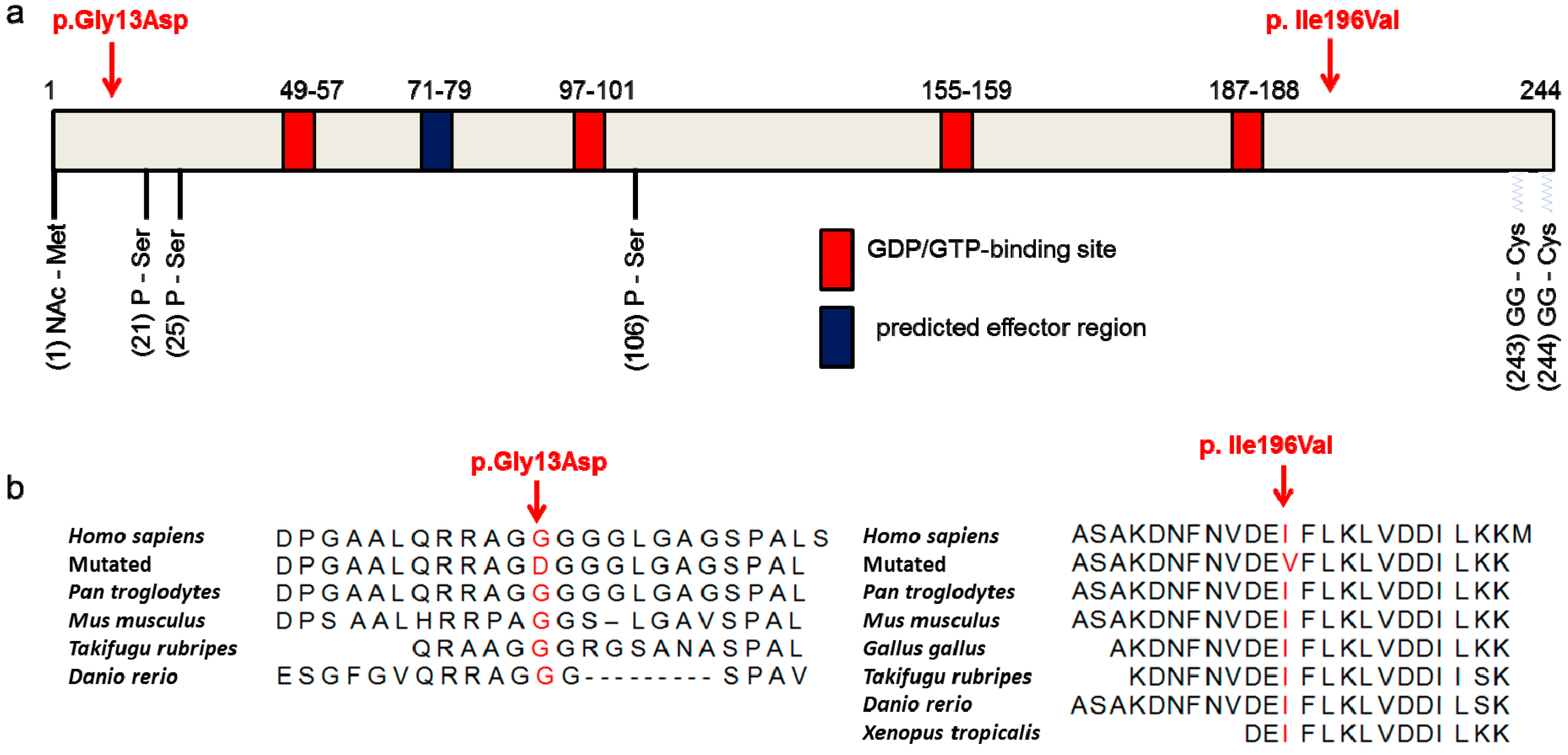
3.4. The p.Ile196Val Mutation Does Not Change the Secondary and Tertiary Structure of RAB12 In Silico
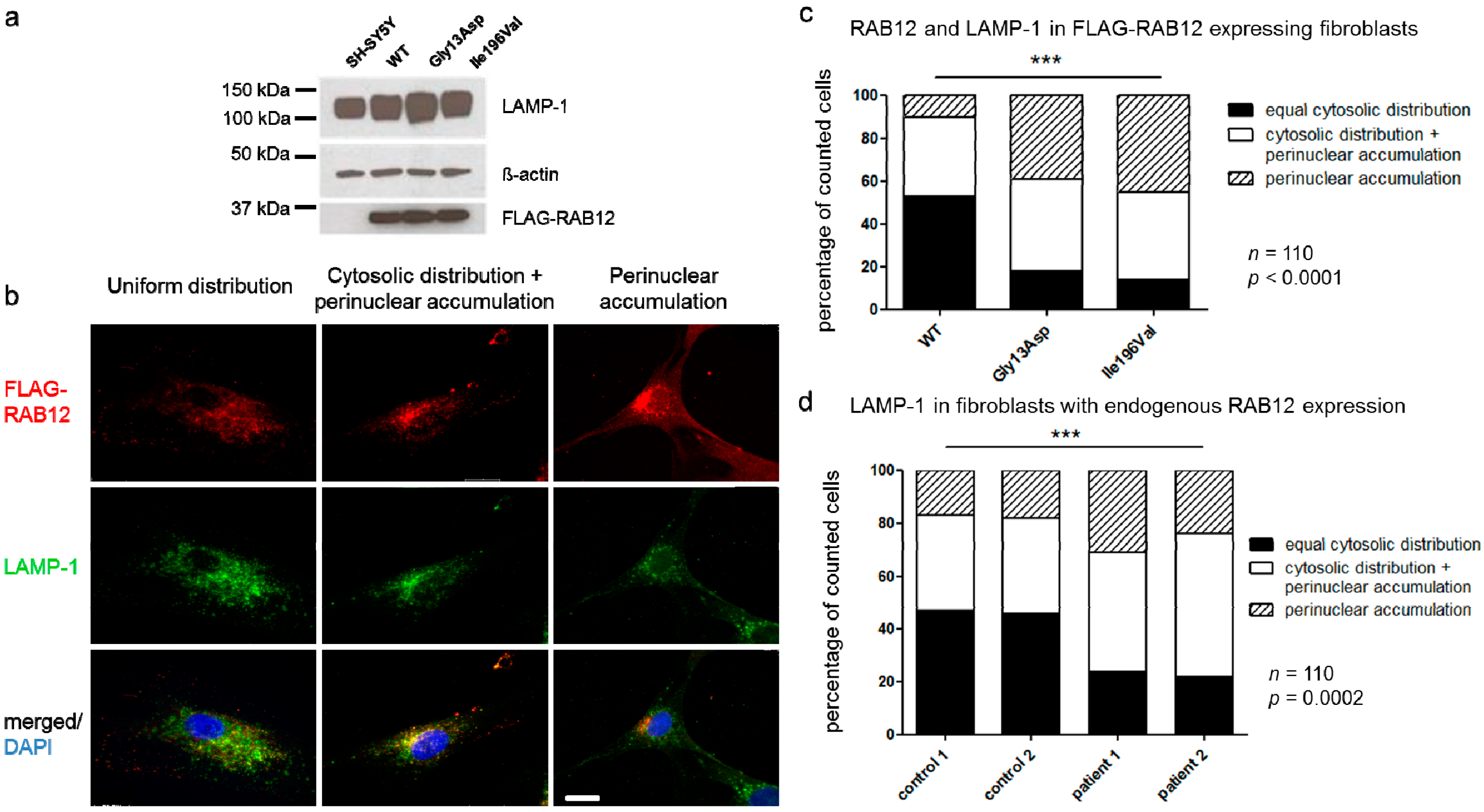
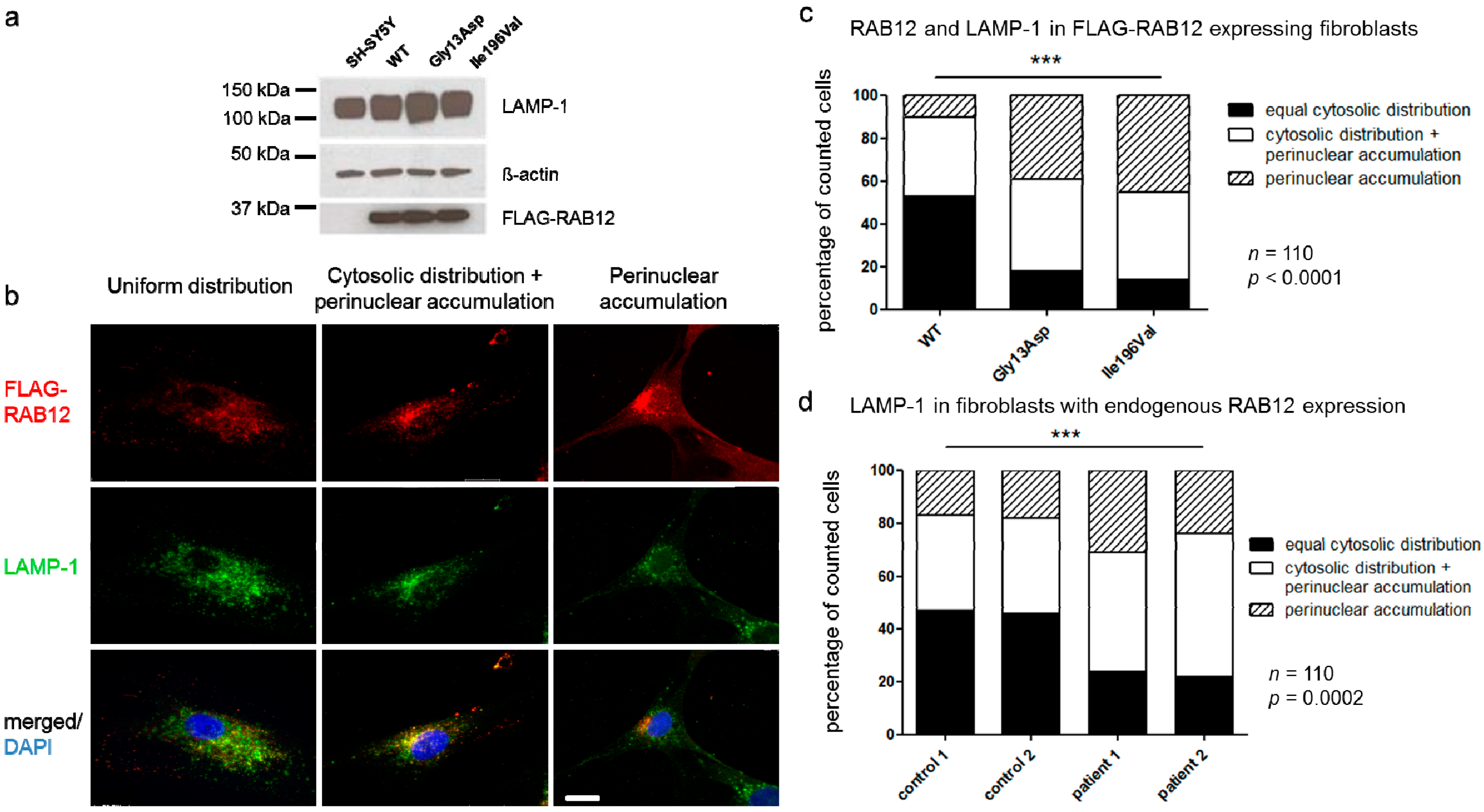
3.5. RAB12 Mutations Alter the Subcellular Localization of RAB12 and Lysosomes
3.6. Soluble TFRC Levels in Patients’ Blood Were Reduced
3.7. Colocalization of RAB12 and TFRC and Degradation of TFRC Was Unchanged in RAB12 Mutants
3.8. Autophagy Is Not Impaired in SH-SY5Y Cells Overexpressing RAB12 Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jankovic, J.; Ashoori, A. Movement disorders in musicians. Mov. Disord. 2008, 23, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Frucht, S.J.; Fahn, S.; Greene, P.E.; O’Brien, C.; Gelb, M.; Truong, D.D.; Welsh, J.; Factor, S.; Ford, B. The natural history of embouchure dystonia. Mov. Disord. 2001, 16, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Altenmuller, E.; Jabusch, H.C. Focal dystonia in musicians: Phenomenology, pathophysiology and triggering factors. Eur. J. Neurol. 2010, 17, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Jabusch, H.C.; Altenmuller, E.; Hagenah, J.; Bruggemann, N.; Lohmann, K.; Enders, L.; Kramer, P.L.; Saunders-Pullman, R.; Bressman, S.B.; et al. Etiology of musician’s dystonia: Familial or environmental? Neurology 2009, 72, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Jabusch, H.; Altenmüller, E.; Enders, L.; Saunders-Pullman, R.; Bressman, S.; Münchau, A.; Klein, C.; Hagenah, J. Phenotypic spectrum of musician’s dystonia: A task-specific disorder? Mov. Disord. 2011, 26, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, C.I.; Furuya, S.; Altenmuller, E. The impact of stress on motor performance in skilled musicians suffering from focal dystonia: Physiological and psychological characteristics. Neuropsychologia 2016, 85, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Enders, L.; Spector, J.T.; Altenmuller, E.; Schmidt, A.; Klein, C.; Jabusch, H.C. Musician’s dystonia and comorbid anxiety: Two sides of one coin? Mov. Disord. 2011, 26, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Altenmuller, E.; Jabusch, H.C. Focal dystonia in musicians: Phenomenology, pathophysiology, triggering factors, and treatment. Med. Probl. Perform. Artist. 2010, 25, 3–9. [Google Scholar]
- Lohmann, K.; Schmidt, A.; Schillert, A.; Winkler, S.; Albanese, A.; Baas, F.; Bentivoglio, A.R.; Borngraber, F.; Bruggemann, N.; Defazio, G.; et al. Genome-wide association study in musician’s dystonia: A risk variant at the arylsulfatase G locus? Mov. Disord. 2014, 29, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Altenmuller, E.; Jabusch, H.C.; Lee, A.; Wiegers, K.; Klein, C.; Lohmann, K. The GAG deletion in Tor1A (DYT1) is a rare cause of complex musician’s dystonia. Parkinsonism Relat. Disord. 2012, 18, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Lohmann, K.; Masuho, I.; Miyamoto, R.; Ferbert, A.; Lohnau, T.; Kasten, M.; Hagenah, J.; Bruggemann, N.; Graf, J.; et al. Mutations in GNAL: A novel cause of craniocervical dystonia. JAMA Neurol. 2014, 71, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.; Lambright, D.G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 2010, 22, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Dupree, P.; Killisch, I.; Lutcke, A.; Zerial, M.; Simons, K. Molecular cloning and subcellular localization of three GTP-binding proteins of the rab subfamily. J. Cell Sci. 1993, 106, 1249–1261. [Google Scholar] [PubMed]
- Matsui, T.; Itoh, T.; Fukuda, M. Small GTPase Rab12 regulates constitutive degradation of transferrin receptor. Traffic 2011, 12, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Fukuda, M. Rab12 regulates mTORC1 activity and autophagy through controlling the degradation of amino-acid transporter PAT4. EMBO Rep. 2013, 14, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Aligianis, I.A.; Johnson, C.A.; Gissen, P.; Chen, D.; Hampshire, D.; Hoffmann, K.; Maina, E.N.; Morgan, N.V.; Tee, L.; Morton, J.; et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 2005, 37, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.R.; Sim, J.C.; McLean, C.; Giannandrea, M.; Galea, C.A.; Riseley, J.R.; Stephenson, S.E.; Fitzpatrick, E.; Haas, S.A.; Pope, K.; et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am. J. Hum. Genet. 2014, 95, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Bras, J.; Cormier-Dequaire, F.; Condroyer, C.; Nicolas, A.; Darwent, L.; Guerreiro, R.; Majounie, E.; Federoff, M.; Heutink, P.; et al. Loss-of-function mutations in RAB39B are associated with typical early-onset Parkinson disease. Neurol. Genet. 2015, 1, e9. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Hagenah, J.; Graf, J.; Lorwin, A.; Vollstedt, E.J.; Peters, E.; Katalinic, A.; Raspe, H.; Klein, C. Cohort Profile: A population-based cohort to study non-motor symptoms in parkinsonism (EPIPARK). Int. J. Epidemiol. 2013, 42, 128–128k. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Jabusch, H.C.; Altenmuller, E.; Hagenah, J.; Bruggemann, N.; Hedrich, K.; Saunders-Pullman, R.; Bressman, S.B.; Kramer, P.L.; Klein, C. Dominantly transmitted focal dystonia in families of patients with musician’s cramp. Neurology 2006, 67, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Noguchi, K.; Fukuda, M. Dennd3 functions as a guanine nucleotide exchange factor for small GTPase Rab12 in mouse embryonic fibroblasts. J. Biol. Chem. 2014, 289, 13986–13995. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yin, K.; Lu, D.; Li, B.; Zhu, D.; Chen, Y.; Zhang, H.; Xu, S.; Chai, J.; Gu, L. Structural insights into a unique Legionella pneumophila effector LidA recognizing both GDP and GTP bound Rab1 in their active state. PLoS Pathog. 2012, 8, e1002528. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bowers, K.J.; Edmond, C.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Article No. 84. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing, Tampa, Florida, 11–17 November 2006; ACM: New York, NY, USA, 2006. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A.; van de Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Structural clues to Rab GTPase functional diversity. J. Biol. Chem. 2005, 280, 15485–15488. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Efergan, A.; Azouz, N.P.; Klein, O.; Noguchi, K.; Rothenberg, M.E.; Fukuda, M.; Sagi-Eisenberg, R. Rab12 regulates retrograde transport of mast cell secretory granules by interacting with the RILP-dynein complex. J. Immunol. 2016, 196, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [PubMed]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.V.; Westaway, S.K.; Morton, J.E.; Gregory, A.; Gissen, P.; Sonek, S.; Cangul, H.; Coryell, J.; Canham, N.; Nardocci, N.; et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 2006, 38, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; MacGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539–2543. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Al-Saif, A.; Al-Semari, A.; Bohlega, S.; Zlitni, S.; Alzahrani, F.; Bavi, P.; Kaya, N.; Colak, D.; Khalak, H.; et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am. J. Hum. Genet. 2008, 83, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Chuang, R.; Marras, C.; Lang, A.E. The curious case of phenocopies in families with genetic Parkinson’s disease. Mov. Disord. 2011, 26, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Shi, L.; Hakenberg, J.; Naughton, B.; Sklar, P.; Zhang, J.; Zhou, H.; Tian, L.; Prakash, O.; Lemire, M.; et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 2016, 34, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Bressman, S.B.; Raymond, D.; Wendt, K.; Saunders-Pullman, R.; De Leon, D.; Fahn, S.; Ozelius, L.; Risch, N. Diagnostic criteria for dystonia in DYT1 families. Neurology 2002, 59, 1780–1782. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Jabusch, H.C.; Altenmuller, E.; Kasten, M.; Klein, C. Challenges of making music: What causes musician’s dystonia? JAMA Neurol. 2013, 70, 1456–1459. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Next Generation Sequencing | Sanger Sequencing | Gene Panel | Total | |
|---|---|---|---|---|
| MD patients | 4 b | 237 | 0 | 241 |
| Relatives of MD patients | 6 a,b | 8 | 0 | 14 |
| Unrelated WD patients | 0 | 54 | 20 b | 74 |
| Other dystonia patients | 0 | 378 | 226 b | 604 |
| PD patients | 0 | 0 | 512 | 512 |
| Healthy controls | 0 | 461 | 0 | 461 |
| Total | 10 | 1138 | 758 | 1906 |
| Variant (NM_001025300.2, NP_001020471.2) | ID | Polyphen-2 (Score) | Mutation Taster (Score) | MutPred (Score) | CADD Score | Allele Frequency in MD Patients | Allele Frequency in Other Dystonia Patients | Allel Frequency in Controls (a) | Allele Frequency in GnomAD All/non-Finnish EUR |
|---|---|---|---|---|---|---|---|---|---|
| c.38G>A p.Gly13Asp | L-3921 | Benign (0.086) | Disease causing (0.994) | Benign (0.266) | 22.4 | 1/482 | 0/492 | 0/1946 | 0.0006/n.a. |
| c.276A>G p.Arg92= rs372618073 | L-3799 L-5334 | n.a. | Disease causing (0.999) | n.a. | 8.2 | 1/482 | 0/1356 | 1/1946 | 0.0003/0.0006 |
| c.391GA>CT p.Asp131Leu (b) | L-5385 | Benign (0.048) | Disease causing (0.999) | Disease causing (0.492) | 25 | 0/482 | 0/1356 | 1/1946 | 0.00003/0.00006 |
| c.442G>A p.Ala148Thr | L-1729 | Probably damaging (0.995) | Disease causing (0.999) | Disease causing (0.654) | 31 | 0/482 | 1/1356 | 0/1946 | n.a. |
| c.520G>A p.Ala174Thr rs149427020 | L-11086 L-11092 L-11132 | Probably damaging (0.977) | Disease causing (0.999) | Disease causing (0.828) | 32 | 0/482 (c) | 0/1184 EUR 3/172 EA | 0/1 946 (c) | 0.0004/0.0064 (d) |
| c.542G>A p.Arg181Gln rs371288995 | L-8527 | Probably damaging (0.970) | Disease causing (0.999) | Benign (0.377) | 25.3 | 0/482 | 1/1356 | 0/1946 | 0.00007/0.0001 |
| c.586A>G p.Ile196Val rs143888944 | L-2381 L-2332 L-3758 L-3781 L-8307 L-9497 | Benign (0.323) | Disease causing (0.999) | Disease causing (0.555) | 16.5 | 3/482 (e) | 3/1356 | 0/1946 | 0.0003/0.0006 |
| c.693G>A p.Pro231= | L-5328 | n.a. | Disease causing (0.999) | n.a. | 10.5 | 0/482 | 0/1356 | 1/1946 | 0.00001/0.00002 |
| ID | Variant | Sex | Age at Onset (Years) | Initial Symptom | Age (Years) | Current Symptoms | Family History | Ethnicity |
|---|---|---|---|---|---|---|---|---|
| L-3921 | c.38G>A p.Gly13Asp | male | 25 | MD | 38 | MD | negative | German |
| L-1729 | c.442G>A p.Ala148Thr | female | 76 | dervical dystonia | 78 | torticollis, essential tremor | negative | German |
| L-11086 | c.520G>A p.Ala174Thr | male | 23 | MD | 33 | task-specific arm dystonia (MD + WD) | negative | South Korean |
| L-11092 | c.520G>A p.Ala174Thr | male | 17 | WD | 41 | torticollis, WD | negative | South Korean |
| L-11132 | c.520G>A p.Ala174Thr | female | 58 | dystonia of the upper lip | 70 | torticollis, oromandibular dystonia | negative | South Korean |
| L-8527 | c.542G>A p.Arg181Gln | female | 58 | cervical dystonia | 59 | torticollis | unknown | German |
| L-2381 | c.586A>G p.Ile196Val | male | 27 | MD | 44 | MD | positive | German |
| L-2332 | c.586A>G p.Ile196Val | male | 31 | MD | 48 | MD | negative | Dutch |
| L-3781 | c.586A>G p.Ile196Val | male | 17 | MD | 39 | MD | positive for dystonia | German |
| L-3758 | c.586A>G p.Ile196Val | female | 56 | cervical dystonia | 67 | torticollis, shoulder elevation, head tremor | positive for Parkinson´s disease | German |
| L-8307 | c.586A>G p.Ile196Val | female | 70 | cervical dystonia | 73 | torticollis, head tremor | positive for Parkinson´s disease/tremor | German |
| L-9497 | c.586A>G p.Ile196Val | female | 41 | WD | 53 | task-specific arm dystonia | positive for Parkinson´s disease/tremor | German |
| Parameter (Reference) | Female Patient with WD (L-2276) | Female Patient with WD (L-2283) | Male Patient with MD (L-2381) |
|---|---|---|---|
| Erythrocytes (Mio/µL) | 4.5 | 4.5 | 4.5 |
| (4.20–5.40) | (4.20–5.40) | (4.5–5.9) | |
| Hemoglobin (g/dL) | 13.6 | 13.8 | 14 |
| (12.0–16.0) | (12.0–16.0) | (14.0–17.5) | |
| Hematocrit (%) | 42 | 40.5 | 43 |
| (37.0–47.0) | (37.0–47.0) | (40–53) | |
| Ferritin (ng/mL) | 31.2 | 28.4 | 276 |
| (10.0–291.0) | (10.0–291.0) | (18–360) | |
| Transferrin (g/L) | 2.4 | 2.6 | 2.3 |
| (2.0–3.8) | (2.0–3.8) | (2.50–3.80) | |
| Fe2+ (µg/dL) | 75.2 | 102.6 | 81 |
| (50.0–170.0) | (50.0–170.0) | (61.5–156.46) | |
| Soluble Transferrin receptor (mg/L) | 0.8 | 0.7 | 1.7 |
| (0.83–1.76) | (0.83–1.76) | (2.20–4.99) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hebert, E.; Borngräber, F.; Schmidt, A.; Rakovic, A.; Brænne, I.; Weissbach, A.; Hampf, J.; Vollstedt, E.-J.; Größer, L.; Schaake, S.; et al. Functional Characterization of Rare RAB12 Variants and Their Role in Musician’s and Other Dystonias. Genes 2017, 8, 276. https://doi.org/10.3390/genes8100276
Hebert E, Borngräber F, Schmidt A, Rakovic A, Brænne I, Weissbach A, Hampf J, Vollstedt E-J, Größer L, Schaake S, et al. Functional Characterization of Rare RAB12 Variants and Their Role in Musician’s and Other Dystonias. Genes. 2017; 8(10):276. https://doi.org/10.3390/genes8100276
Chicago/Turabian StyleHebert, Eva, Friederike Borngräber, Alexander Schmidt, Aleksandar Rakovic, Ingrid Brænne, Anne Weissbach, Jennie Hampf, Eva-Juliane Vollstedt, Leopold Größer, Susen Schaake, and et al. 2017. "Functional Characterization of Rare RAB12 Variants and Their Role in Musician’s and Other Dystonias" Genes 8, no. 10: 276. https://doi.org/10.3390/genes8100276
APA StyleHebert, E., Borngräber, F., Schmidt, A., Rakovic, A., Brænne, I., Weissbach, A., Hampf, J., Vollstedt, E.-J., Größer, L., Schaake, S., Müller, M., Manzoor, H., Jabusch, H.-C., Alvarez-Fischer, D., Kasten, M., Kostic, V. S., Gasser, T., Zeuner, K. E., Kim, H.-J., ... Lohmann, K. (2017). Functional Characterization of Rare RAB12 Variants and Their Role in Musician’s and Other Dystonias. Genes, 8(10), 276. https://doi.org/10.3390/genes8100276