Abstract
We report on two brothers with visual impairment, and non-syndromic alopecia in the elder proband. The parents were first-degree Pakistani cousins. Whole exome sequencing of the elder brother and parents, followed by Sanger sequencing of all four family members, led to the identification of the variants responsible for the two phenotypes. One variant was a homozygous nonsense variant in the inhibitory subunit of the cone-specific cGMP phosphodiesterase gene, PDE6H:c.35C>G (p.Ser12*). PDE6H is expressed in the cones of the retina, which are involved in perception of color vision. This is the second report of a homozygous PDE6H:c.35C>G variant causing incomplete achromatopsia (OMIM 610024), thus strongly supporting the hypothesis that loss-of-function variants in PDE6H cause this visual deficiency phenotype. The second variant was a homozygous missense substitution in the lysophosphatidic acid receptor 6, LPAR6:c.188A>T (p.Asp63Val). LPAR6 acts as a G-protein-coupled receptor involved in hair growth. Biallelic loss-of-function variants in LPAR6 cause hypotrichosis type 8 (OMIM 278150), with or without woolly hair, a form of non-syndromic alopecia. Biallelic LPAR6:c.188A>T was previously described in five families from Pakistan.
1. Introduction
Consanguineous marriage, a union between two individuals who are related as second degree cousins or closer, is rooted in cultural and socio-economic factors and is estimated to affect one fifth of the world population [1,2]. Progenies of consanguineous unions are at increased risk compared to the general population for inheriting recessive disorders, due to the risk of being homozygous by descent for a recessive disease-causing allele. We report on a consanguineous family in which two distinct autosomal recessive conditions segregated, one involving visual impairment that was initially believed to be Leber congenital amaurosis, and the other, congenital alopecia. Identification of the two genetic conditions needed extensive clinical follow-up and was only successful after whole exome sequencing (WES).
In early childhood, Leber congenital amaurosis and achromatopsia are two types of congenital retinal dystrophies that overlap clinically, both presenting with visual impairment and nystagmus [3]. Achromatopsia is characterized by deficient color vision, low visual acuity, photophobia, and nystagmus. In complete achromatopsia there is complete lack of function of all of the three types of retinal cones, responsible for blue, green, and red color vision. Thus, the color vision is absent and rods only mediate visual impulses. In incomplete achromatopsia one or two cone types have residual function, allowing partial color vision [4]. Achromatopsia is an autosomal recessive disorder resulting from dysfunction of one of six genes: CNGB3, CNGA3, GNAT2, PDE6C, PDE6H [4], and ATF6 [5]. Achieving an accurate diagnosis in children with retinal dystrophies is important and usually involves genetic testing.
Autosomal recessive forms of alopecia are caused by variants in six genes: HR, DSG4, LIPH, DSC3, DSP, and LPAR6 [6]. Variants in LPAR6 cause hypotrichosis type 8 (OMIM 278150), a type of non-syndromic alopecia, with or without woolly hair and diffuse progressive hair loss, usually beginning in early childhood [7,8].
We report here on a consanguineous Pakistani family in which two rare variants, PDE6H and LPAR6, are segregated.
2. Experimental Section
2.1. Clinical Presentation
A 15-year-old boy (Figure 1a, II:1) was the first child of first-degree cousins from Pakistan. He was treated for neonatal hyperbilirubinemia; the perinatal period was otherwise normal. Visual contact, however, was first established at 10 weeks (normal <6 weeks). From four months he was observed to develop horizontal nystagmus with involuntary rhythmic horizontal head movements, suggestive of spasmus nutans [9]. He had strabismus and the quality of visual contact was questioned. Spasmus nutans can overlap with the more severe neurological condition of opsoclonus-myoclonus syndrome [10], but follow-up examinations with cerebral MRI, EEG, and screening for malignancy gave normal results. General psychomotor development was normal. Myopia (10 diopters) and astigmatism were diagnosed at eight months. At 11 months, the involuntary head movements had decreased, although a fine nystagmus with small amplitude and high frequency persisted. A precise diagnosis was still missing and at 2.5 years his condition was changed to being loosely labeled “congenital nystagmus with myopia”, complicated by a hitherto unresolved deficiency of hair growth. Since the shaving of his scalp in the neonatal period, his hair had never regrown leaving him bald-headed.
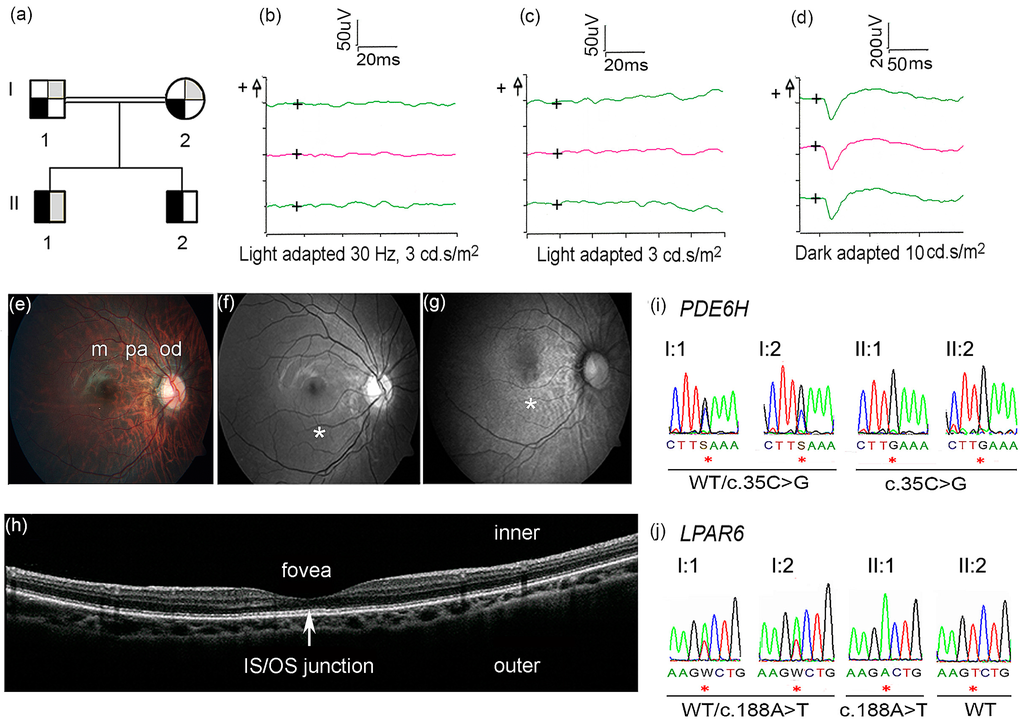
Figure 1.
(a) Family pedigree showing segregation of incomplete achromatopsia (IA) and alopecia. Black symbol refers to IA, grey symbol refers to alopecia; ¼ filled symbols = carrier, ½ filled symbols = affected. The genotype of the PDE6H and LPAR6 are the following: I:1 WT/c.35C>G; WT/c.188A>T; I:2 WT/c.35C>G; WT/c.188A>T; II:1 c.35C>G/c.35C>G; c.188A>T/ c.188A>T; II:2 c.35C>G/c.35C>G; WT/WT. Ophthalmological examinations (b–h) were performed in the right eye of Patient II:1. (b–d) ERG x-axis = time in milliseconds (ms), y-axis = response in microvolt (uV); the “+” symbol (on the left side of y-axis) indicate positive voltage; the “+” symbols inside the ERG graphs indicate the origin of the axes; red lines indicate median values of two measurements (green lines); (b) Cone ERG showed almost absent flicker response at 30 Hz; (c) Cone ERG showed almost absent single flash response; (d) Dark adapted ERG showed normal rod response; (e) Color fundus photography showed normal macula (m) and mild to moderate myopic changes in the peripapillary area (pa) and in the optic disc (od); (f) Red-free fundus photography showed normal macula (*); (g) Fundus autofluorescence images showed mottling in the macula (*); (h) Optical coherence tomography showed normal distribution of retinal layers, without disruption of the inner segment/outer segment junction (arrow); (i) Sanger sequencing showed homozygosity for PDE6H c.35C>G in Patient II:1 and II:2 and heterozygosity in I:1 and I:2; (j) Sanger sequencing showed homozygosity for LPAR6 c.188A>T in Patient II:1, heterozygosity in I:1 and I:2, and wild-type (WT) sequence in Patient II:2 (the reverse sequence is shown in the figure).
Deficient red-green color vision and profound weak-sightedness were disclosed at nine years, but blue-yellow color vision was normal (Hardy, Rand, Rittler Color Vision Test and HRR Pseudoisochromatic Plate Test, 4th ed., Richmond Products, Inc., Albuquerque, NM, USA). Photophobia was not present. The results from repeated follow-up eye examinations are summarized in Table 1. A clinical diagnosis of Leber congenital amaurosis was eventually proposed. An electroretinogram (ERG) was performed at 11 and 15 years with the patient awake using an Espion E3 Electroretinography System (Diagnosys LLC, Littleton, MA, USA) in accordance with the International Society for Clinical Electrophysiology of Vision (iscev.org). The cone ERG showed an almost absent flicker response at 30 Hz (Figure 1b) and almost absent single flash response (Figure 1c). Importantly, scotopic responses were normal (Figure 1d) and, therefore, the diagnosis of Leber congenital amaurosis was discarded [11]. Color fundus photography showed mild to moderate myopic changes of the optic disc and peripapillary area (Figure 1e). Red-free fundus photography showed normal macula (Figure 1f). Fundus autofluorescence photography showed a mottled pattern in the macula (Figure 1g). The peripheral retina was normal. Optical coherence tomography at age 15 showed a normal foveal pit and normal distribution of retinal layers, without signs of disruption of the inner segment/outer segment junction (Figure 1h).

Table 1.
Comparison of the ophthalmological presentation of the five patients reported with biallelic c.35C>G variant in PDE6H.
The patient was referred to one of the authors (PS) for assessment of a possible syndrome comprising the combination of ocular and hair abnormalities. When dissecting the expanded family history, it became clear that these two features segregated as separate inherited phenotypes, as the mother’s sister’s two daughters had alopecia, without visual impairment. It was, therefore, concluded that the patient had inherited two autosomal recessive conditions.
The younger brother, a 10-year-old boy (Figure 1a, II:2) presented at 18 months with myopia (eight diopters), horizontal nystagmus of the left eye, and strabismus treated with Botulinum toxin injections over the years. Examination showed mild to moderate myopic changes of the optic disc and peripapillary area. Discreet, small disruptions of the retinal pigment epithelium in the central macula, only visible by ophthalmoscopy, were observed. At nine years, an ERG, with the patient awake, showed almost absent photopic responses and normal scotopic responses. There was no deterioration in visual acuity over the years (Table 1). At 10 years nystagmus was no longer detectable. His ophthalmological findings were similar to those in Patient II:1. He did not have alopecia.
2.2. Materials and Methods
The study was conducted in accordance with the Declaration of Helsinki, and the Regional Ethical Committee approved the research project (REK 2010/1152-1). The parents of the patients signed a written informed consent for the genetic analyses and for publishing the results and photos of the patients.
Whole Exome Sequencing (WES) and Data Analysis
DNA from peripheral leukocytes from Patient II:1 and his parents (I:1, I:2) was sheared using a Covaris sonicator (Covaris, Woburn, MA, USA) to produce fragments with an average size of 200 bp. Paired-end Illumina adapters (Illumina, Inc., San Diego, CA, USA) were ligated to the fragments according to the manufacturer’s recommendations. Exome capture was performed with the SureSelect Human All Exon kit v5 (Agilent Technologies, Santa Clara, CA, USA). The exome-captured library was sequenced on an Illumina HiSeq2000 with 100 bp paired end reads. Reads that did not pass Illumina’s standard filter were removed prior to alignment. The remaining reads were aligned to the reference human genome (GRCh37/hg19), using the Burrows-Wheeler aligner tool [12]. PCR duplicates were identified using Picard (broadinstitute.github.io/picard). Approximately 95% of the reads mapped uniquely to the reference sequence yielding an average of 125× coverage per targeted base. Joint variant calling was performed using the Unified Genotyper in the Genome Analysis Tool kit (GATK 2.5-2) [13]. Variants were annotated with snpEff [14] and Variant Effect Predictor using annotations GRCh 37.64 and Ensembl 71, respectively. The Integrative Genomic Viewer [15] was used for data visualization.
The annotated variant files were filtered and analyzed using the software FILTUS 0.99-91 [16]. Variants in genes known to generate false positive signals in exome sequencing were discarded [17], together with variants with minor allele frequency higher than 0.01 according to the 1000 Genome Project [18], the Exome Variant Server (evs.gs.washington.edu/EVS) and an in-house database of 443 exomes of mixed ethnicity. We focused on variants resulting in missense, nonsense, frameshift, and small insertion/deletions in order to find causative variants. Allele frequency was also assessed in the Exome Aggregate database (ExAc, exac.broadinstitute.org), which contains data from more than 60,000 human exomes. Finally, we used the autozygosity functionality of FILTUS to detect regions of homozygosity (ROH) in the patients’ genome. The lengths of identified ROHs were reported in megabases and in centimorgan (according to the Decode recombination map of the human genome [19]).
3. Results
Based on the family pedigree (Figure 1a), WES data from Patient II:1 and the parents were analyzed according to a recessive mode of inheritance (Table S1). Variants in 23 genes were detected after data filtering (Table S2). Homozygous variants identified in PDE6H (NM_006205.2, OMIM 601190) c.35C>G, p.Ser12* and LPAR6 (NM_005767.5, OMIM 609239) c.188A>T, and p.Asp63Val (Figure 1i–j)) were considered to be likely disease-causing because of their established involvement in incomplete achromatopsia and alopecia, respectively [8,20,21,22,23]. These variants were validated by Sanger sequencing. We proved the heterozygosity of the parents and homozygosity of Patient II:1 for both variants, whereas Patient II:2 was homozygous for PDE6H c.35C>G and wild-type for LPAR6 c.188A (Figure 1i–j).
Autozygosity analysis of the patient revealed that both PDE6H and LPAR6 were surrounded by regions of homozygosity (ROH), of lengths 21 Mb (34 cM) and 9 Mb (10 cM), respectively (Figure 2a,b). Such ROH sizes are typical for autozygous segments resulting from first cousin marriage, and suggest that both PDE6H:c.35C>G and LPAR6:c.188A>T are in an autozygous state in the patient, inherited via both parents from one of their shared grandparents.
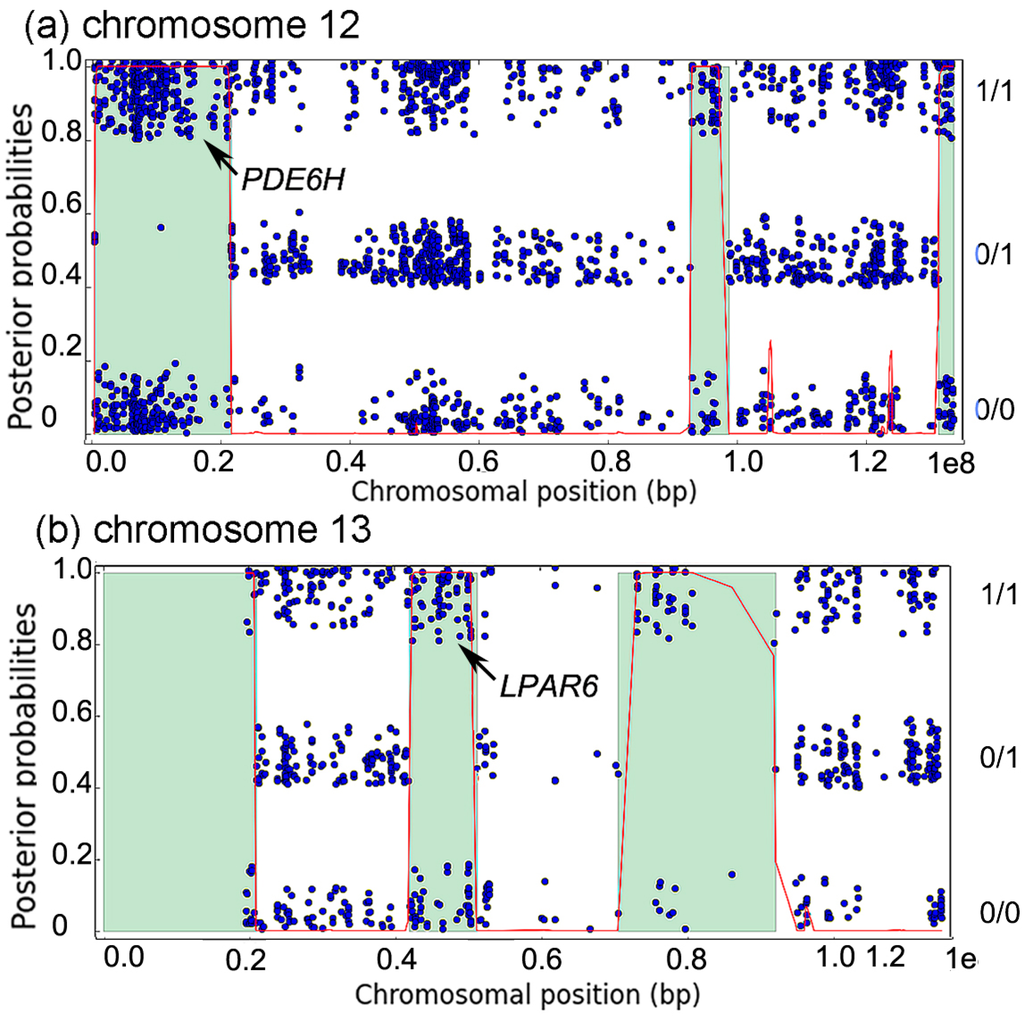
Figure 2.
(a,b) Autozygosity analysis of chromosomes 12 and 13 in patient II:1. Each blue dot represents a variant. It is placed in the lower band if homozygous for the reference allele (0/0), in the middle band if heterozygous (0/1), and in the top band if homozygous for a non-reference allele (1/1). The red curve shows posterior probability of autozygosity, and predicted autozygous regions are colored in light green. The autozygous regions containing PDE6H (chromosome 12) and LPAR6 (chromosome 13) are indicated by the arrows.
In the ExAc database (exac.broadinstitute.org), containing more than 60,000 human exomes data), the PDE6H c.35C>G variant has been identified in heterozygosity in nine out of 61,228 individuals (seven European non-Finnish, one Finnish, one African), but in none of the 16,590 alleles of South Asian individuals. The LPAR6 c.188A>T variant is not present in the ExAc database. Sanger sequencing of 180 in-house Pakistani controls did not reveal the PDE6H c.35C>G variant. The Pakistani controls were not analyzed for LPAR6 c.188A>T, because the variant was reported among Pakistani [8,20,21,23].
The DNA variants identified in the study have now been reported to the ClinVar database (ncbi.nlm.nih.gov/clinvar/): SCV000256082 PDE6H c.35C>G, p.Ser12*; SCV000256083 LPAR6 c.188A>T, p.Asp63Val.
4. Discussion
For the proband reported here we describe the co-existence of a rare ophthalmological diagnosis eventually pinpointed as incomplete achromatopsia and congenital alopecia, and incomplete achromatopsia in a younger brother. The genetic variants causing the two separate clinical phenotypes were documented using WES.
It was not straightforward to establish the correct ophthalmological diagnosis. Patient II:1 was initially suspected of having spasmus nutans bordering opsoclonus-myoclonus, before being labeled “congenital nystagmus with myopia”, and was subsequently thought to have Leber congenital amaurosis, carrying a more pessimistic prognosis than achromatopsia. Congenital hypotrichosis with juvenile macular dystrophy (HJMD, OMIM 601553), due to biallelic variants in CDH3 [24], manifests with both features present in Patient 1, the alopecia and the ophthalmological disease. However, HJMD was ruled out in Patient 1 because, from the family history, it was evident that the two conditions segregated independently. WES eventually revealed the true cause of the retinal dystrophy.
This is the second confirmatory report of a PDE6H variant causing incomplete achromatopsia. Interestingly, in both reports the variant is c.35C>G, p.Ser12*, but it arises in different ethnicities: South Asian in our family and Caucasian in the first study, which describes three patients from two families, sharing a common haplotype around the variant [22].
PDE6H encodes the γ subunit (PDEγ) of the cyclic guanosine monophosphate (cGMP) cone phosphodiesterase (PDE) enzyme, essential for the phototransduction [25]. The c.35C>G, p.12Ser* nonsense variant causes lack of functional PDEγ, affecting the cone PDE in the phototransduction, possibly leaving cGMP-gated channels permanently closed [22].
Immunohistochemistry on mouse retinal sections using a PDE6H antibody stained all cones [26]. However, patients with PDE6H c.35C>G have preserved S-cone function and disturbed M- and L-cone functions (Table 1) [22]. The preserved S-cone function in the absence of PDEγ suggests possible compensation for the loss of PDE6H in the PDE. This was demonstrated in PDE6H knockout mice, in which no photoreceptor dysfunction was recorded in the absence of PDE6H, due to functional complementation by the rod PDE6G [26].
Incomplete achromatopsia caused by defective PDE6H presents a rather homogeneous ophthalmological profile [22]. The two siblings in this report, and the three patients previously-described homozygous for PDE6H c.35C>G and incomplete achromatopsia, presented in early childhood with strong red-green color vision deficiency and normal blue-yellow color vision (Table 1). On ERG they all had normal scotopic, but severely reduced photopic, signals. Visual acuity was persistently poor, but did not deteriorate.
Approximately 60% of marriages in Pakistan are estimated to be consanguineous [27] and expose the progeny to higher risk of homozygosity by descent for a recessive disease-causing allele. In this Pakistani family Patient II:1 had two autosomal recessive conditions: incomplete achromatopsia and alopecia (Figure S1).
LPAR6 is a G protein coupled receptor for lysophosphatidic acid (LPA), expressed in the inner root sheath of the hair follicle. Molecular modeling indicated that p.Asp63Val in the transmembrane helices 2 domain alters LPAR6 conformation and inhibits its binding to LPA, a key factor for inner root sheath differentiation and hair development [23]. Individuals with the LPAR6 c.188A>T, p.Asp63Val had a variable degree of alopecia, some with predominantly woolly hair and some with only sparse hair [8,20,21,23]. Interestingly, the variant has only been identified within the Pakistani population [7,8,23] but is not reported in the ExAc database, suggesting that it is not distributed too widely within the Pakistani population in which it may have originated.
5. Conclusions
This study illustrates the dilemma faced when determining how to best distinguish amongst rare congenital retinal dystrophies. Mode of inheritance and clinical evaluation over time supplemented with ERG examination will allow the ophthalmologist to rule out some diseases, but timely establishment of the exact diagnosis requires genetic testing.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4425/7/8/41/s1, Figure S1: Patient 1 at the age of 13, alopecia is evident, Table S1: Analysis of the WES data of Patient II:1 and his parents, according to a recessive mode of inheritance (variants after filtering are listed), Table S2: Number of genetic variants obtained after WES data filtering
Acknowledgments
We are grateful to the family for participating to our study. We are also grateful to Simon Rayner for language revision. The Norwegian Sequencing Centre (NSC), a national technology platform supported by the ‘Functional Genomics and Infrastructure’ programs of the Research Council of Norway and the Southern and Eastern Norway Regional Health Authorities at Oslo University Hospital, Oslo, Norway, provided the sequencing service. Aase og Knut Tønjums oftalmologiske fond supported Christeen Ramane J. Pedurupillay.
Author Contributions
Christeen Ramane J. Pedurupillay, Magnus Dehli Vigeland, analyzed WES data; Christeen Ramane J. Pedurupillay verified WES findings; Christeen Ramane J. Pedurupillay and Muhammad Ansar provided and analyzed ethnic matched controls; Erlend Christoffer Sommer Landsend and Petter Strømme investigated the patients and drafted the clinical description. Christeen Ramane J. Pedurupillay and Doriana Misceo drafted the manuscript; Eirik Frengen, Doriana Misceo and Petter Strømme designed the study and revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bittles, A. Consanguinity and its relevance to clinical genetics. Clin. Genet. 2001, 60, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Hamamy, H. Consanguineous marriages: Preconception consultation in primary health care settings. J. Community Genet. 2012, 3, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K.; Lopez, I.; den Hollander, A.I.; Allikmets, R.; Cremers, F.P. Genetic testing for retinal dystrophies and dysfunctions: Benefits, dilemmas and solutions. Clin. Experiment. Ophthalmol. 2007, 35, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Remmer, M.H.; Rastogi, N.; Ranka, M.P.; Ceisler, E.J. Achromatopsia: A review. Curr. Opin. Ophthalmol. 2015, 26, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Zobor, D.; Chiang, W.C.; Weisschuh, N.; Staller, J.; Menendez, I.G.; Chang, S.; Beck, S.C.; Garrido, M.G.; Sothilingam, V.; et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 2015, 47, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y. Congenital hair loss disorders: Rare, but not too rare. J. Dermatol. 2012, 39, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Pasternack, S.M.; von Kugelgen, I.; Al Aboud, K.; Lee, Y.A.; Ruschendorf, F.; Voss, K.; Hillmer, A.M.; Molderings, G.J.; Franz, T.; Ramirez, A.; et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008, 40, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Wajid, M.; Ishii, Y.; Shapiro, L.; Petukhova, L.; Gordon, D.; Christiano, A.M. Disruption of P2RY5, an orphan G protein-coupled receptor, underlies autosomal recessive woolly hair. Nat. Genet. 2008, 40, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Gottlob, I.; Wizov, S.S.; Reinecke, R.D. Spasmus Nutans—A Long-Term Follow-Up. Investig. Ophthalmol. Vis. Sci. 1995, 36, 2768–2771. [Google Scholar] [PubMed]
- Pike, M. Opsoclonus-myoclonus syndrome. Handb. Clin. Neurol. 2013, 112, 1209–1211. [Google Scholar] [PubMed]
- Pennesi, M.E.; Stover, N.B.; Stone, E.M.; Chiang, P.W.; Weleber, R.G. Residual Electroretinograms in Young Leber Congenital Amaurosis Patients with Mutations of AIPL1. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8166–8173. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Vigeland, M.D.; Gjøtterud, K.S.; Selmer, K.K. FILTUS: A desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics 2016. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, K.V.F.; Adams, D.; Mason, C.E.; Sincan, M.; Tifft, C.; Toro, C.; Boerkoel, C.F.; Gahl, W.; Markello, T. Detecting false-positive signals in exome sequencing. Hum. Mutat. 2012, 33, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Kong, A.; Thorleifsson, G.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Walters, G.B.; Jonasdottir, A.; Gylfason, A.; Kristinsson, K.T.; et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 2010, 467, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Azeem, Z.; Jelani, M.; Naz, G.; Tariq, M.; Wasif, N.; Naqvi, S.K.U.H.; Ayub, M.; Yasinzai, M.; Wali, A.; Ali, G.; et al. Novel mutations in G protein-coupled receptor gene (P2RY5) in families with autosomal recessive hypotrichosis (LAH3). Hum. Genet. 2008, 123, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Habib, R.; Mir, H.; Naz, G.; Ayub, M.; Shafique, S.; Yamin, T.; Ali, N.; Basit, S.; Wasif, N.; et al. Mutations in the LPAR6 and LIPH genes underlie autosomal recessive hypotrichosis/woolly hair in 17 consanguineous families from Pakistan. Clin. Exp. Dermatol. 2011, 36, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Coppieters, F.; Meire, F.; Schaich, S.; Roosing, S.; Brennenstuhl, C.; Bolz, S.; van Genderen, M.M.; Riemslag, F.C.; Lukowski, R.; et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am. J. Hum. Genet. 2012, 91, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.I.; Muhammad, D.; Jan, A.; Ali, R.H.; Hassan, M.; Ahmad, W.; Rashid, S. In silico analysis of missense mutations in LPAR6 reveals abnormal phospholipid signaling pathway leading to hypotrichosis. PLoS ONE 2014, 9, e104756. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, E.; Bergman, R.; Richard, G.; Lurie, R.; Shalev, S.; Petronius, D.; Shalata, A.; Anbinder, Y.; Leibu, R.; Perlman, I.; et al. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P-cadherin. Nat. Genet. 2001, 29, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Lamb, T.D.; Pugh, E.N. Phototransduction, dark adaptation, and rhodopsin regeneration the proctor lecture. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5138–5152. [Google Scholar] [CrossRef] [PubMed]
- Brennenstuhl, C.; Tanimoto, N.; Burkard, M.; Wagner, R.; Bolz, S.; Trifunovic, D.; Kabagema-Bilan, C.; Paquet-Durand, F.; Beck, S.C.; Huber, G.; et al. Targeted ablation of the Pde6h gene in mice reveals cross-species differences in cone and rod phototransduction protein isoform inventory. J. Biol. Chem. 2015, 290, 10242–10255. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Bittles, A.H. The prevalence and demographic characteristics of consanguineous marriages in Pakistan. J. Biosoc. Sci. 1998, 30, 261–275. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).