
Genome Analysis of a Novel Broad Host Range Proteobacteria Phage Isolated from a Bioreactor Treating Industrial Wastewater


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sensitivity Test for Several Bacterial Strains
2.2. Transmission Electron Microscopy
2.3. DNA Purification and Sequencing
2.4. Genome Assembly and Analysis
3. Results and Discussion
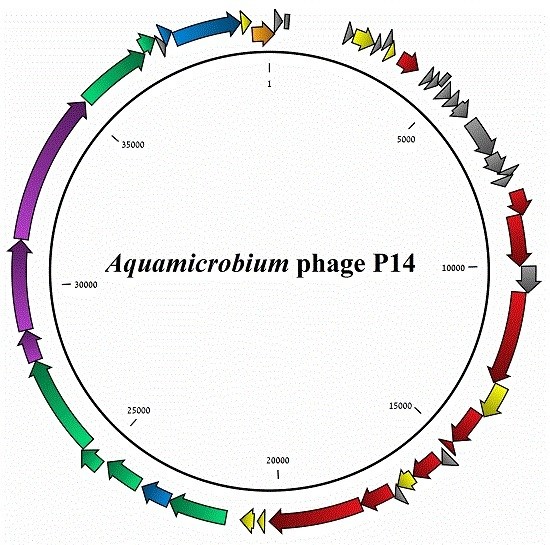
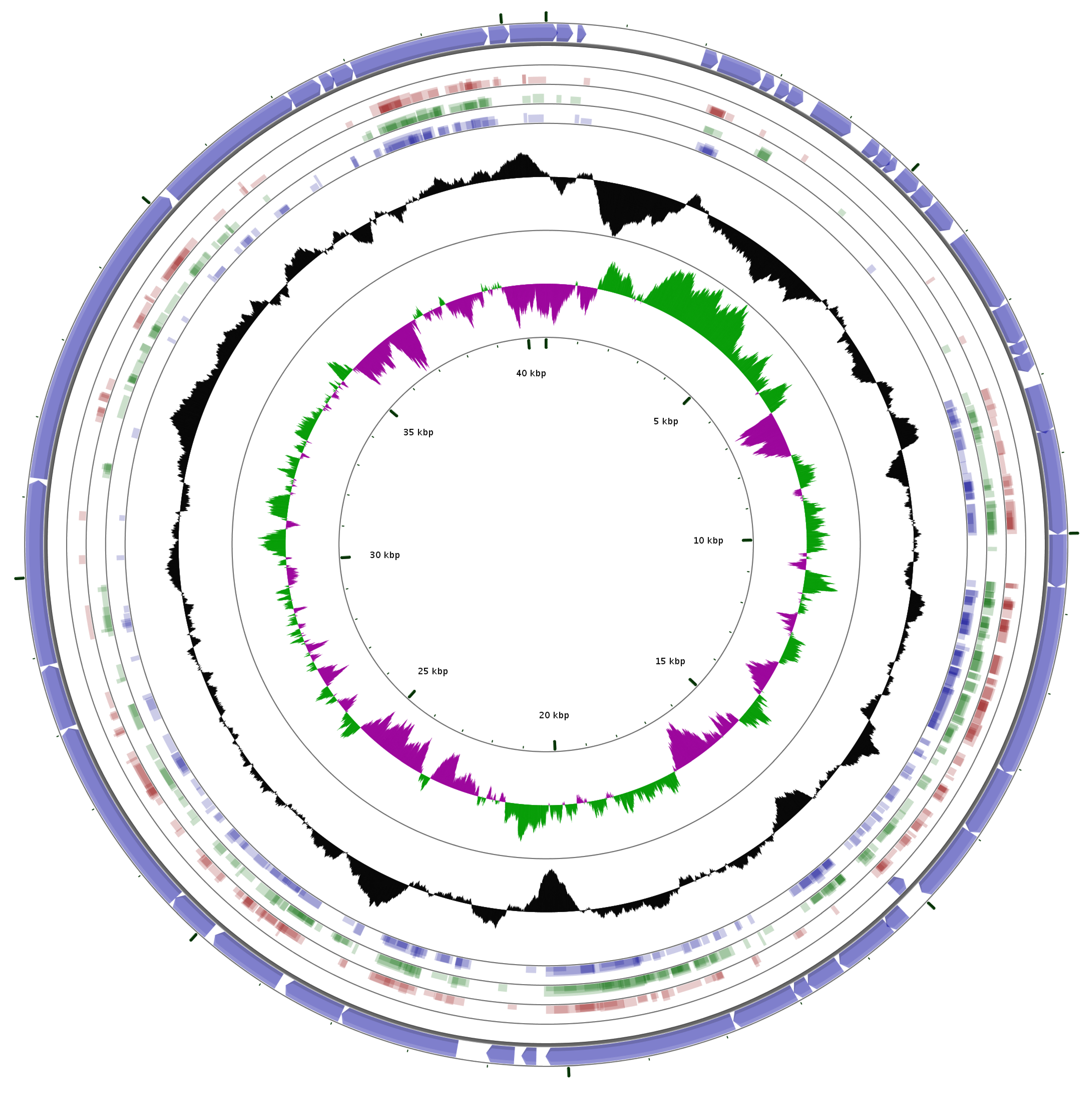
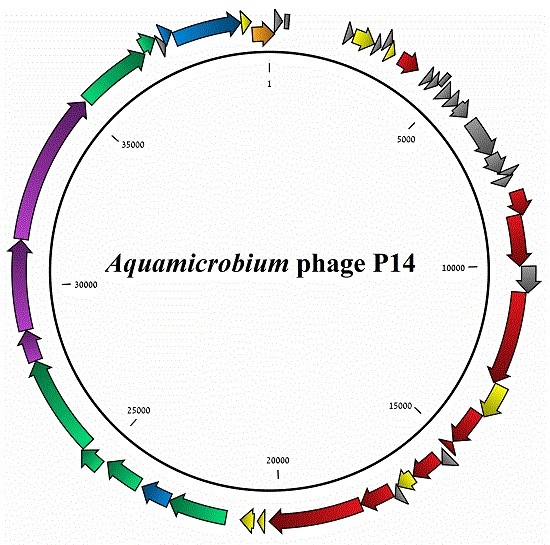
3.1. General Features of the Phage Genome
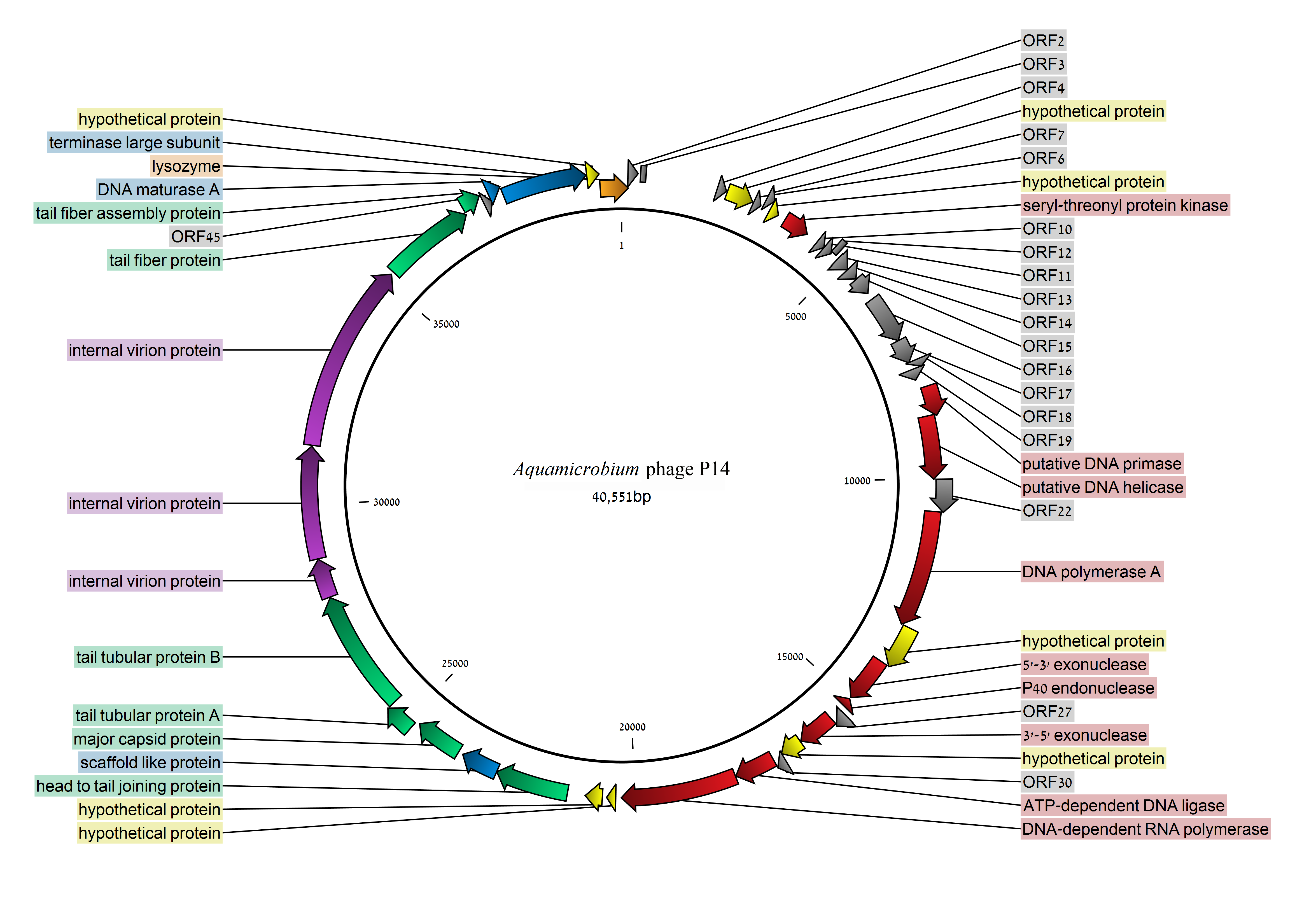
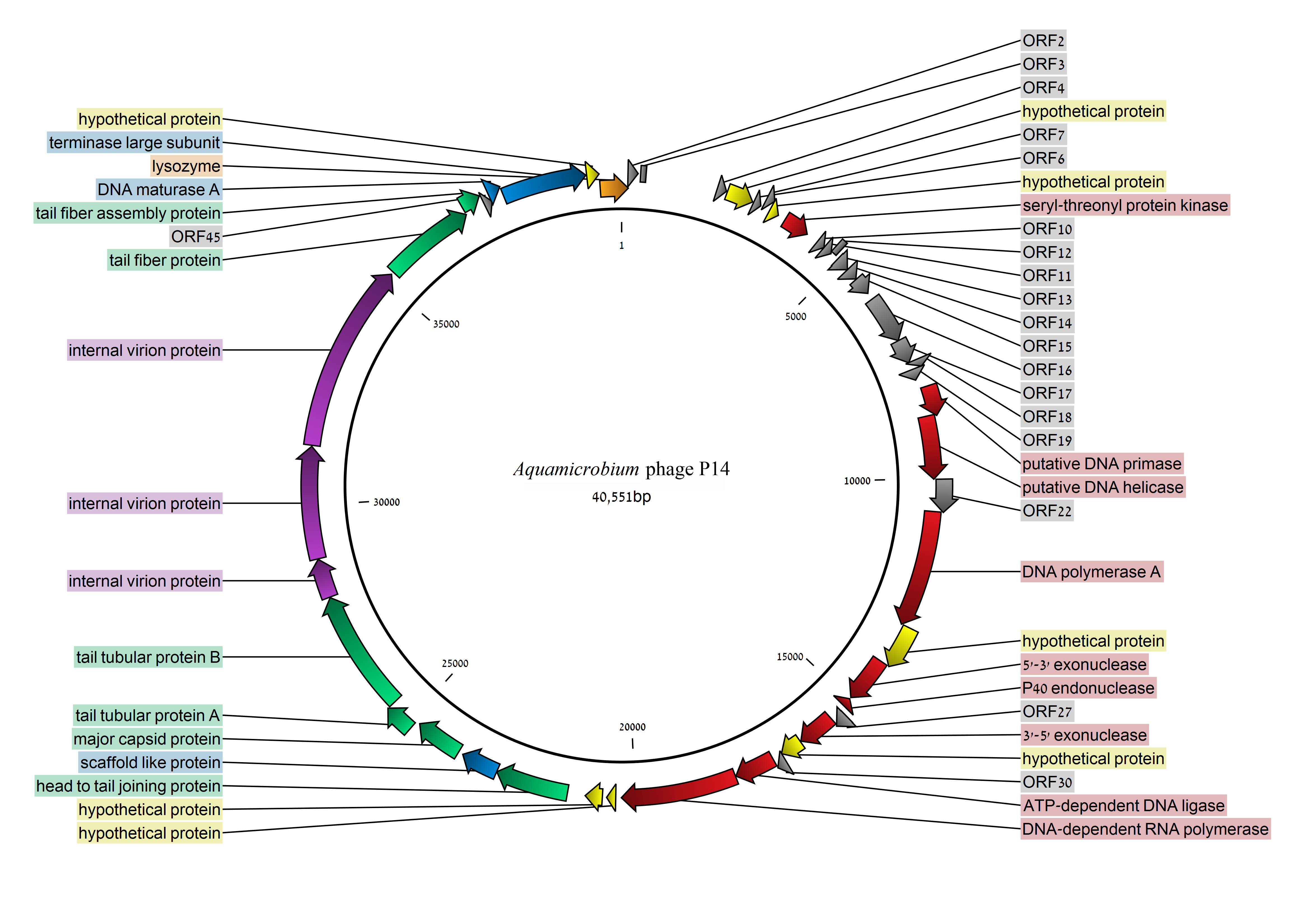
3.2. Coding Sequences Organization
3.3. Early Class
3.4. DNA Replication and Repair (Middle Genomic Region)
3.5. Packaging Related Genes (Late Genomic Region)
3.6. Internal Virion Genes
3.7. Phage Capsid and Tail Genes
3.8. Lysozyme
3.9. Seryl/threonyl Protein Kinase
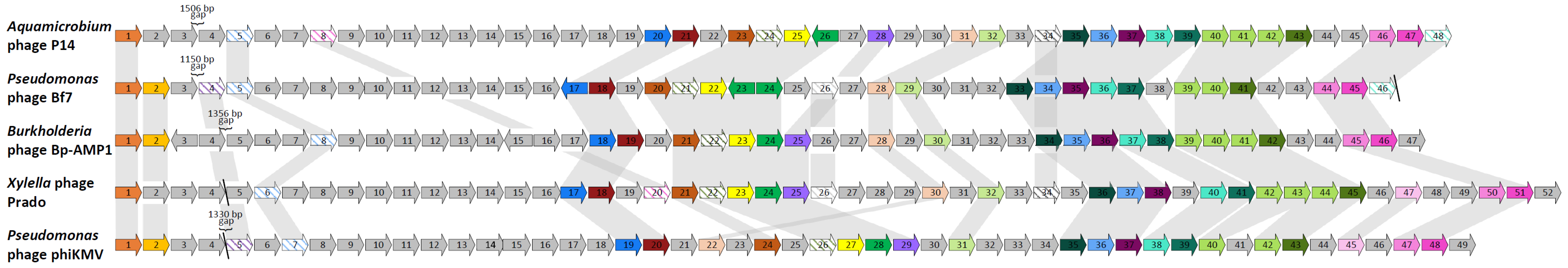
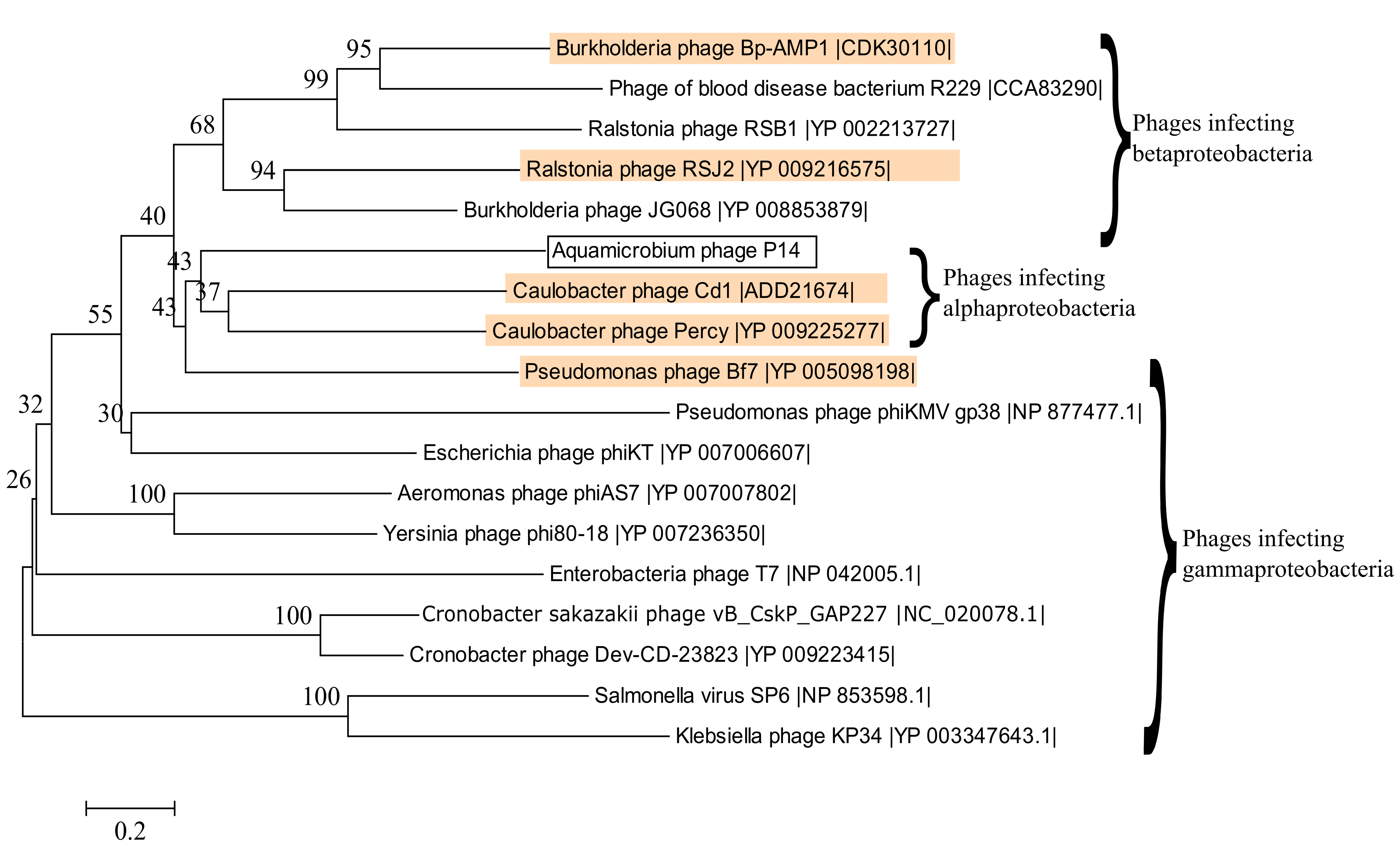
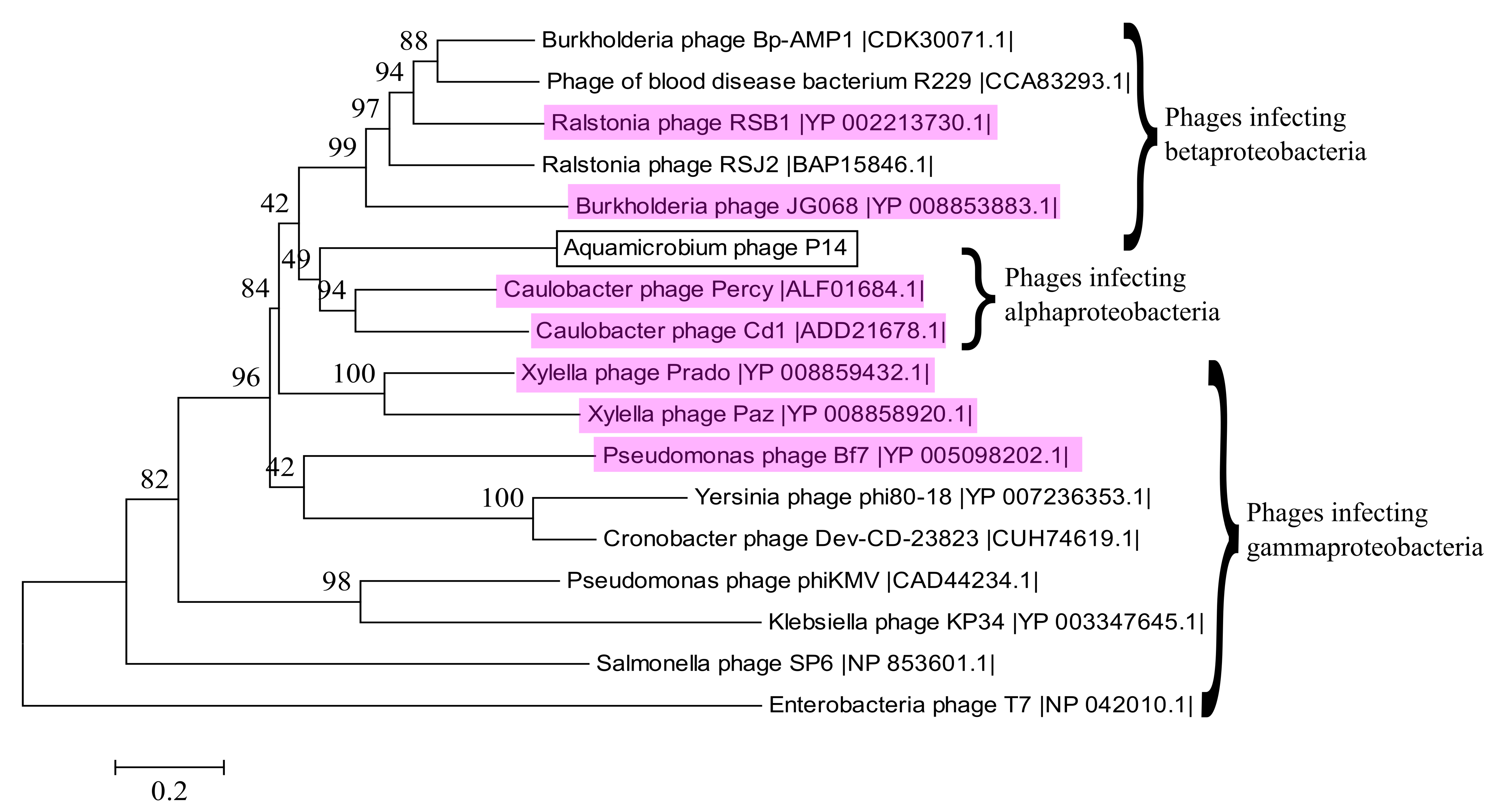
3.10. Alignment to Other Phages
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rohwer, F. Global Phage Diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef]
- Johnke, J.; Cohen, Y.; de Leeuw, M.; Kushmaro, A.; Jurkevitch, E.; Chatzinotas, A. Multiple Micro-Predators Controlling Bacterial Communities in the Environment. Curr. Opin. Biotechnol. 2014, 27, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Hantula, J.; Kurki, A.; Vuoriranta, P.; Bamford, D.H. Ecology of Bacteriophages Infecting Activated Sludge Bacteria. Appl. Environ. Microbiol. 1991, 57, 2147–2151. [Google Scholar] [PubMed]
- Ewert, D.L.; Paynter, M.J.B. Enumeration of Bacteriophages and Host Bacteria in Sewage and the Activated-Sludge Treatment Process. Appl. Environ. Microbiol. 1980, 39, 576–583. [Google Scholar] [PubMed]
- Khan, M.A.; Satoh, H.; Katayama, H.; Kurisu, F.; Mino, T. Bacteriophages Isolated from Activated Sludge Processes and their Polyvalency. Water Res. 2002, 36, 3364–3370. [Google Scholar] [CrossRef]
- Otawa, K.; Lee, S.H.; Yamazoe, A.; Onuki, M.; Satoh, H.; Mino, T. Abundance, Diversity, and Dynamics of Viruses on Microorganisms in Activated Sludge Processes. Microb. Ecol. 2007, 53, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, W.T. Determination of Virus Abundance, Diversity and Distribution in a Municipal Wastewater Treatment Plant. Water Res. 2009, 43, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, O.H.; Kushmaro, A.; Brenner, A. Bacteriophage Predation Regulates Microbial Abundance and Diversity in a Full-Scale Bioreactor Treating Industrial Wastewater. ISME J. 2009, 4, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, O.H.; Kushmaro, A. Bacteriophage Ecology in Environmental Biotechnology Processes. Curr. Opin. Biotechnol. 2011, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Van Houten, R.T.; Borger, A.R.; Eikelboom, D.H.; Fan, Y. Minimization of Excess Sludge Production for Biological Wastewater Treatment. Water Res. 2003, 37, 4453–4467. [Google Scholar] [CrossRef]
- Withey, S.; Cartmell, E.; Avery, L.M.; Stephenson, T. Bacteriophages—Potential for Application in Wastewater Treatment Processes. Sci. Total Environ. 2005, 339, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Soddell, J.; Kurtbke, D. Fighting Foam with Phages? Water Sci. Technol. 2002, 46, 511–518. [Google Scholar] [PubMed]
- Petrovski, S.; Seviour, R.J.; Tillett, D. Characterization of the Genome of the Polyvalent Lytic Bacteriophage GTE2, which has Potential for Biocontrol of Gordonia-, Rhodococcus-, and Nocardia-Stabilized Foams in Activated Sludge Plants. Appl. Environ. Microbiol. 2011, 77, 3923–3929. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.C.; Huang, J.Y.; Ramaswami, D. Decomposition of Exopolysaccharide Slime by a Bacteriophage Enzyme. Water Res. 1988, 22, 1185–1188. [Google Scholar] [CrossRef]
- Bura, R.; Cheung, M.; Liao, B.; Finlayson, J.; Lee, B.; Droppo, I.; Leppard, G.; Liss, S. Composition of Extracellular Polymeric Substances in the Activated Sludge Floc Matrix. Water Sci. Technol. 1998, 37, 325–333. [Google Scholar] [CrossRef]
- Lu, T.K.; Collins, J.J. Dispersing Biofilms with Engineered Enzymatic Bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [PubMed]
- Goldman, G.; Starosvetsky, J.; Armon, R. Inhibition of Biofilm Formation on UF Membrane by use of Specific Bacteriophages. J. Membr. Sci. 2009, 342, 145–152. [Google Scholar] [CrossRef]
- Hagens, S.; Loessner, M.J. Application of Bacteriophages for Detection and Control of Foodborne Pathogens. Appl. Microbiol. Biotechnol. 2007, 76, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Carlton, R.; Noordman, W.; Biswas, B.; De Meester, E.; Loessner, M. Bacteriophage P100 for Control of Listeria monocytogenes in Foods: Genome Sequence, Bioinformatic Analyses, Oral Toxicity Study, and Application. Regul. Toxicol. Pharmacol. 2005, 43, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Guenther, S.; Huwyler, D.; Richard, S.; Loessner, M.J. Virulent Bacteriophage for Efficient Biocontrol of Listeria monocytogenes in Ready-to-Eat Foods. Appl. Environ. Microbiol. 2009, 75, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kocharunchitt, C.; Ross, T.; McNeil, D. Use of Bacteriophages as Biocontrol Agents to Control Salmonella Associated with Seed Sprouts. Int. J. Food Microbiol. 2009, 128, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Baranowski, E.; Ruiz-Jarabo, C.M.; Domingo, E. Evolution of Cell Recognition by Viruses. Science 2001, 292, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G. Ecology of Prokaryotic Viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed]
- Sime-Ngando, T. Environmental Bacteriophages: Viruses of Microbes in Aquatic Ecosystems. Front. Microbial. 2014, 5, 355. [Google Scholar] [CrossRef] [PubMed]
- Souza, K.A.; Ginoza, H.S.; Haight, R.D. Isolation of a Polyvalent Bacteriophage for Escherichia coli, Klebsiella pneumoniae, and Aerobacter aerogenes. J. Virol. 1972, 9, 851–856. [Google Scholar] [PubMed]
- Karnovsky, M.J. A Formaldehyde Glutaraldehyde Fixative of High Osmolality for use in Electron Microscopy. J. Cell Biol. 1965, 27, 137–139. [Google Scholar]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2015, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, Snoscan and snoGPS Web Servers for the Detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G. Application of a Time-Delay Neural Network to Promoter Annotation in the Drosophila melanogaster Genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega, Accurate Alignment of very Large Numbers of Sequences. Mult. Seq. Alignment Methods 2014, 1079, 105–116. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A Comparative Genomics Tool for Circular Genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A Better Web Interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Zafar, N.; Mazumder, R.; Seto, D. CoreGenes: A Computational Tool for Identifying and Cataloging “Core” Genes in a Set of Small Genomes. BMC Bioinform. 2002, 3, 12. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for In-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Bacteriophage Taxonomy. Microbiol. Aust. 2011, 32, 90–94. [Google Scholar]
- Lavigne, R.; Burkal’tseva, M.V.; Robben, J.; Sykilinda, N.N.; Kurochkina, L.P.; Grymonprez, B.; Jonckx, B.; Krylov, V.N.; Mesyanzhinov, V.V.; Volckaert, G. The Genome of Bacteriophage φKMV, a T7-Like Virus Infecting Pseudomonas aeruginosa. Virology 2003, 312, 49–59. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, J.; Niu, Y.D.; Cui, N.; Ma, Y.; Cao, F.; Jin, L.; Li, Z.; Xu, Y. Isolation and Characterization of a “phiKMV-Like” Bacteriophage and its Therapeutic Effect on Mink Hemorrhagic Pneumonia. PLoS ONE 2015, 10, e0116571. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Ceyssens, P.J.; Dunon, V.; Ackermann, H.W.; Van Vaerenbergh, J.; Maes, M.; De Proft, M.; Lavigne, R. Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, Belonging to the “phiKMV-Like Viruses”. Appl. Environ. Microbiol. 2011, 77, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Lerma, R.A.; Tidwell, T.J.; Cahill, J.L.; Rasche, E.S.; Kuty Everett, G.F. Complete Genome Sequence of Caulobacter crescentus Podophage Percy. Genome Announc. 2015, 3, e01373-15. [Google Scholar] [CrossRef] [PubMed]
- Bhunchoth, A.; Phironrit, N.; Leksomboon, C.; Chatchawankanphanich, O.; Kotera, S.; Narulita, E.; Kawasaki, T.; Fujie, M.; Yamada, T. Isolation of Ralstonia solanacearum-infecting Bacteriophages from Tomato Fields in Chiang Mai, Thailand, and their Experimental use as Biocontrol Agents. J. Appl. Microbiol. 2015, 118, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.H.; Abdu, A.H.; Schobert, M.; Dennis, J.J. Genomic Characterization of JG068, a Novel Virulent Podovirus Active Against Burkholderia cenocepacia. BMC Genom. 2013, 14, 574. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.N.; Lin, J.W.; Weng, S.F.; Tseng, Y.H. Genomic Characterization of the Intron-Containing T7-Like Phage phiL7 of Xanthomonas campestris. Appl. Environ. Microbiol. 2009, 75, 7828–7837. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.E.; Bruce, S.A.; Murray, K. Molecular Cloning of the DNA Ligase Gene from Bacteriophage T4: II. Amplification and Preparation of the Gene Product. J. Mol. Biol. 1979, 132, 493–505. [Google Scholar] [CrossRef]
- Ahern, S.J.; Das, M.; Bhowmick, T.S.; Young, R.; Gonzalez, C.F. Characterization of Novel Virulent Broad-Host-Range Phages of Xylella fastidiosa and Xanthomonas. J. Bacteriol. 2014, 196, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Prevelige, P.E.; Fane, B.A. Building the machines: Scaffolding protein functions during bacteriophage morphogenesis. In Viral Molecular Machines; Rossmann, M.G., Rao, V.B., Eds.; Springer: New York, NY, USA, 2012; pp. 325–350. [Google Scholar]
- Canchaya, C.; Fournous, G.; Chibani-Chennoufi, S.; Dillmann, M.; Brüssow, H. Phage as Agents of Lateral Gene Transfer. Curr. Opin. Microbiol. 2003, 6, 417–424. [Google Scholar] [CrossRef]
- Garcia-Doval, C.; Van Raaij, M.J. Structure of the Receptor-Binding Carboxy-Terminal Domain of Bacteriophage T7 Tail Fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Sajben-Nagy, E.; Maróti, G.; Kredics, L.; Horváth, B.; Párducz, Á.; Vágvölgyi, C.; Manczinger, L. Isolation of New Pseudomonas tolaasii Bacteriophages and Genomic Investigation of the Lytic Phage BF7. FEMS Microbiol. Lett. 2012, 332, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Hayes, S.J.; Zaman, S.; Lieman, K.; Rolando, M.; Hardies, S.C. Improved Isolation of Undersampled Bacteriophages: Finding of Distant Terminase Genes. Virology 2004, 329, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Klumpp, J.; Bishop-Lilly, K.A.; Rajavel, M.; Willner, K.M.; Butani, A.; Henry, M.; Biswas, B.; Li, M.; Albert, M.J. Whole Genome Sequencing and Comparative Genomic Analyses of Two Vibrio cholerae O139 Bengal-Specific Podoviruses to Other N4-Like Phages Reveal Extensive Genetic Diversity. Virol. J. 2013, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Remenant, B.; De Cambiaire, J.; Cellier, G.; Jacobs, J.M.; Mangenot, S.; Barbe, V.; Lajus, A.; Vallenet, D.; Medigue, C.; Fegan, M. Ralstonia syzygii, the Blood Disease Bacterium and some Asian R. solanacearum Strains Form a Single Genomic Species Despite Divergent Lifestyles. PLoS ONE 2011, 6, e24356. [Google Scholar] [CrossRef] [PubMed]
- Gatedee, J.; Kritsiriwuthinan, K.; Galyov, E.E.; Shan, J.; Dubinina, E.; Intarak, N.; Clokie, M.R.; Korbsrisate, S. Isolation and Characterization of a Novel Podovirus which Infects Burkholderia pseudomallei. Virol. J. 2011, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Korbsrisate, S.; Withatanung, P.; Lazar Adler, N.; Clokie, M.R.; Galyov, E.E. Temperature Dependent Bacteriophages of a Tropical Bacterial Pathogen. Front. Mcrobiol. 2014, 5, 599. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
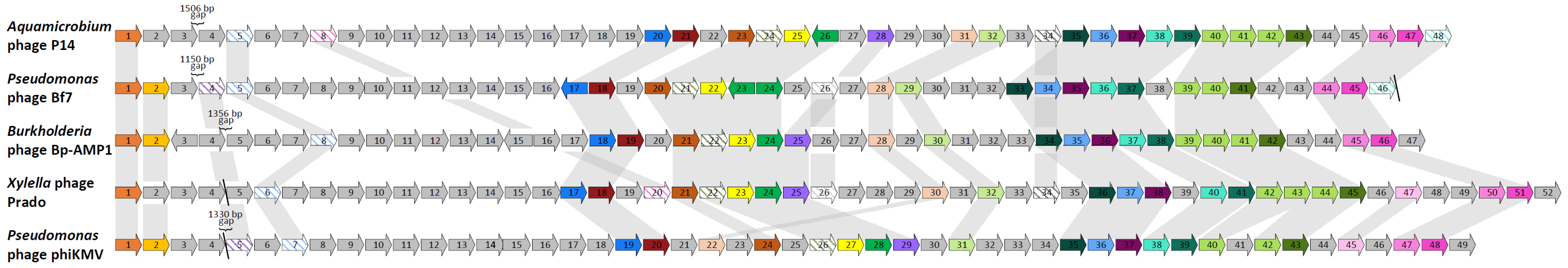{kind=link}
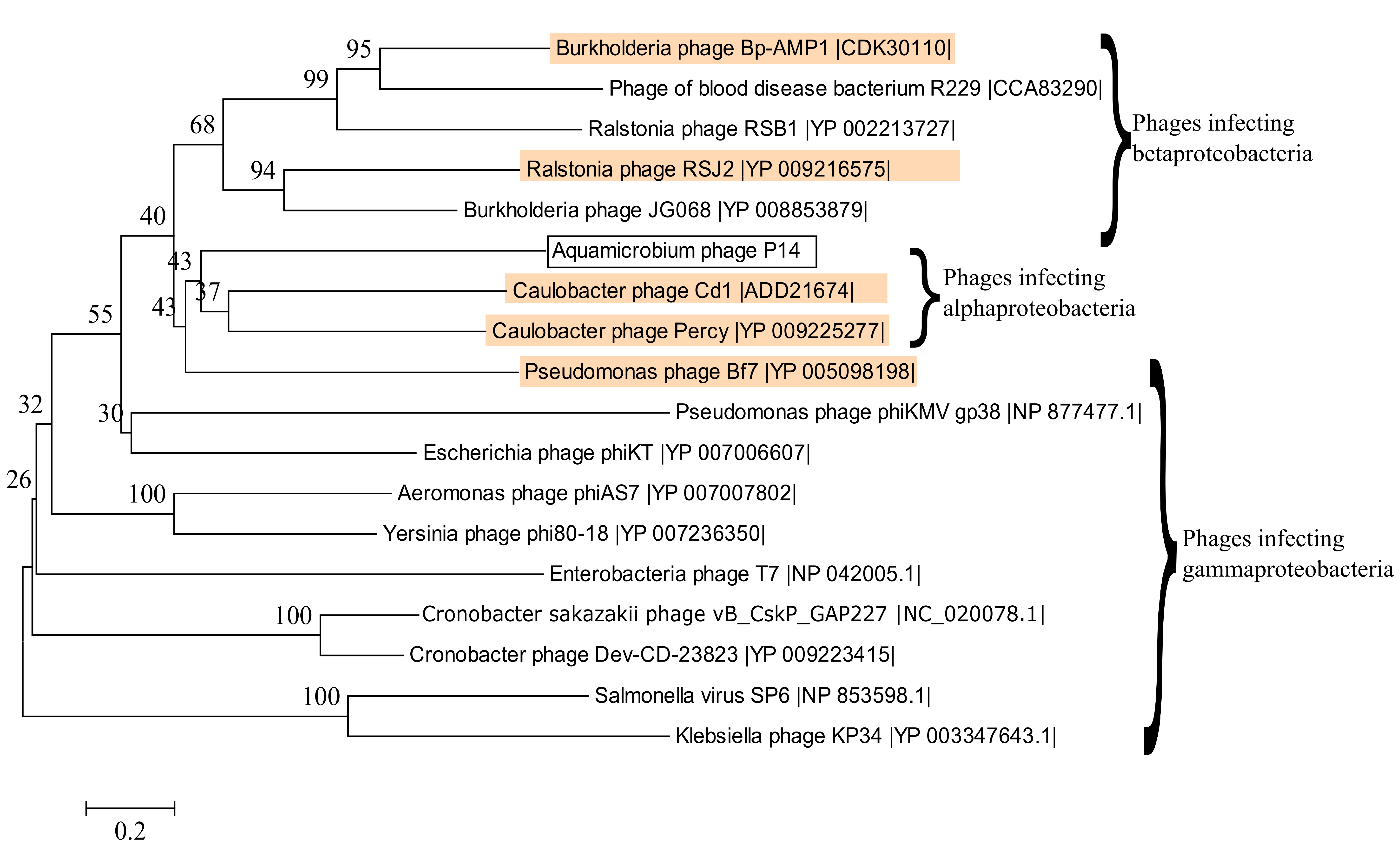{kind=link}
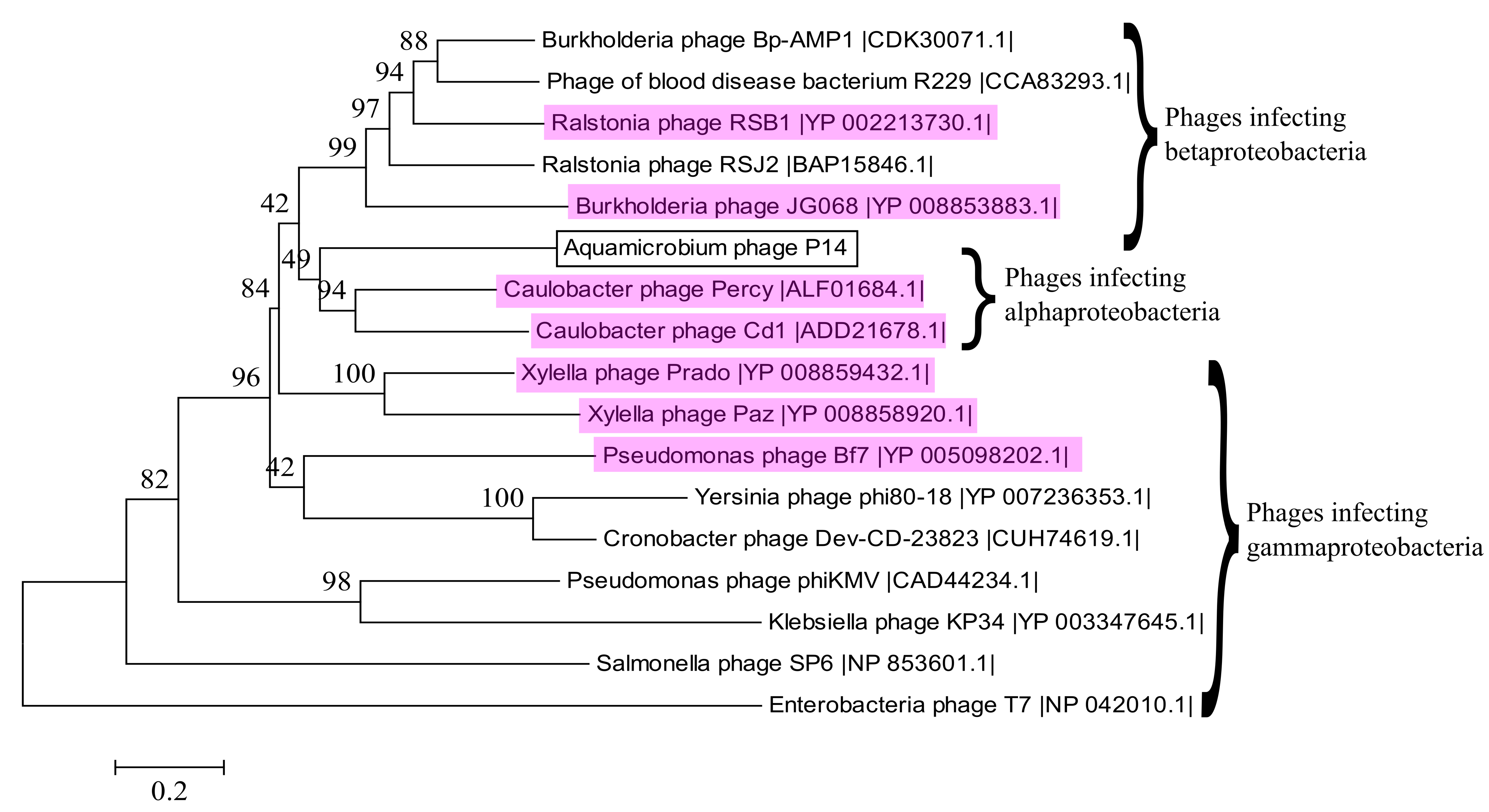{kind=link}
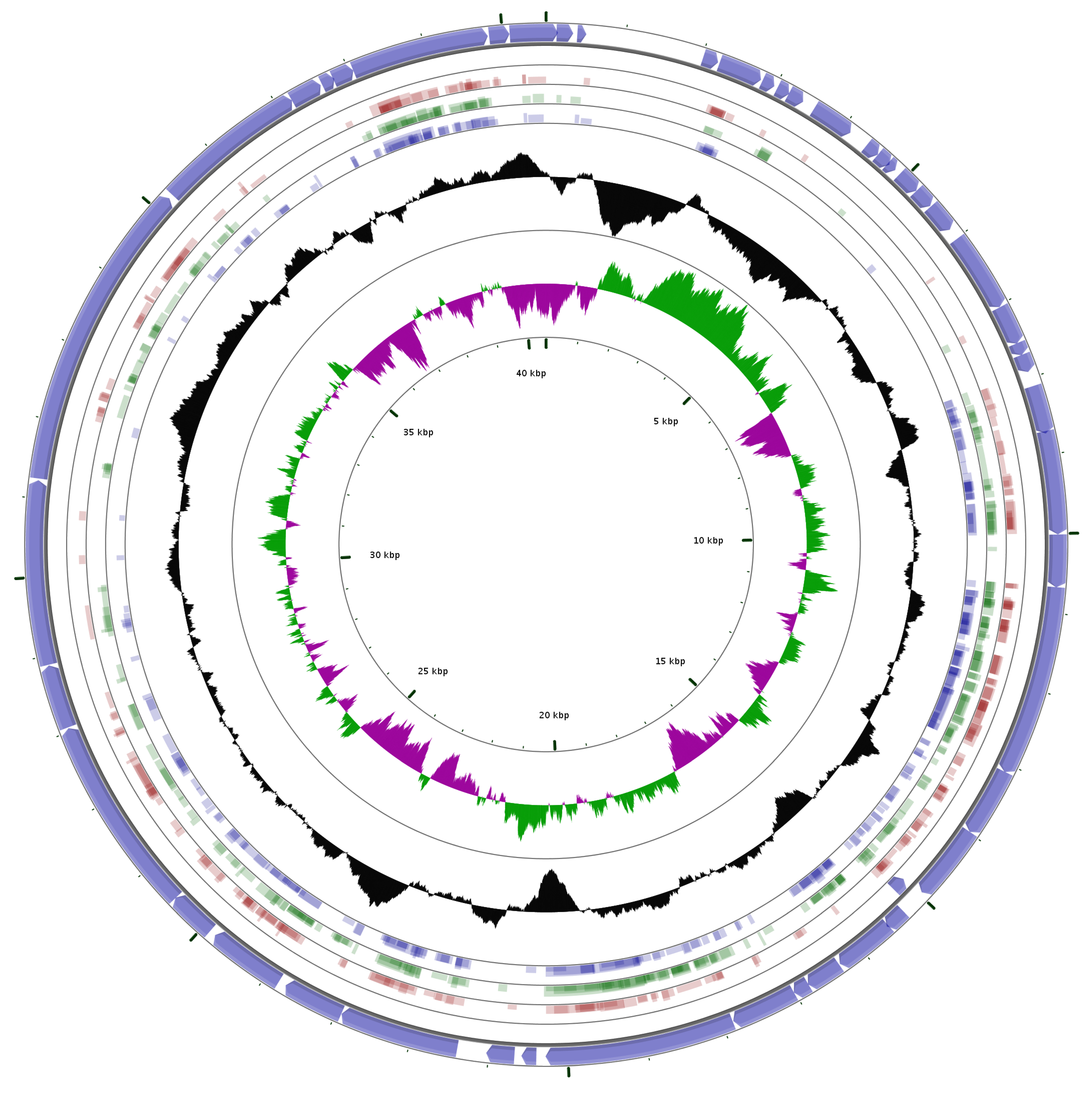{kind=link}
| ORF | Start | End | Strand | Protein Length | Blastx Match | E-Value | Source | Accession No. |
|---|---|---|---|---|---|---|---|---|
| ORF1 | 40092 | 149 | + | 202 | lysozyme | 1.00 × 10−24 | Serratia marcescens | WP_043138231.1 |
| putative lysozyme | 4.00 × 10−22 | Pseudomonas phage Bf7 | YP_005098158.1 | |||||
| ORF2 | 133 | 342 | + | 69 | none | |||
| ORF3 | 395 | 508 | + | 37 | none | |||
| ORF4 | 2014 | 2211 | + | 65 | none | |||
| ORF5 | 2232 | 2795 | + | 187 | hypothetical protein | 2.00 × 10−39 | Pseudomonas phage | YP009151803.1 |
| ORF6 | 2818 | 2982 | + | 54 | none | |||
| ORF7 | 3027 | 3191 | + | 54 | none | |||
| ORF8 | 3188 | 3394 | + | 68 | hypothetical protein | 1.00 × 10−18 | Xylella phage Prado | YP008859401.1 |
| ORF9 | 3559 | 4107 | + | 182 | seryl/threonyl protein kinase | 7.00 × 10−5 | Erwinia phage FE44 | YP008766718.1 |
| ORF10 | 4367 | 4549 | + | 60 | none | |||
| ORF11 | 4551 | 4742 | + | 63 | none | |||
| ORF12 | 4718 | 4843 | + | 41 | none | |||
| ORF13 | 4919 | 5215 | + | 98 | none | |||
| ORF14 | 5218 | 5475 | + | 85 | none | |||
| ORF15 | 5486 | 5863 | + | 125 | none | |||
| ORF16 | 6011 | 7024 | + | 337 | none | |||
| ORF17 | 7028 | 7534 | + | 168 | none | |||
| ORF18 | 7531 | 7692 | + | 53 | none | |||
| ORF19 | 7689 | 7919 | + | 76 | none | |||
| ORF20 | 8101 | 8727 | + | 208 | putative DNA primase | 2.00 × 10−53 | Burkholderia phage Bp-AMP4 | CDL65258.1 |
| ORF21 | 8697 | 10010 | + | 437 | putative DNA helicase | 4.00 × 10−146 | Caulobacter phage Cd1 | ADD21652.1 |
| ORF22 | 10012 | 10680 | + | 222 | none | |||
| ORF23 | 10677 | 13109 | + | 810 | DNA polymerase A family protein | 0.0 | Burkholderia pseudomallei BDU 2 | KGV49475.1 pfam00476 |
| putative DNA polymerase | 0.0 | Ralstonia phage RSJ2 | BAP15824.1 | |||||
| ORF24 | 13106 | 13984 | + | 292 | hypothetical protein | 8.00 × 10−60 | Burkholderia sp. 2002721687 | AJY44207.1 |
| hypothetical protein | 1.00 × 10−46 | Pseudomonas phage Bf7 | YP005098178.1 | |||||
| ORF25 | 13984 | 14958 | + | 324 | putative exonuclease | 3.00 × 10−76 | Burkholderia phage Bp-AMP1 | CDK30092.1 |
| ORF26 | 15181 | 14993 | − | 62 | DNA endonuclease P40 | 3.00 × 10−17 | Xanthomonas phage phiL7 | YP002922654.1 |
| ORF27 | 15292 | 15573 | + | 93 | none | |||
| ORF28 | 15552 | 16331 | + | 259 | RNase H superfamily protein | 3.00 × 10−119 | Burkholderia cepacia | WP_060050861.1 pfam13482 |
| DNA exonuclease | 2.00 × 10−102 | Burkholderia phage JG068 | YP008853863.1 | |||||
| ORF29 | 16345 | 16809 | + | 154 | hypothetical protein | 4.00 × 10−67 | Pseudomonas composti | WP061238191.1 |
| hypothetical protein | 3.00 × 10−29 | Mycobacterium phage Omega | NP818474.1 | |||||
| ORF30 | 16806 | 17015 | + | 69 | none | |||
| ORF31 | 17015 | 17860 | + | 281 | ATP-dependent DNA ligase | 8.00 × 10−67 | Xylella phage Prado | YP008859411.1 |
| ORF32 | 17869 | 20283 | + | 804 | DNA-dependent RNA polymerase | 0.0 | Caulobacter phage Percy | ALF01667.1 |
| ORF33 | 20400 | 20588 | + | 62 | hypothetical protein | 8.00 × 10−5 | Caulobacter phage Percy | ALF01668.1 |
| ORF34 | 20677 | 21033 | + | 118 | hypothetical protein | 1.00 × 10−14 | Caulobacter phage Cd1 | ADD21663.1 |
| ORF35 | 21404 | 22930 | + | 508 | head-to-tail joining protein | 2.00 × 10−172 | Caulobacter phage Percy | ALF01671.1 |
| ORF36 | 22934 | 23722 | + | 262 | hypothetical protein | 2.00 × 10−21 | Burkholderia thailandensis MSMB43 | EIP87426.1 |
| scaffold-like protein | 7.00 × 10−20 | Caulobacter phage Cd1 | ADD21667.1 | |||||
| ORF37 | 23820 | 24815 | + | 331 | major capsid protein | 2.00 × 10−98 | Caulobacter phage Cd1 | ADD21668.1 |
| ORF38 | 24897 | 25502 | + | 201 | tail tuber protein A | 4.00 × 10−48 | Burkholderia phage Bp-AMP1 | CDK30105.1 |
| ORF39 | 25499 | 28045 | + | 848 | tail tubular protein B | 0.0 | Caulobacter phage Cd1 | ADD21670.1 |
| ORF40 | 28057 | 28875 | + | 272 | hypothetical protein | 1.00 × 10−10 | Caulobacter phage Cd1 | ADD21671.1 |
| internal virion protein | 7.00 × 10−7 | Xylella phage Prado | YP008859423.1 | |||||
| ORF41 | 28885 | 31227 | + | 780 | hypothetical protein | 2.00 × 10−43 | Caulobacter phage Cd1 | ADD21672.1 |
| internal virion protein | 1.00 × 10−38 | Xylella phage Paz | YP008858912.1 | |||||
| ORF42 | 31250 | 35209 | + | 1319 | internal virion protein | 0.0 | Caulobacter phage Cd1 | ADD21673.1 |
| ORF43 | 35267 | 37195 | + | 642 | tail fiber protein | 5.00 × 10−32 | Caulobacter phage Cd1 | ADD21674.1 |
| ORF44 | 37195 | 37605 | + | 136 | tail fiber assembly protein | 3.00 × 10−24 | Mediterranean phage uvMED | BAQ90231.1 |
| ORF45 | 37625 | 37795 | + | 56 | none | |||
| ORF46 | 37782 | 38057 | + | 91 | putative DNA maturase A | 1.00 × 10−10 | Caulobacter phage Cd1 | ADD21677.1 |
| ORF47 | 38057 | 39814 | + | 585 | terminase large subunit | 0.0 | Caulobacter phage Percy | ALF01684.1 |
| ORF48 | 39831 | 40088 | + | 85 | hypothetical protein | 4.00 × 10−4 | Pseudomonas phage Bf7 | YP005098203.1 |
| Phage | Hosts | CoreGenes3.0 Correlation | Genome Length | GC Content | Classification |
|---|---|---|---|---|---|
| Aquamicrobium phage P14 | Aquamicrobium strains and Alcaligenaceae strains | - | 40,551 bp | 57.8% | Podoviridae phiKMV-like phages |
| Burkholderia phage Bp-AMP1 (HG793132.1) | Burkholderia pseudomallei strains | 47.92% | 42,409 bp | 61.8% | Podoviridae |
| Burkholderia phage Bp-AMP4 (HG796221.1) | Burkholderia pseudomallei | 47.92% | 42,112 bp | 61.8% | Podoviridae |
| Caulobacter phage CD1 (GU393987.1) | Caulobacter crescentus | 54.17% | 41,581 bp | 61.2% | Podoviridae phiKMV-like phages |
| Caulobacter phage Percy (NC_029092.1) | Caulobacter crescentus | 47.27% | 44,773 bp | 60.9% | Podoviridae phiKMV-like phages |
| Erwinia phage FE44 (NC_022744.1) | Erwinia carotovora | 22.92% | 39,860 bp | 48.6% | Podoviridae T7likevirus |
| Pseudomonas phage Bf7 (NC_016764.1) | Pseudomonas strains | 47.92% | 40,058 bp | 58.4% | Podoviridae phiKMV-like phages |
| Ralstonia phage RSJ2 (NC_028988.1) | Ralstonia solanacearum strains | 50.00% | 44,360 bp | 60.9% | Podoviridae |
| Xanthomonas phage phiL7 (NC_012742.1) | Xantomonas campestris | 20.83% | 44,080 bp | 55.6% | Siphoviridae |
| Xylella phage Paz (NC_022982.1) | Xylella fastidiosa and Xantomonas | 47.92% | 43,869 bp | 60.2% | Podoviridae phiKMV-like phages |
| Xylella phage Prado (NC_022987.1) | Xylella fastidiosa and Xantomonas | 52.08% | 43,940 bp | 63.0% | Podoviridae phiKMV-like phages |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Leeuw, M.; Baron, M.; Brenner, A.; Kushmaro, A. Genome Analysis of a Novel Broad Host Range Proteobacteria Phage Isolated from a Bioreactor Treating Industrial Wastewater. Genes 2017, 8, 40. https://doi.org/10.3390/genes8010040
De Leeuw M, Baron M, Brenner A, Kushmaro A. Genome Analysis of a Novel Broad Host Range Proteobacteria Phage Isolated from a Bioreactor Treating Industrial Wastewater. Genes. 2017; 8(1):40. https://doi.org/10.3390/genes8010040
Chicago/Turabian StyleDe Leeuw, Marina, Maayan Baron, Asher Brenner, and Ariel Kushmaro. 2017. "Genome Analysis of a Novel Broad Host Range Proteobacteria Phage Isolated from a Bioreactor Treating Industrial Wastewater" Genes 8, no. 1: 40. https://doi.org/10.3390/genes8010040
APA StyleDe Leeuw, M., Baron, M., Brenner, A., & Kushmaro, A. (2017). Genome Analysis of a Novel Broad Host Range Proteobacteria Phage Isolated from a Bioreactor Treating Industrial Wastewater. Genes, 8(1), 40. https://doi.org/10.3390/genes8010040