1. Introduction
Modern molecular biology has been revolutionized with the emergence of high-throughput experimental technologies such as microarrays and next-generation DNA sequencing. While many exciting discoveries have been made by data-driven analysis of such whole-genome data sets, an important problem that many biologists face everyday is how to interpret such large-scale data sets. A typical output from such a high-throughput experiment is a list of genes that are observed to be associated with a certain phenotype, such as those differentially expressed in tumors compared to normal tissues. In contrast to the easiness in obtaining the gene list, the bottleneck usually lies in understanding the meaning of the genes and generating new testable hypotheses with the hope to reveal the underlying molecular cause of the phenotype.
Biologists are knowledge-driven. A principled way to interpret such gene lists is to compare them with a database of well-annotated gene sets, such as biological pathways. For example, one of the most widely used approach, Over Representation Analysis (ORA) [
1], counts the number of common genes shared by an input gene set and each annotated gene set and applies a statistical test, such as the cumulative hyper-geometric test, to calculate the statistical significance of the overlap. A
p-value cutoff, e.g., 0.05, is then applied to select the annotated gene sets that share a statistically significant overlap with the input gene set. ORA is very easy to implement, and the idea behind it is straightforward to biologists. A popular extension of ORA, known as the Gene Set Enrichment Analysis (GSEA) [
2], tries to eliminate the need for an ad hoc cutoff (e.g., expression fold change), which is often used in defining the input gene set. GSEA works by ranking all genes in the genome according to, say, level of differential expression, and tests whether any annotated gene set is ranked unexpectedly high or low through a running-sum statistic. While GSEA is becoming more popular, it is sensitive to noise and may report too many pathways that are conceptually hard to comprehend by biologists. In addition, GSEA is not applicable in cases where a completely ranked gene list is unavailable. As a result, ORA is still widely used by biologists.
Both ORA and GSEA depend on the availability of trusted gene annotations, such as gene ontology (GO) or metabolic pathways, which limits their applicability to only well annotated species. In addition, gene annotations in databases such as GO or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway may be strongly biased by some classes of genes or phenotypes that are popular targets, such as cancer. More importantly, it is becoming more and more well known that such enrichment-based analyses, including both ORA and GSEA, have very low discriminative power, as they treat genes as independent functional units. In reality, genes function in a highly coordinated way. For example, two gene sets may share few genes but can be involved in similar functional pathways, or they can represent two sub-modules of the same pathway. Common enrichment-based analysis may not be able to detect the relationship between gene sets.
To address the aforementioned issues, several studies have proposed the use of biological networks, such as protein–protein interaction (PPI) networks, as a more unbiased tool to investigate the biological meaning of gene sets [
3,
4,
5,
6]. The rationale is that genes that are located within a short distance in the same network are likely involved in similar biological processes. As such networks are typically obtained from high-throughput experiments, they are less likely to be biased by existing knowledge and can probably provide better coverage to different classes of genes and phenotypes. Furthermore, network-based analysis allows the relationship between genes to be explicitly modeled, instead of treating them as independent entities. While conceptually interesting, such methods have had limited success because high-throughput biological networks are usually very noisy, and still have many missing edges. Furthermore, the results obtained by such analyses, often in the form of PPI subnetworks, can be difficult to interpret because functional connections to biological processes are missing.
Another strategy, which seems to be more successful in practice, is to combine both biological networks and pathways in an analysis. For example, Alexeyenko et al. proposed a network-based method to investigate the associations between input gene sets and annotated gene sets by counting the number of network links between members of two gene sets [
7]. Later, Glaab et al. proposed an algorithm called EnrichNet, which extends the method of [
7] to include gene pairs that are not necessarily direct neighbors but are within close proximity in the network [
8]. These approaches take gene correlations and interactions into consideration and agree with the fact that genes function in a coordinated way, which is a meaningful improvement over ORA. However, EnrichNet only provides scores to measure the functional associations and does not provide information about the statistical significance of the scores.
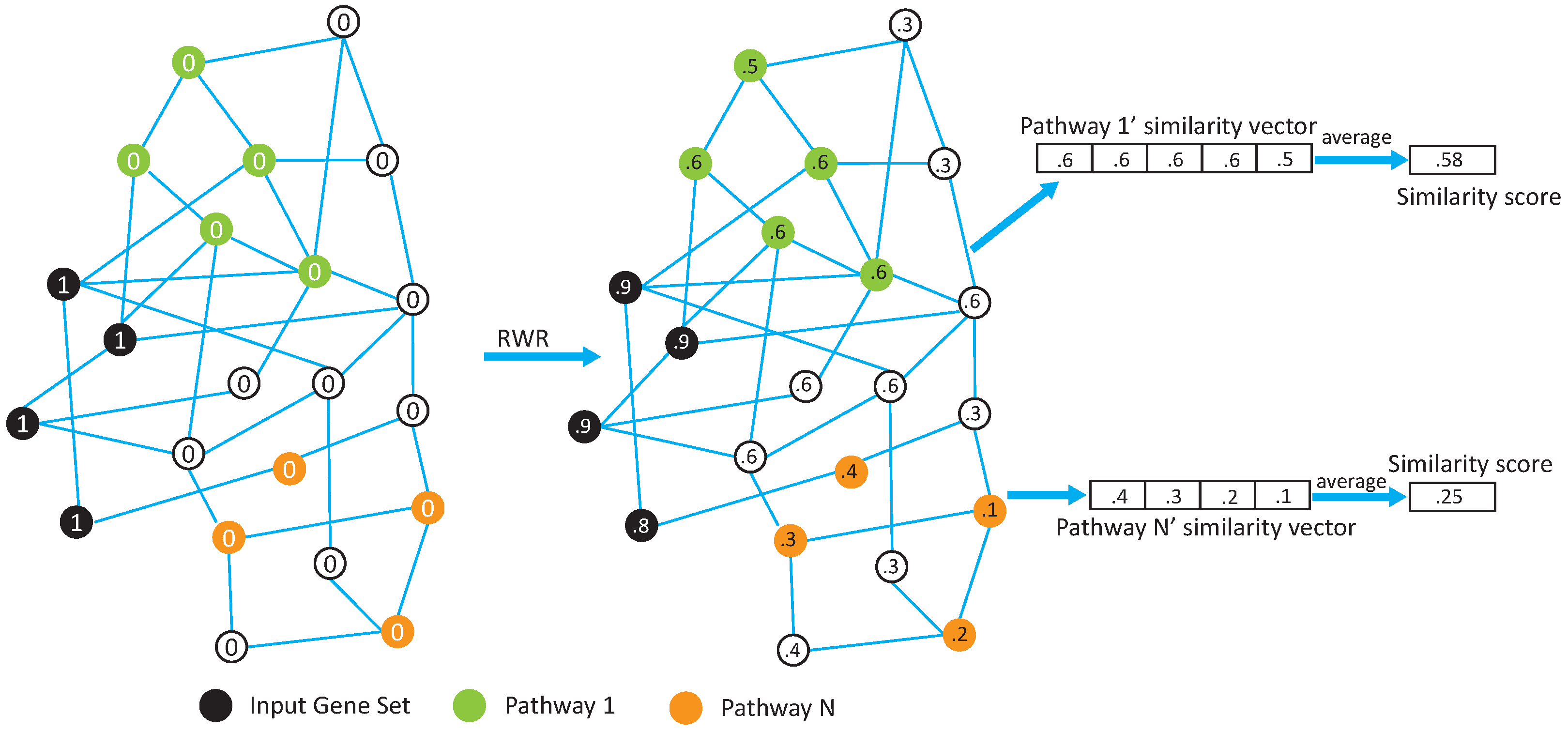
In this paper, we propose two network-based pathway enrichment analysis algorithms, NetPEA and NetPEA’, for conducting a network-based pathway enrichment analysis. Our algorithms consider not only shared genes but also gene–gene interactions. The two algorithms share some common features to identify hidden relationships between an input gene set and pathways, but each uses a different randomization strategy to evaluate statistical significance and, as a result, emphasize different pathway properties.
The remainder of this paper is organized as follows. We present the details of the two algorithms and the data sets in
Section 2. In
Section 3, we present the test results of our methods on multiple data sets and discuss the significance of our finding. We conclude with some remarks for future improvements in
Section 4.
3. Results and Discussion
Validation of associations between genes sets is difficult because of the lack of ground truth and the biases inherent in different evaluation standards. To evaluate the performance of our methods and have a fair comparison with the existing ones, we adopted and designed multiple evaluation methods.
3.1. Validation Using KEGG Pathways as Input Genes
To validate that our algorithms can indeed identify the most relevant pathways, we first used KEGG pathways as input genes to identify the most significantly associated KEGG pathway for each pathway. The rationale is that each pathway should have a closer relationship with itself. Indeed, NetPEA ranks each pathway itself as the most enriched pathway with a very significant z-score. Moreover, some between-pathway associations found by NetPEA are also reasonable. For example, the top three pathways associated with “DNA replication” are “DNA replication”, ”mismatch repair”, and “nucleotide excision repair”, while the top three pathways associated with “chemokine signaling” are “hemokine signaling”, “cytokine–cytokine receptor interaction”, and “gap junction”.
On the other hand, NetPEA’ ranks the pathway itself as the most enriched pathway only for 62% pathways, and ranks the pathway itself in the top 10% for 91% of pathways. The deviation from the ground truth is because NetPEA’ is intended to explore hidden pathways by de-emphasizing pathways that are considered significant simply because of their overlaps with the input gene set. Indeed, for a number of cases where the input gene set itself is not ranked as the top gene set, we found evidence of the association between the reported top gene set and the input gene set, such as “maturity onset diabetes of the young” and “methane metabolism”. Note also that the association between the two pathways is not significant according to NetPEA.
3.2. Validation Using GSEA Outputs as Benchmarks
For four data sets that we have access to the coupled microarrays and ranked gene lists, we applied GSEA to rank the pathways and we use the rankings as benchmarks. GSEA is a benchmark widely used to validate gene set rankings, and [
8] uses it to check pathway rankings. While the results may be biased, it provides partial evidence that our algorithms achieve better performances. Here we calculate Spearman correlation coefficients between each mentioned method and GSEA.
Table 1 shows that for each input gene set, the largest correlation coefficient is from NetPEA or NetPEA’, which means that our algorithms gain more support on pathway rankings and are better than ORA and EnrichNet.
3.3. Evaluation Based on Number of Enriched Pathways
We apply our algorithms, NetPEA and NetPEA’, to each of the data sets mentioned in
Section 2.4. Meanwhile, we run ORA on these input gene sets and compare the significant pathways found by the three methods.
3.3.1. NetPEA vs. ORA
Table 2 shows significant pathways only reported by NetPEA but not present in the results for ORA. For most of cases (11/14), ORA does not identify any pathway that is not found by NetPEA. For common significant pathways discovered by both methods, we define
,
and
by the ratios of their
p-values.
means the
p-value ratio (NetPEA/ORA) is less than 0.001;
represents the ratio greater than 1000; otherwise it is
. Strikingly, no pathways fall into the range
. Overall, NetPEA can successfully identify nearly all significant pathways reported by ORA. Moreover, NetPEA reports many significant pathways not found by ORA. The superiority of NetPEA over ORA can be explained by the fact that NetPEA not only considers the pathway enrichment caused by common genes but also takes gene interactions into account. Through the gene interactions, some pathways not enriched in ORA are elevated to be significant. In
Section 3.6, we will show that these additional pathways are biologically meaningful.
3.3.2. NetPEA’ vs. ORA
As NetPEA’ is devised to complement NetPEA, it provides new significant pathway information that is not present in the results of ORA or NetPEA. Compared to information in
Table 2, the number of common significant pathways decreases for each input gene set (
Table 3). At the same time, NetPEA’ produces some significant pathways not present in ORA. This difference is because of the network randomization in NetPEA’, which eliminates some significant pathways in ORA or NetPEA with loose gene interactions and lifts insignificant ones in ORA with close gene interactions. As shown clearly in
Section 3.4, these pathways are often preserved between different experiments for the same disease, signifying the importance of the pathways for the disease.
3.4. Evaluation Using Cross-Data Stability Analysis
It is well known that the agreement is often poor between different high throughput experiments concerning the same disease performed by different groups. Among other reasons, this is because many of the genes identified by these experiments are caused by downstream effects, which can vary significantly among experiments. It is reasonable to assume that if indeed we can find true causal genes/pathways, agreement between experiments will be improved. Therefore, for the genes identified from the two breast cancer data sets and the two lung cancer data sets, we compare the significant pathways from different data sets reported by NetPEA, NetPEA’ and ORA. We calculate the ratios of common significant pathways to the total number of unique significant pathways identified from the two experiments as well as the
p-values of the overlap under the hypergeometric distribution using 208 total KEGG pathways as background.
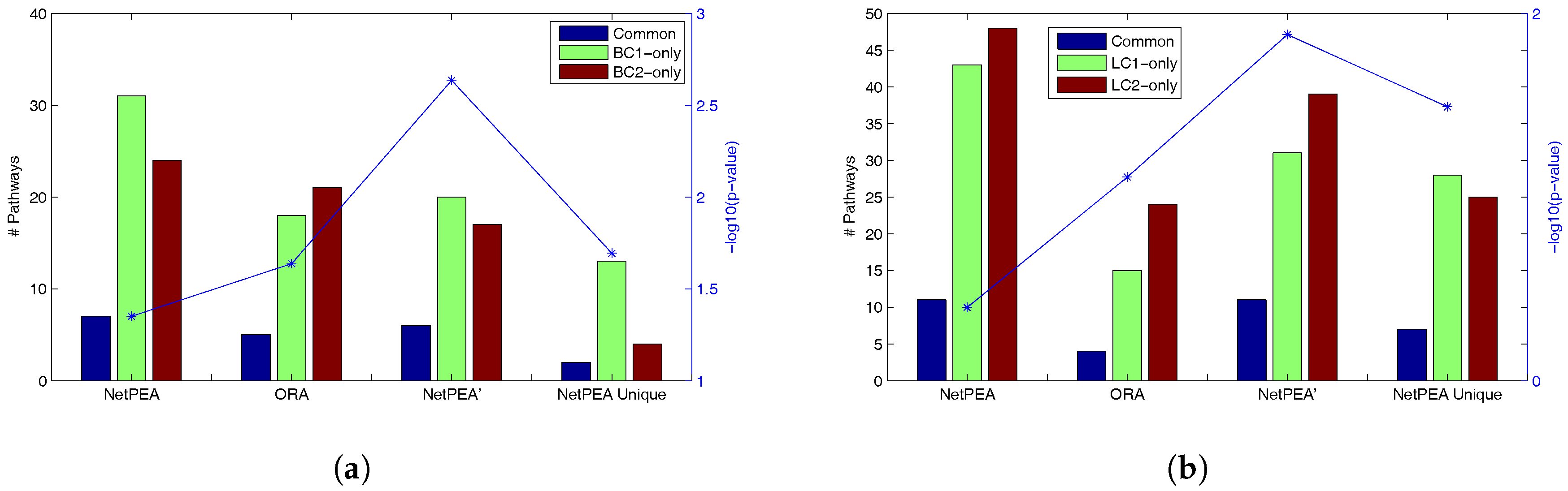
Table 4 shows that NetPEA can find more common significant pathways than ORA and NetPEA’ as a result of the increased number of significant pathways reported by NetPEA, which partially suggest that the additional pathways reported by NetPEA are reasonable. Remarkably, while NetPEA’ reports much fewer pathways than NetPEA, the number of common pathways remains almost unchanged. As shown in
Figure 2, the statistical significance of overlap between the pathways detected from different data sets is the highest in NetPEA’, compared to NetPEA and ORA. Therefore, we believe that the pathways reported by NetPEA’ may have a greater chance of containing the pathways that are directly associated with the phenotype instead of downstream effects. In addition, while the overlap of pathways detected by NetPEA is not as significant as in ORA, this is mainly due to the increased number of detected pathways and the limited number of candidate pathways as background. Since almost all pathways reported by ORA are also reported by NetPEA, we removed the ORA-detected pathways from NetPEA results and reanalyzed the overlap. As shown in
Figure 2 (“NetPEA unique”), the pathways found by NetPEA but not by ORA do have an increased level of overlap compared to ORA, suggesting that the additional pathways identified by NetPEA are biologically relevant. Collectively, the results suggest that our algorithms have an advantage in interpreting the results of high throughput experiments performed by different groups and can potentially discover the key pathways underlying the diseases.
3.5. Pathways Cross Verification Analysis
Checking whether pathways ranked at the top by one method are also ranked at the top by other methods can provide additional confidence to biologists and help biologists to narrow down new hypotheses to test. Here we use two cross verification methods, positive cross verification and negative cross verification, to compare our algorithms with ORA, EnrichNet and GSEA. For positive cross verification, we examine how many pathways out of the top 20 by one method appear in the top 20 pathways determined by all the other methods. For negative cross verification, we checked how many pathways out of the top 20 by one method are ranked below the top 100 by the other three methods. We verify NetPEA and NetPEA’ separately because if we verify them together they may vote for each other, which would provide biased, favorable results for our algorithms.
3.5.1. NetPEA
Table 5 shows that ORA receives the most recognition and reports only one pathway that is not agreed by others, which is understandable because ORA is the most conservative method and most of its results are also reported by NetPEA and EnrichNet. For the two network-based approaches, they receive similar results on positive cross verification, while NetPEA is better than the counterpart with less negative results. Moreover, the only pathway of negative verification result of NetPEA is “taste transduction” for diabetes, which has been reported previously [
20]. The pathways of EnrichNet’s negative verification result include “thyroid cancer”, “basal cell carcinoma”, “melanogenesis”, “endometrial cancer” and “hedgehog signaling”. Our limited literature search does not reveal enough evidence of their associations with diabetes. GSEA is the one receiving the least recognition as it exploits whole microarrays and, from a methodology point of view, it is far away from the other three methods. Its negative cross verification results include “olfactory transduction”, “mismatch repair” and “snare interactions in vesicular transport” for diabetes. These associations claimed by GSEA are hard to understand. Therefore, NetPEA has an advantage over other methods to rank meaningful pathways at the top.
3.5.2. NetPEA’
Compared with
Table 5,
Table 6 shows that NetPEA’ receives less support than NetPEA. This is reasonable since NetPEA’ eliminates some pathways that are an important part of ORA. On the other hand, we conclude that NetPEA’ shares some similarities with GSEA because the results of positive verification of GSEA increase while its negative verification results decrease.
3.6. Novel Pathways
As shown in
Section 3.3, our algorithms usually report more significant pathways than ORA. A careful inspection of these additional pathways suggests that many of them are biologically relevant and important. Here we only discuss a few of these pathways.
For the diabetes down-regulated input gene set, NetPEA ranks the pathway “glycerolipid metabolism”, as 5th with a significant
z-score 3.5 (
p-value =
), and NetPEA’ ranks it as 1st with a significant
z-score 7.9 (
p-value =
). The same pathway has a
p-value 0.12 in ORA and is ranked 37th by EnrichNet. Extensive literature review shows that “glycerolipid metabolism” plays an important role in the pathogenesis of obesity and type 2 diabetes [
21,
22].
Another good example is the Leukemia up-regulated input gene set, where NetPEA’ ranks the pathway “chronic myeloid leukemia” 4th with a significant
z-score 3.12 (
p-value =
). ORA ranks it 63rd with an insignificant p-value, and EnrichNet ranks it 77th. This pathway is missed by NetPEA (
z-score = 0.23). For the Leukemia down-regulated gene set, both NetPEA and NetPEA’ rank “folate biosynthesis” as the most significant pathway (
z-score = 5.0 and 6.5 respectively), while the same pathway is ranked 22nd in EnrichNet and has a
p-value 0.05 in ORA. A search through the literature confirms that the relationship between the pathway, “folate biosynthesis”, and leukemia can be verified [
23].
Other verifiable significant associations that are identified by our methods but missed by both EnrichNet (rank > 30) and ORA (p-value > 0.05) include “pathways in cancer” for the p53 up-regulated gene set, “steroid hormone biosynthesis” and “sphingolipid metabolism” in Parkinson’s disease, “natural killer cell mediated cytotoxicity” in diabetes, as well as “MAPK signaling”, “ERBB signaling”, “PPAR signaling”, “focal adhesion” and “ECM receptor interaction” for various cancer gene sets, to mention a few.




{kind=link}
{kind=link}