
Transcriptomic Analysis of the Endangered Neritid Species Clithon retropictus: De Novo Assembly, Functional Annotation, and Marker Discovery


 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and RNA Preparation
2.3. Library Construction and Illumina Sequencing
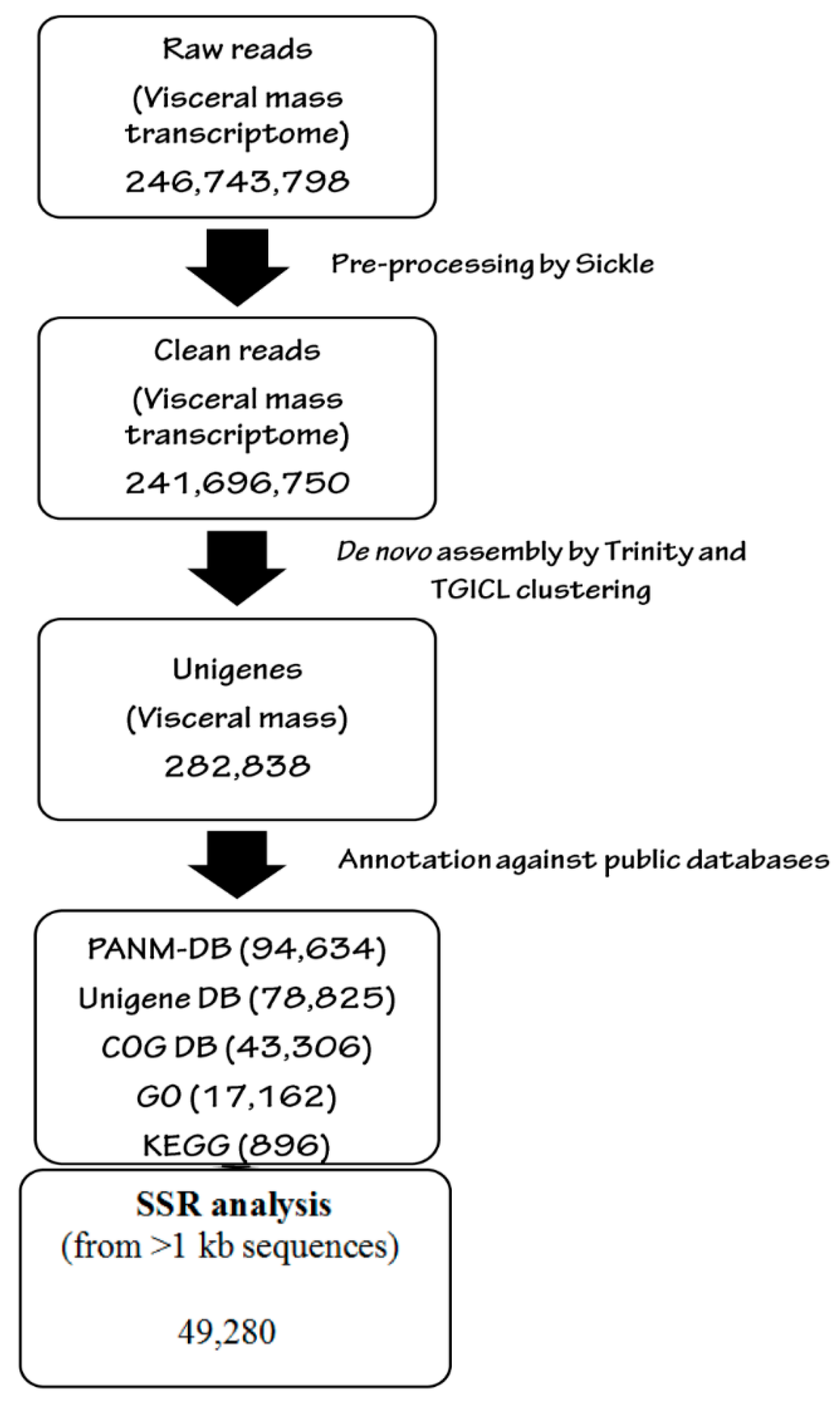
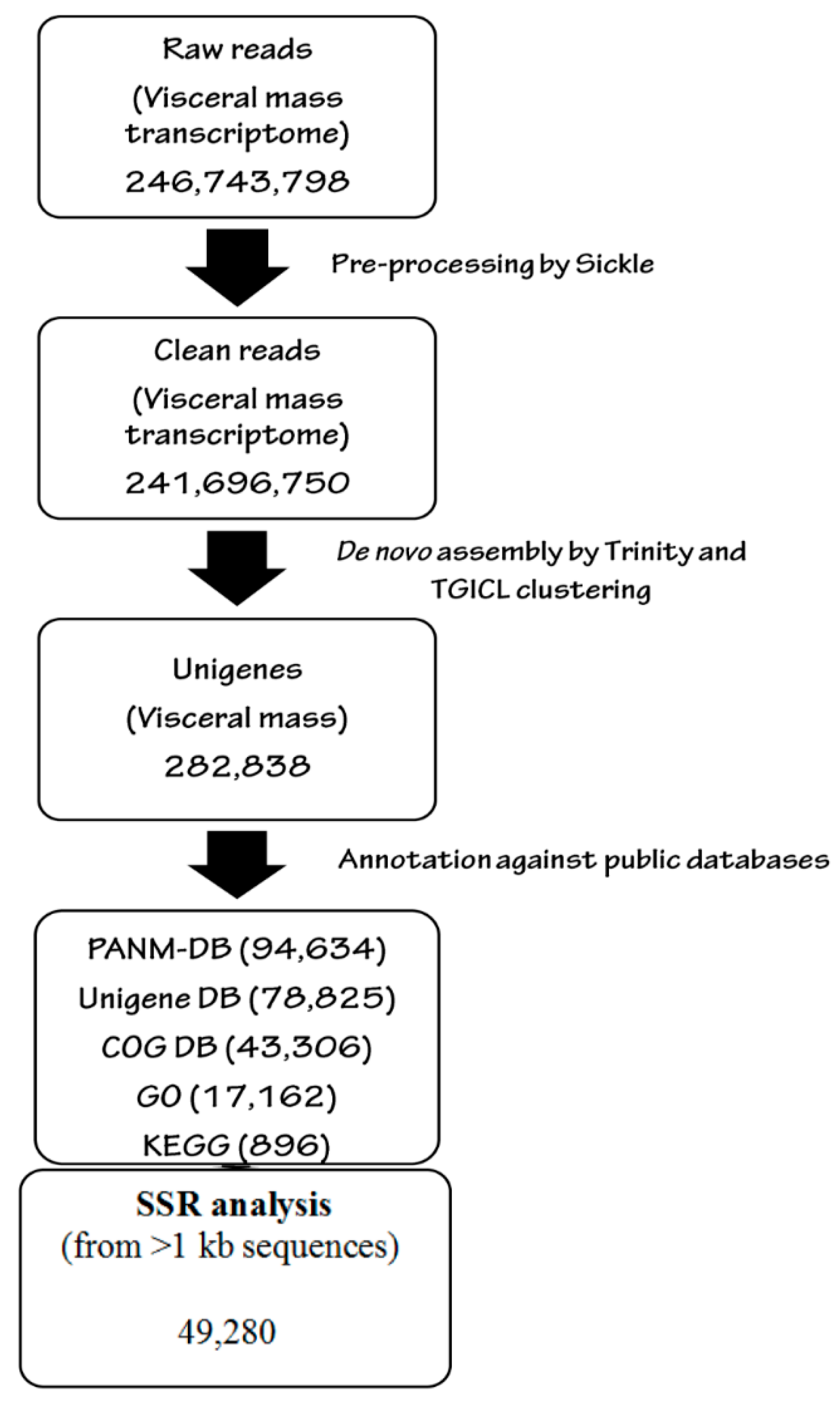
2.4. Quality Control of Reads and de novo Analysis
2.5. Transcriptomic Annotation
2.6. Identification of Simple Sequence Repeats (SSRs)
3. Results and Discussion
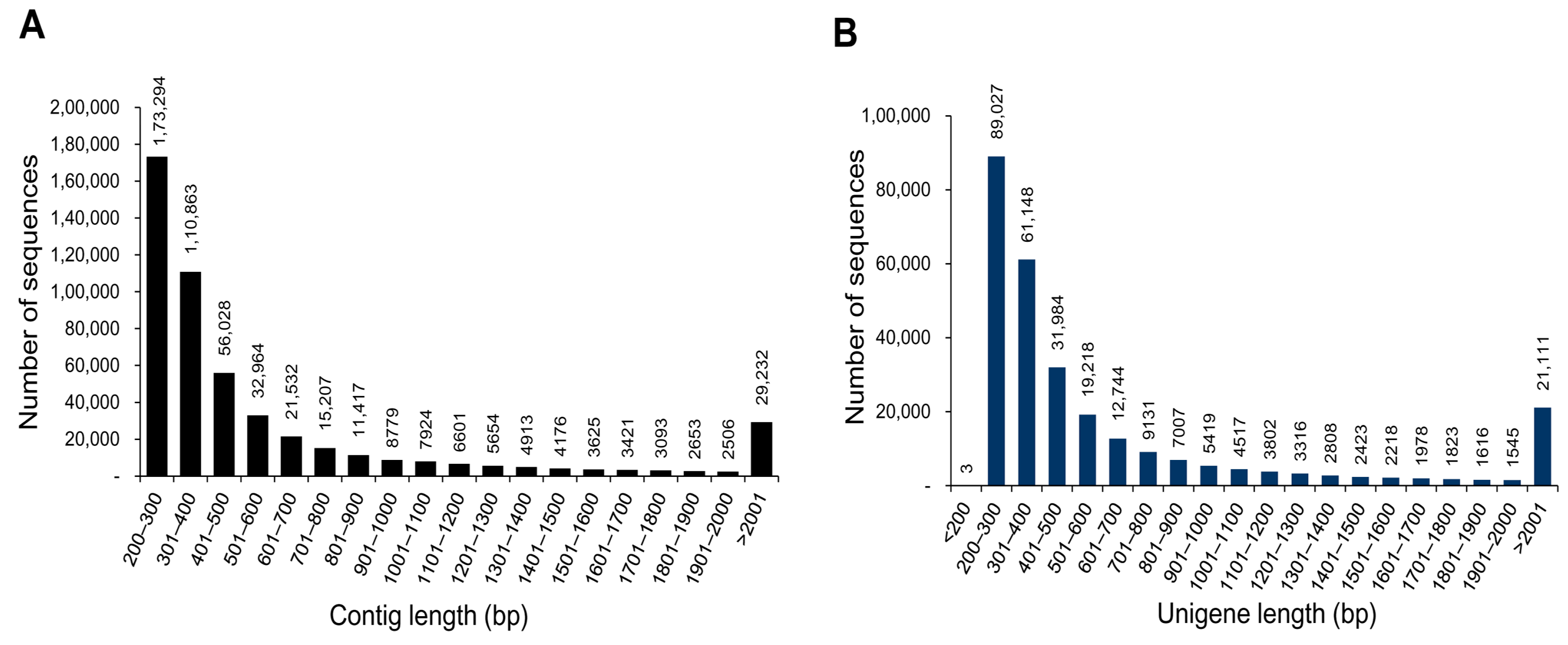
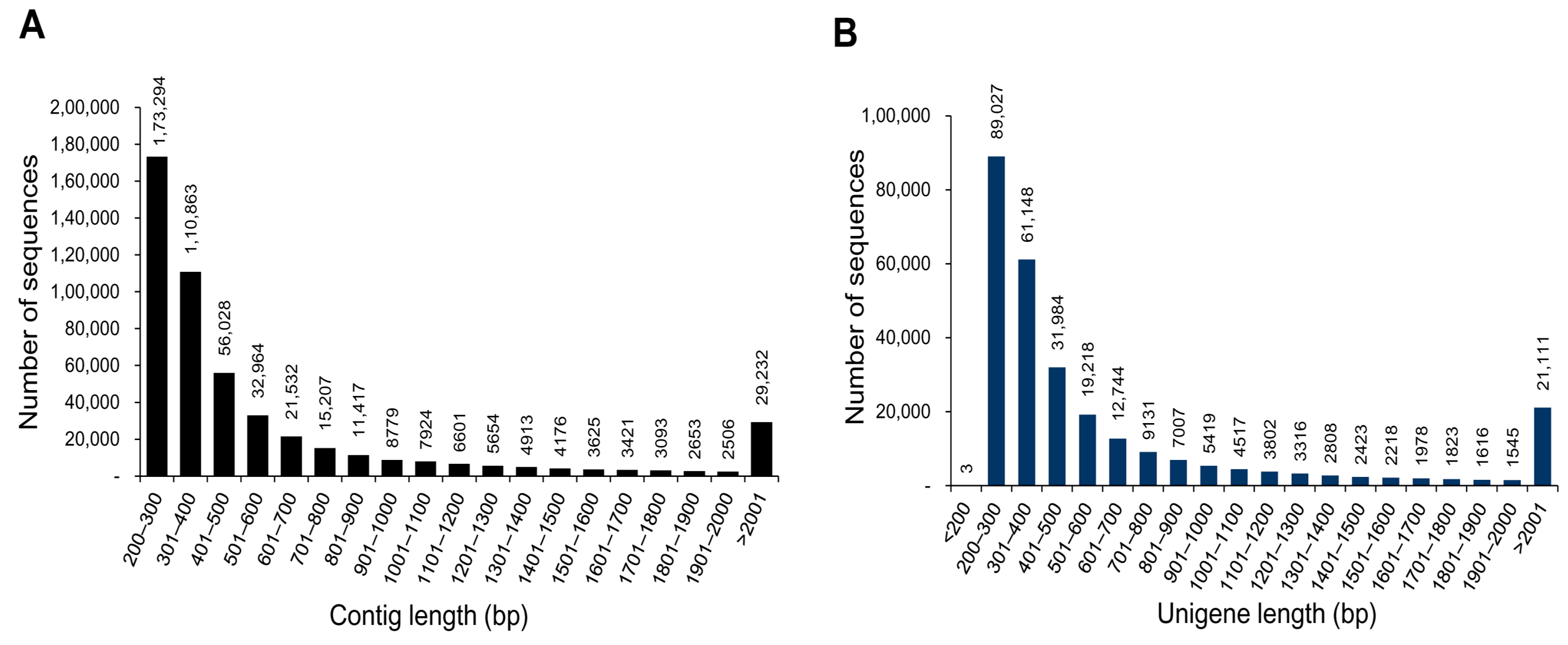
3.1. Illumina Reads and Sequence Assembly
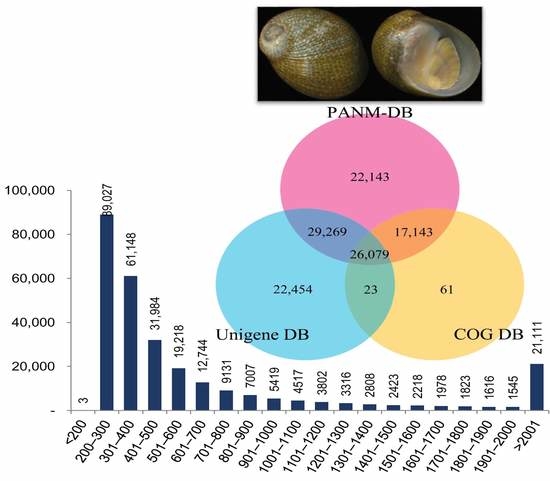
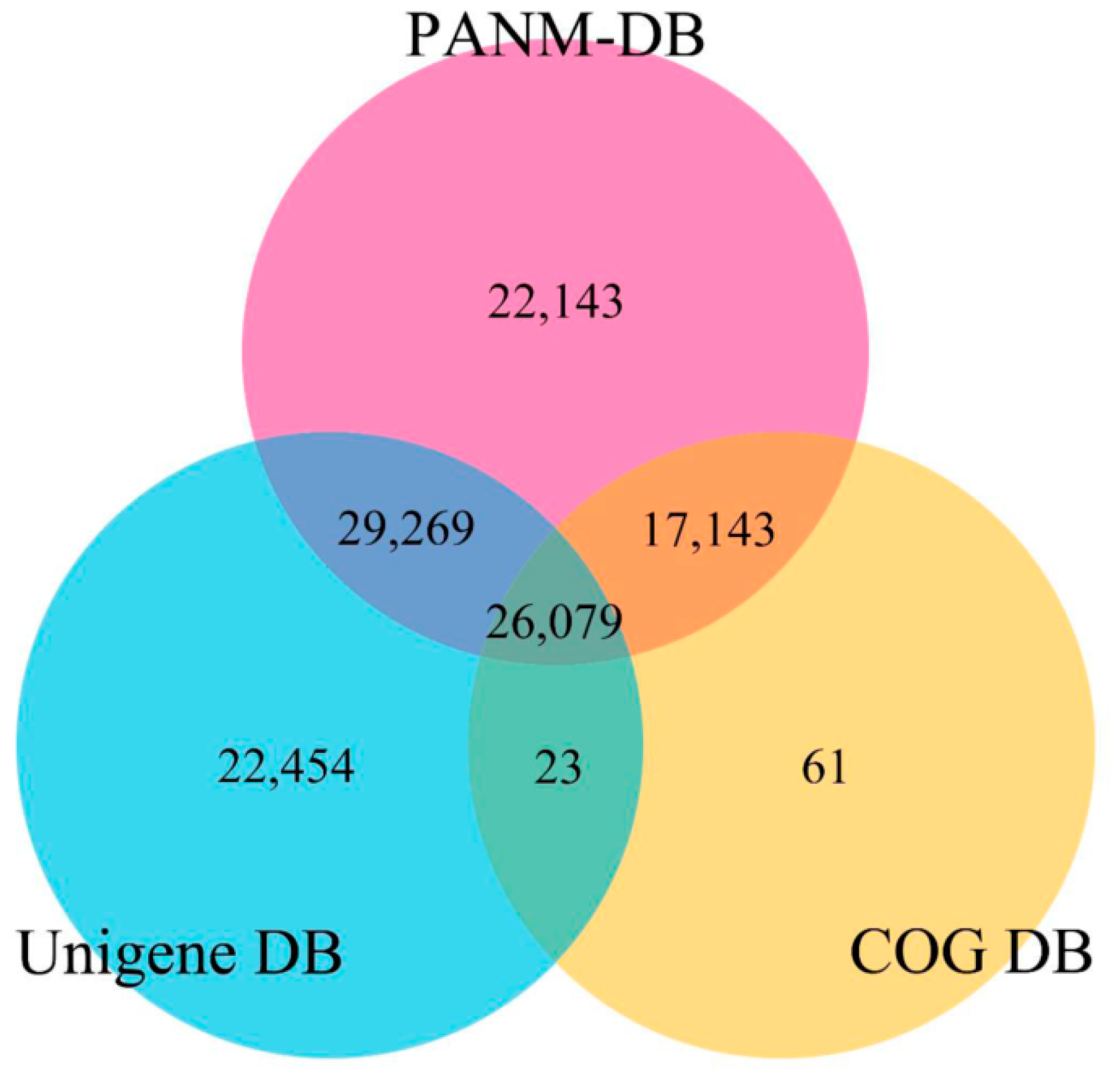
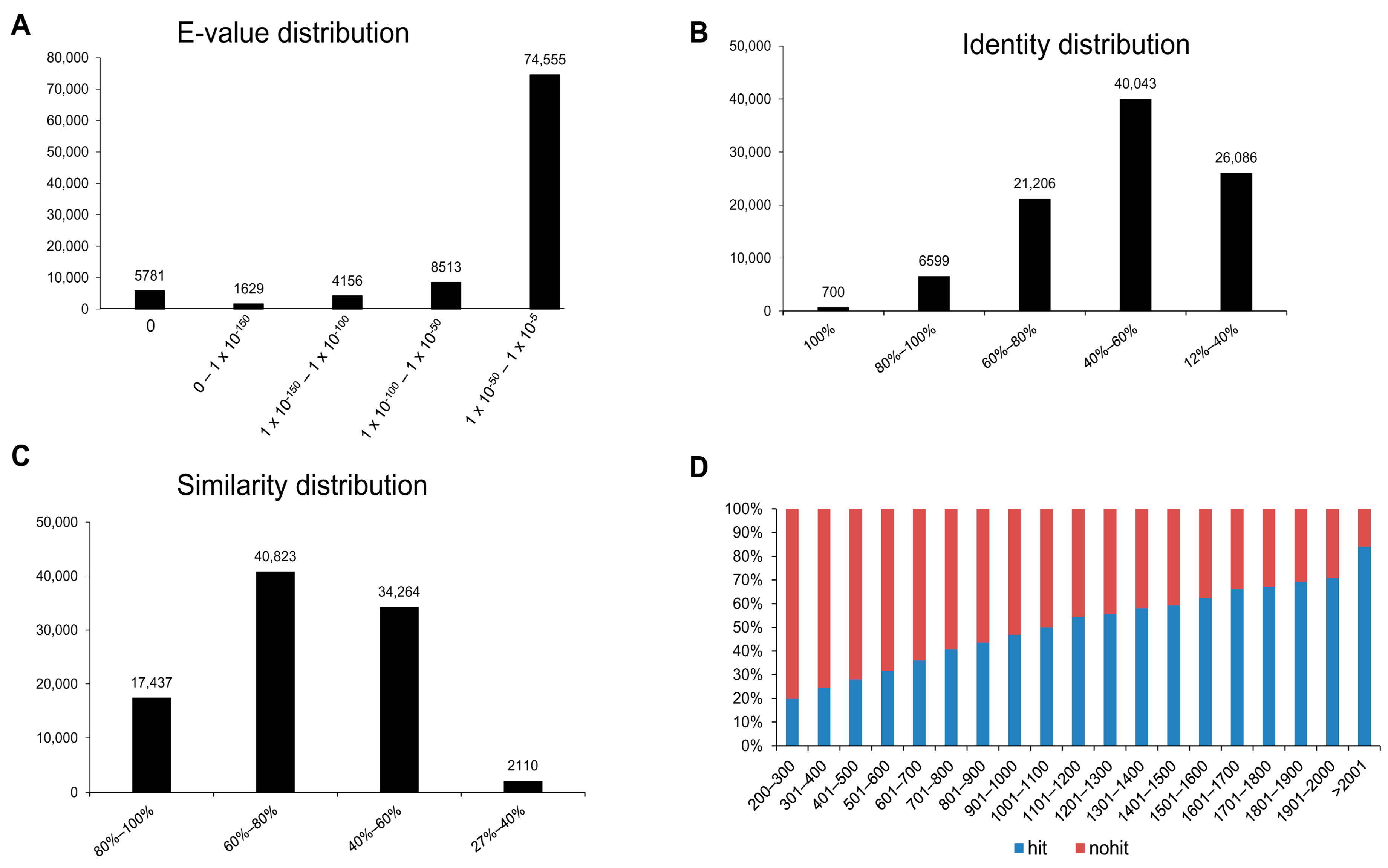
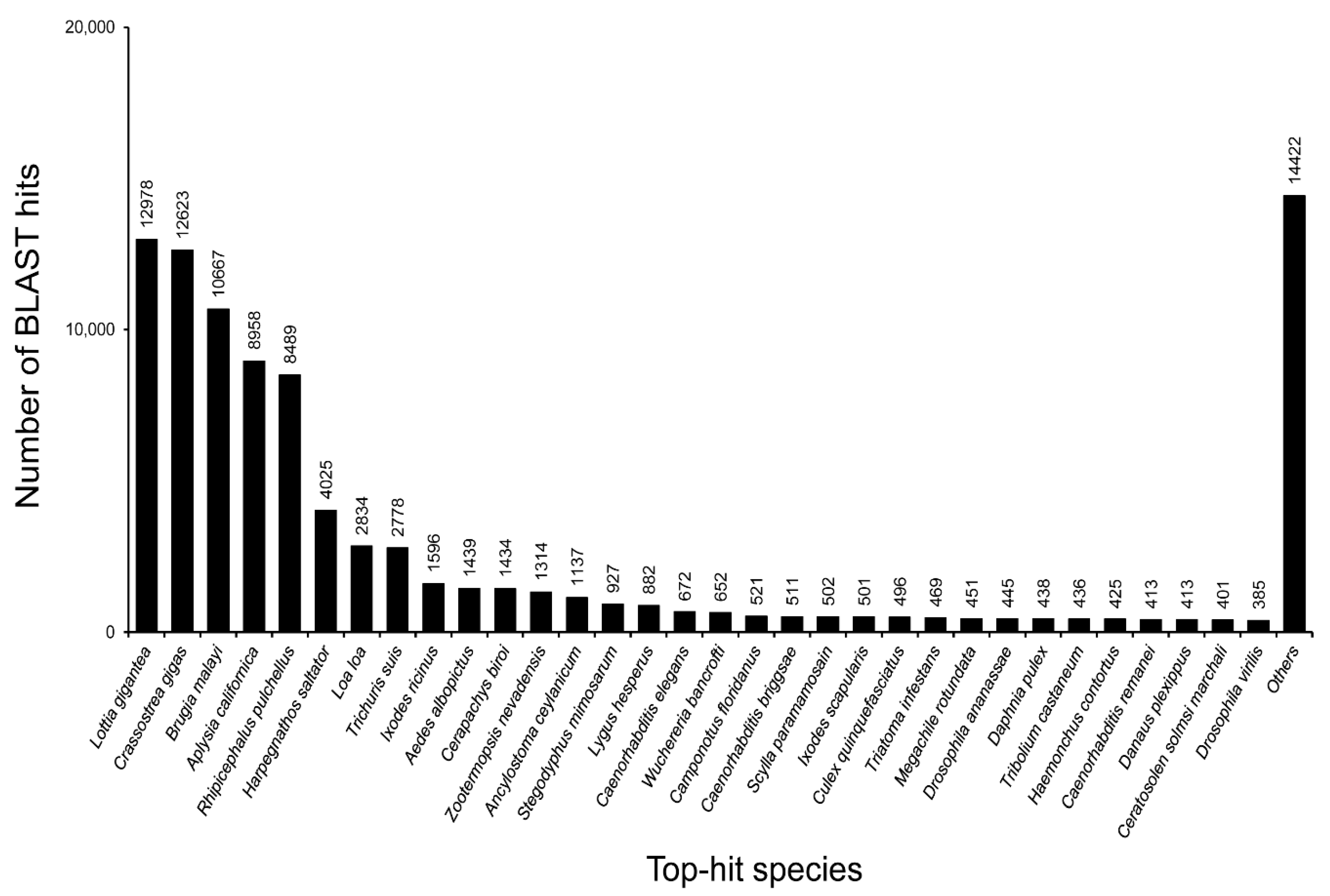
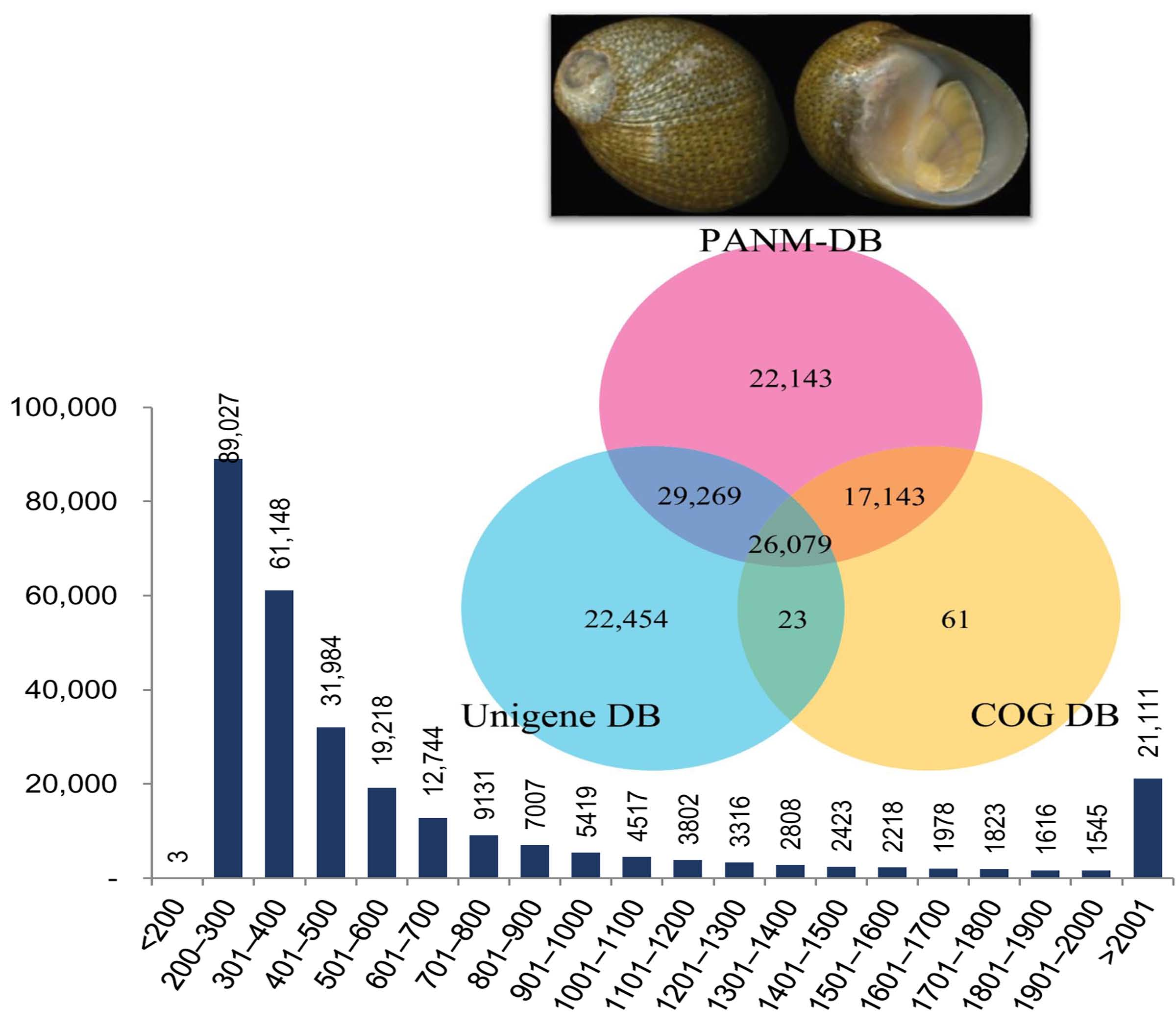
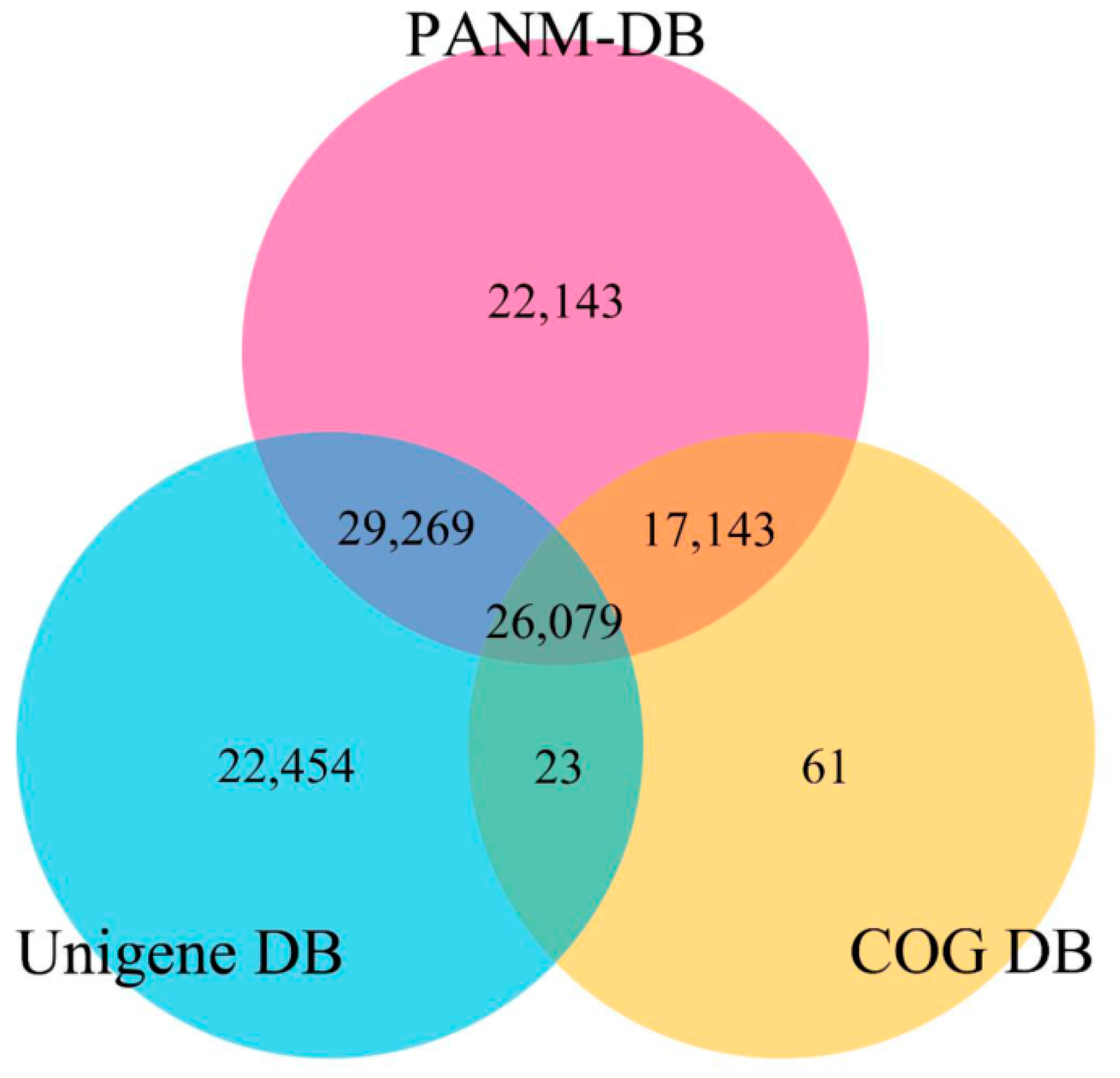
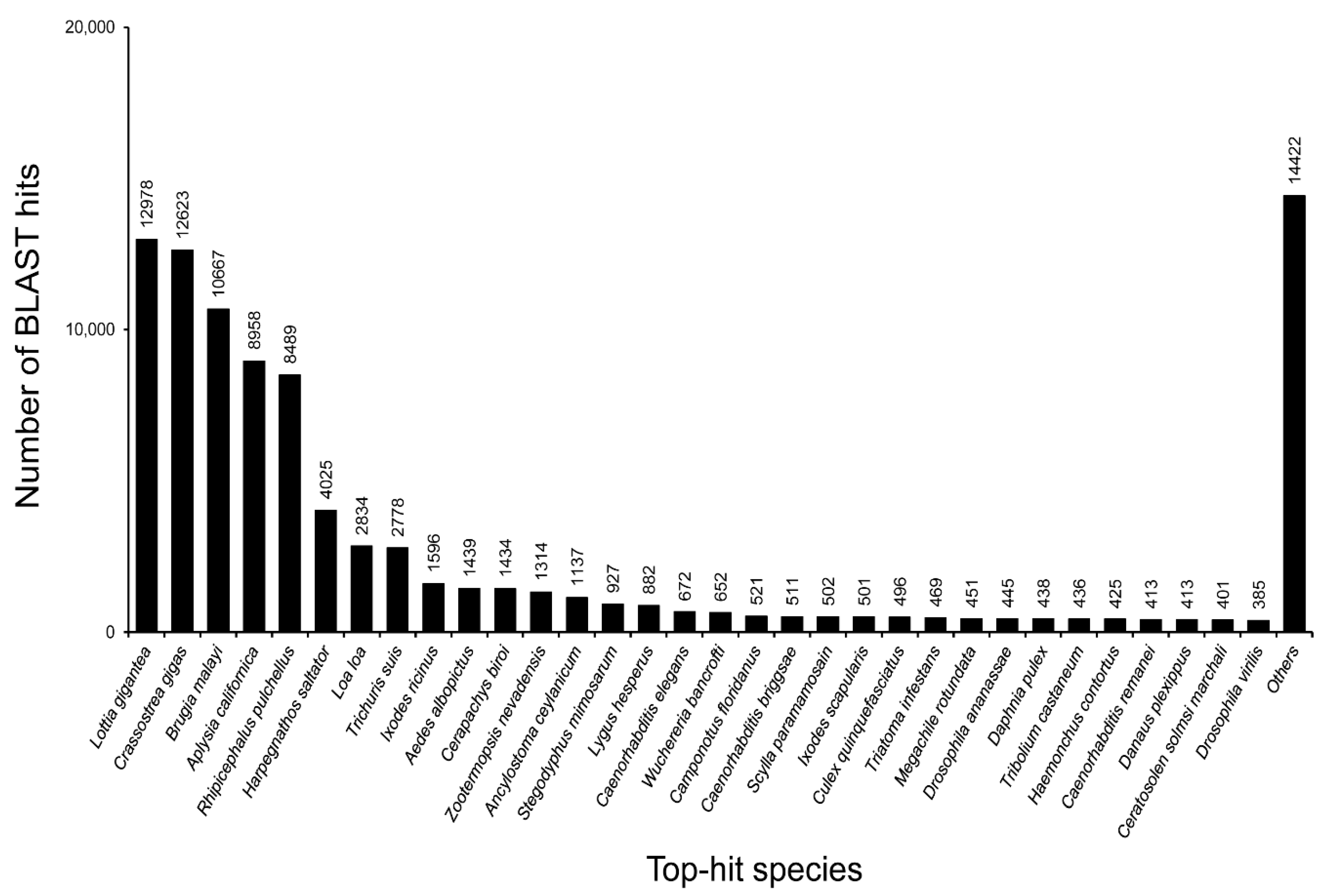
3.2. Annotation of Unigenes and BLAST-Based Homology Search
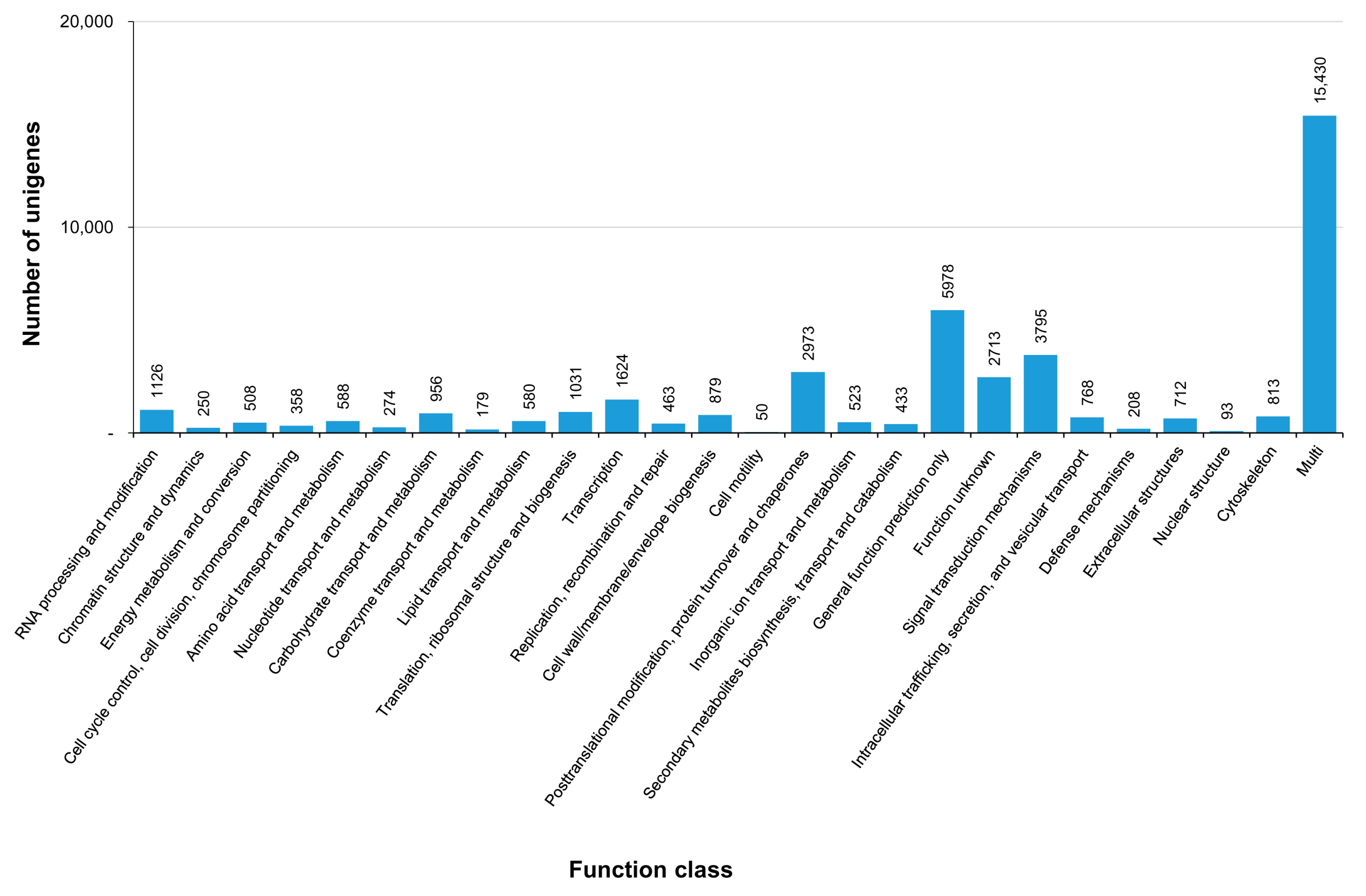
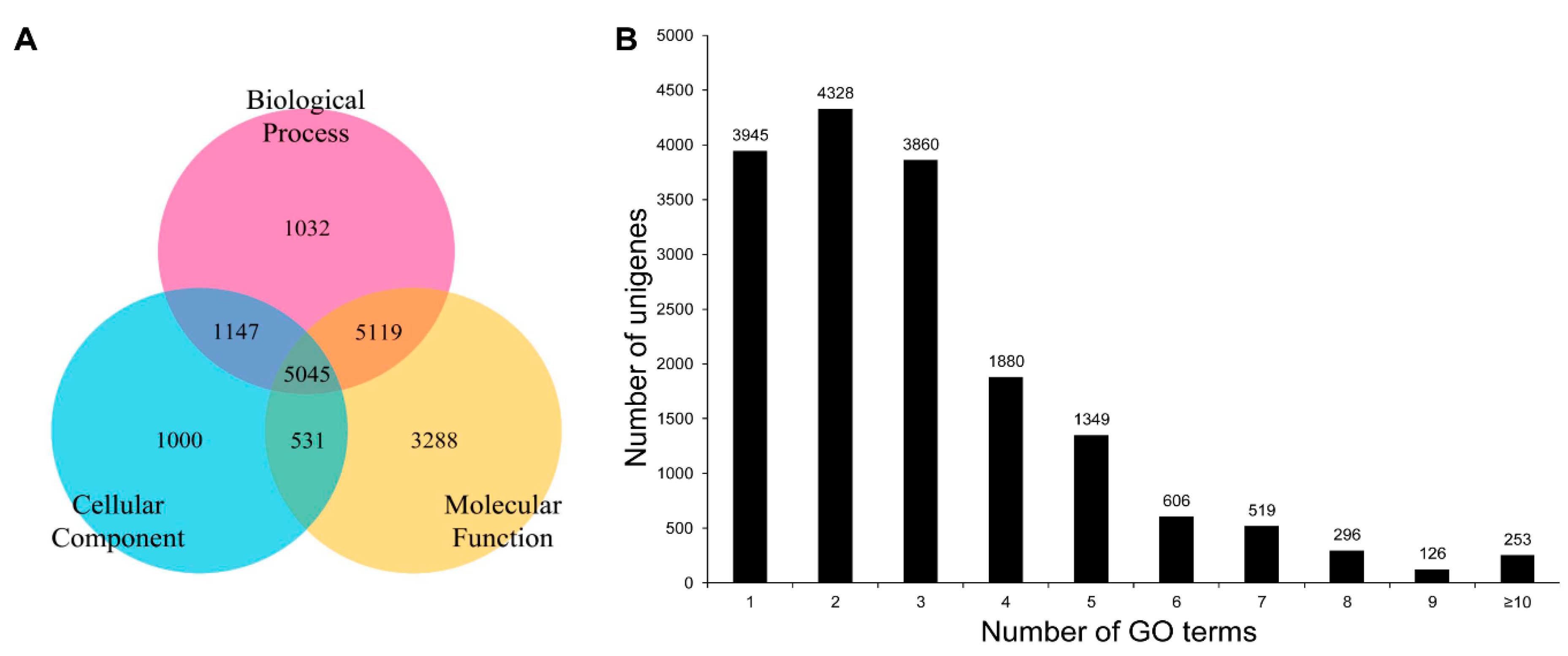
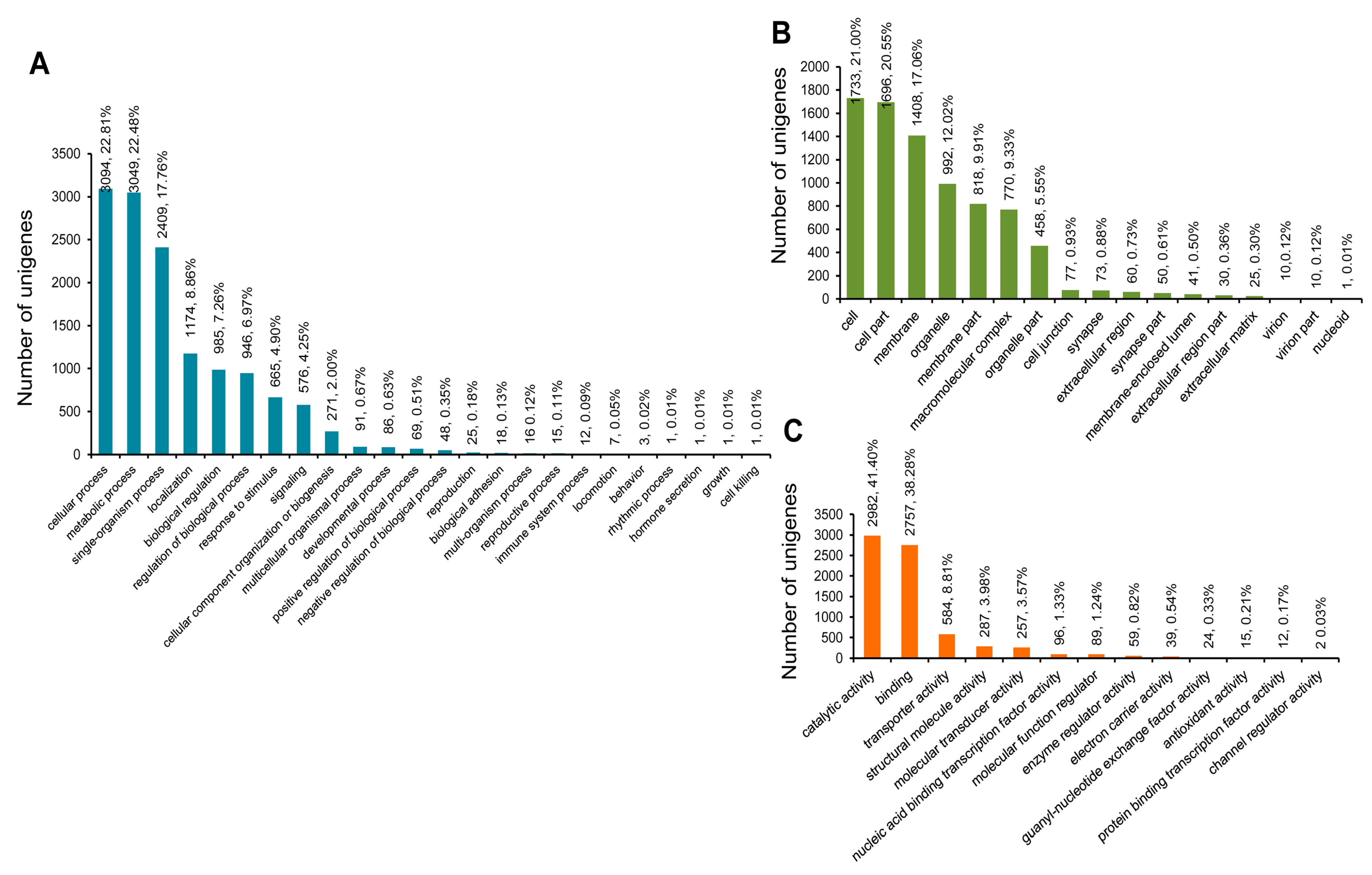
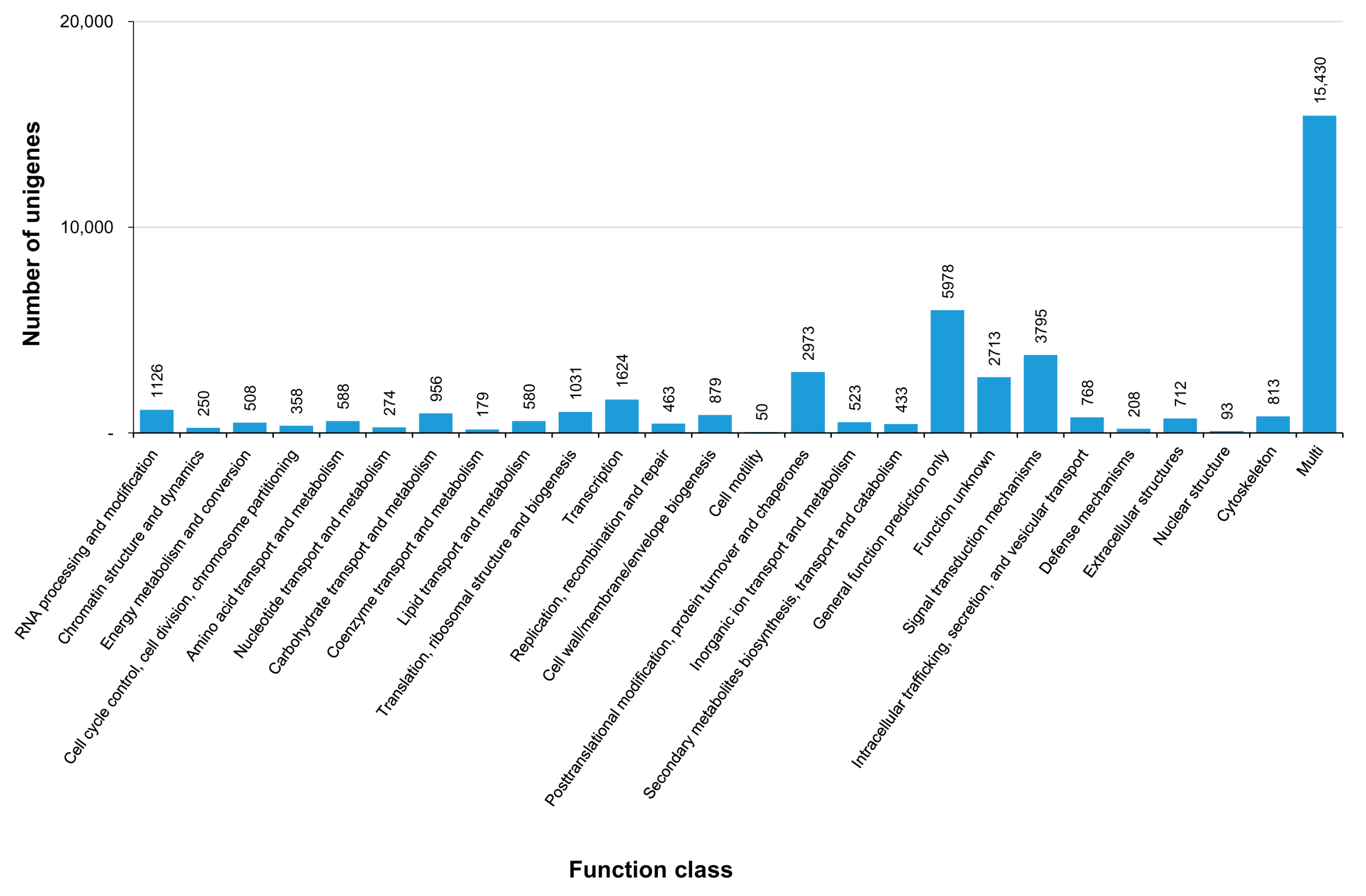
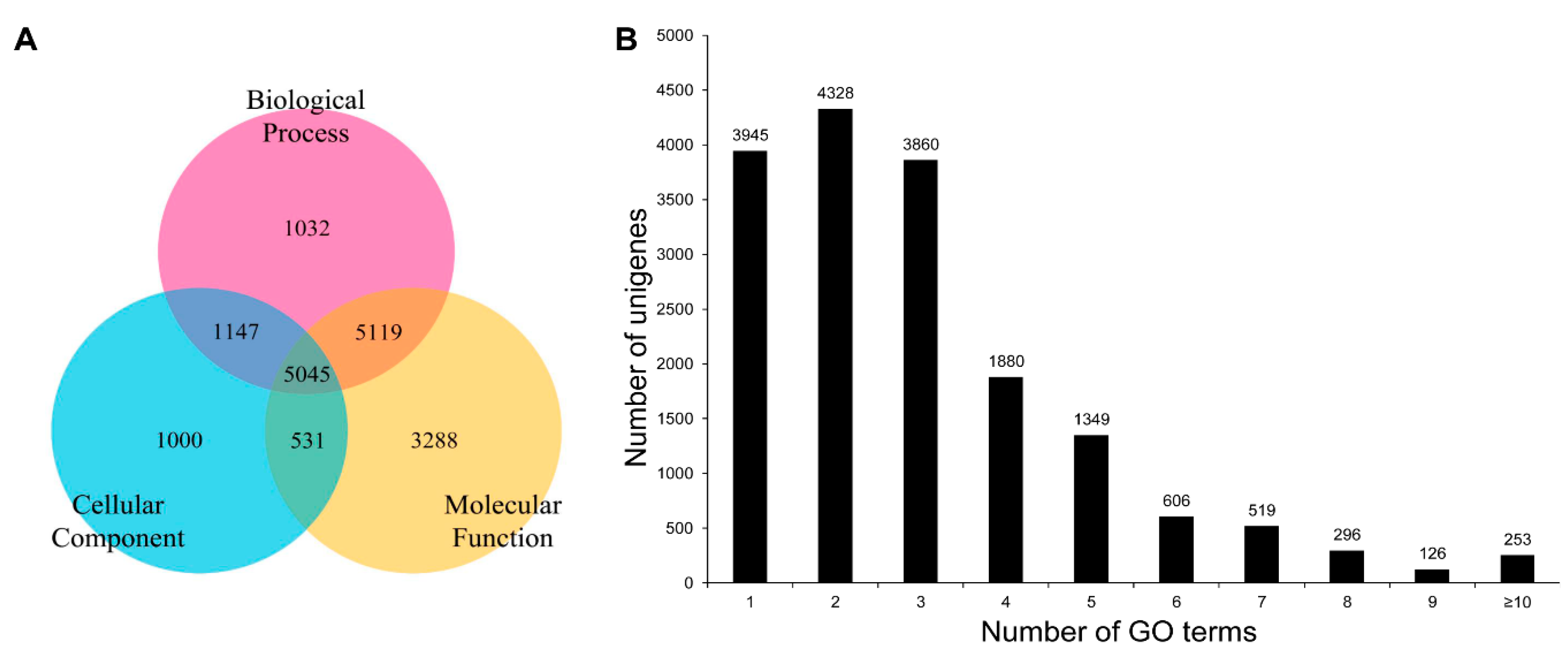
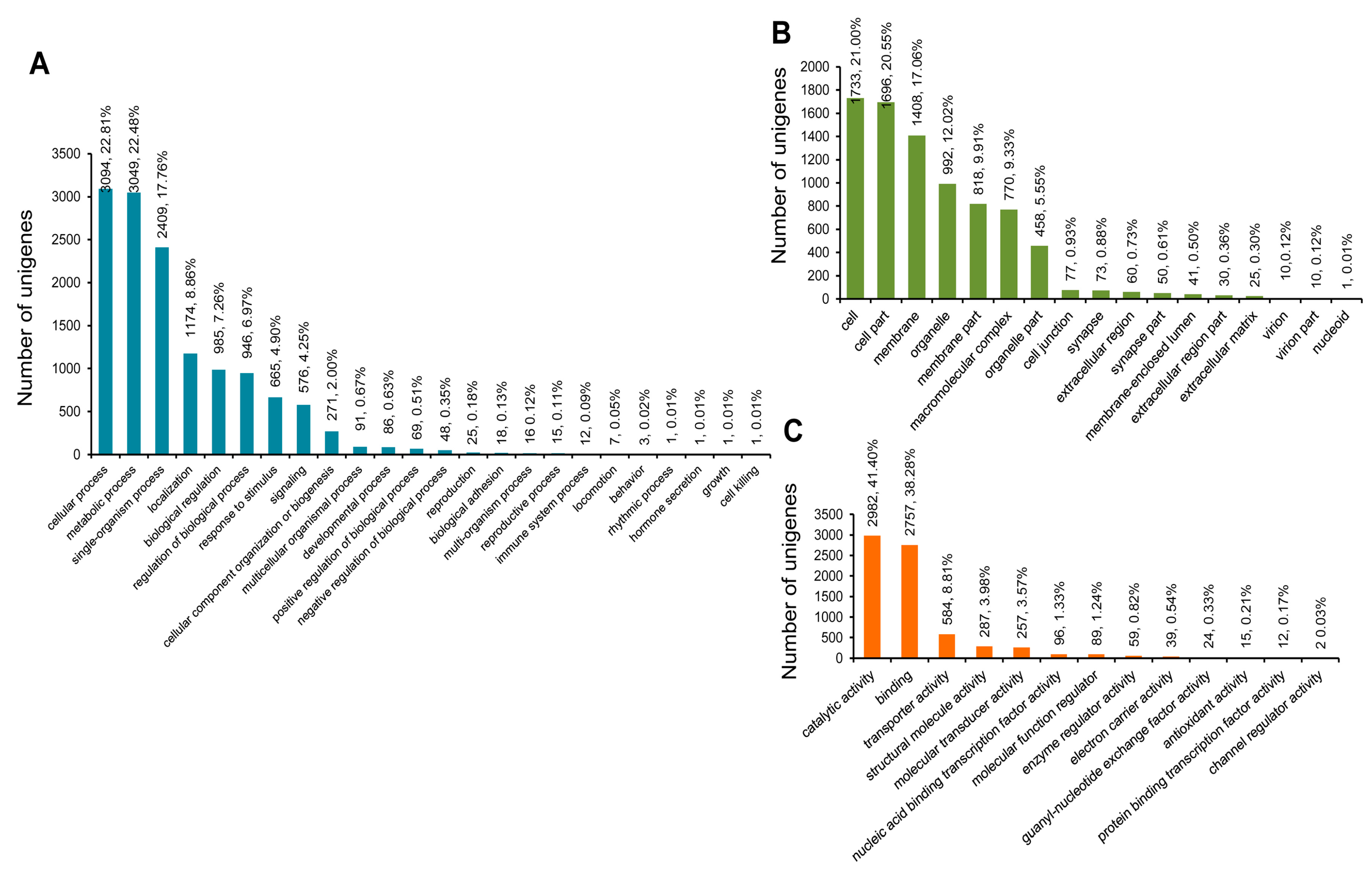
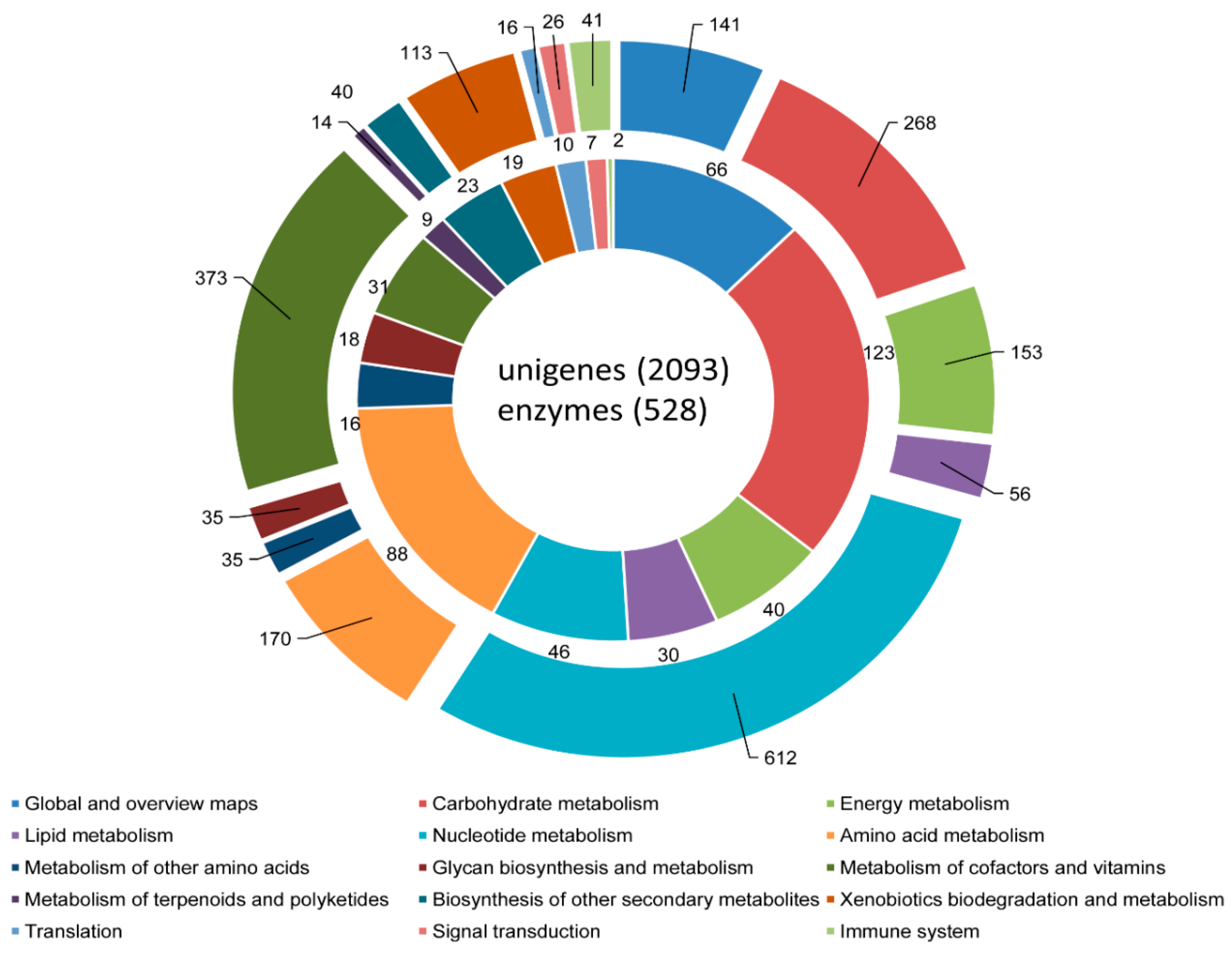
3.3. COG, GO, and KEGG Classifications
3.4. InterProScan Analysis for Conserved Protein Domains
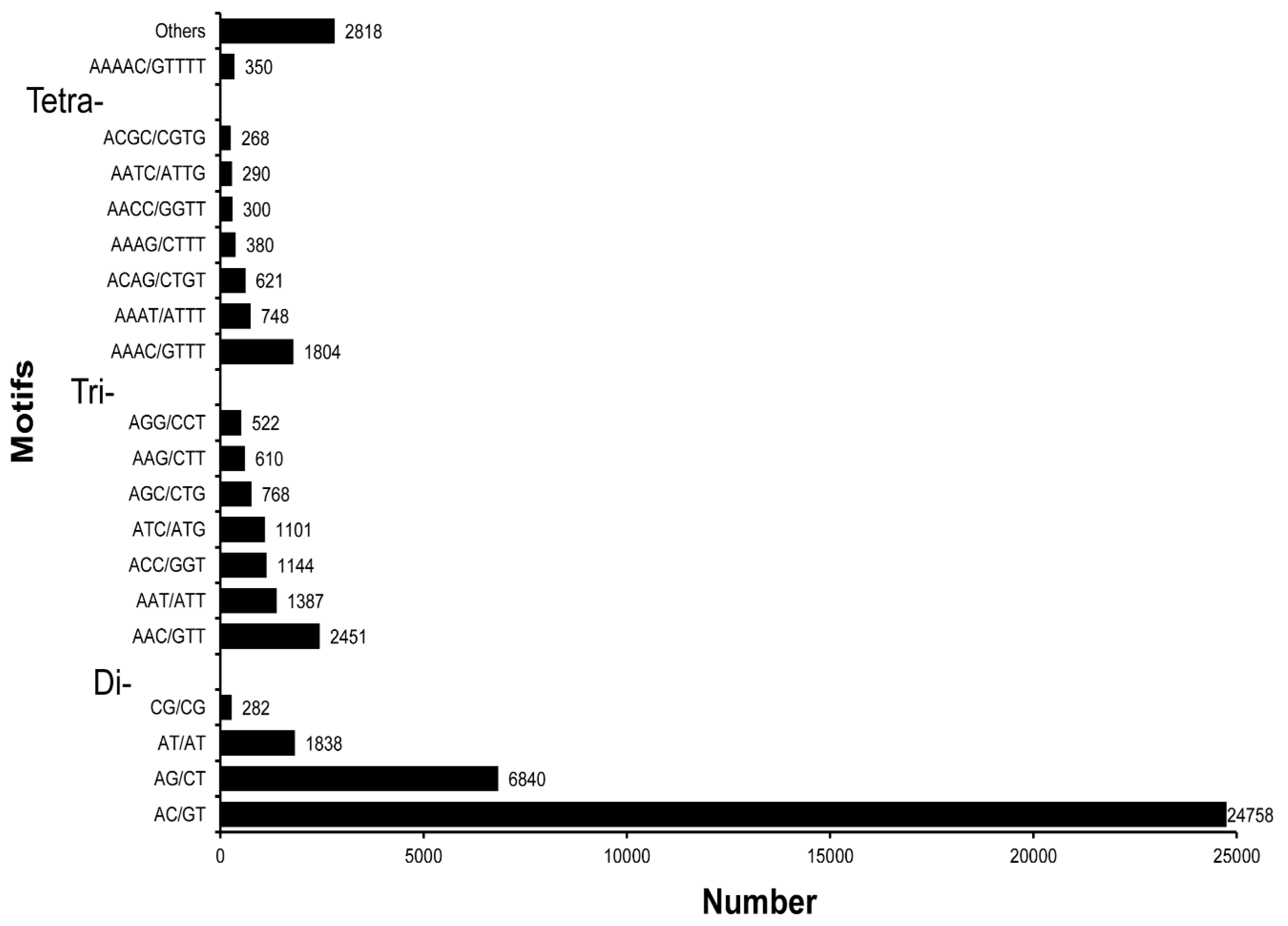
3.5. SSR Characterization and Primer Identification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neubauer, T.; Schneider, S.; Bohme, M.; Prieto, J. First records of freshwater rissooidean gastropods from the paleogene of South-East Asia. J. Mollusc. Stud. 2012, 78, 275–282. [Google Scholar] [CrossRef]
- Chukwuka, C.O.; Ejere, V.C.; Asogwa, C.N.; Nnamonu, E.I.; Okeke, O.C.; Odii, E.I.; Ugwu, G.C.; Okanya, L.C.; Levi, C.A. Eco-physiological adaptation of the land snail Achatina achatina (Gastropoda: Pulmonata) in tropical agro-ecosystem. J. Basic Appl. Zool. 2014, 67, 48–57. [Google Scholar] [CrossRef]
- Hayes, K.A.; Cowie, R.H.; Jorgensen, A.; Schultheib, R.; Albrecht, C.; Thiengo, S.C. Molluscan studies in evolutionary biology: Apple snails (Gastropoda: Ampullariidae) as a system for addressing fundamental questions. Am. Malac. Bull. 2009, 27, 47–58. [Google Scholar] [CrossRef]
- Amin, S.; Prentis, P.J.; Gilding, E.K.; Pavasovic, A. Assembly and annotation of a non-model gastropod (Nerita melanotragus) transcriptome: A comparison of de novo assemblers. BMC Res. Not. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Tomiyama, K. Niche segregation of coexisting two freshwater snail species Semisulcospira libertine (Gould, Prosobranchia: Pleuroceridae) and Clithon retropictus Martens, Prosobranchia: Neritidae). Jpn. J. Malacol. 2000, 59, 135–147. [Google Scholar]
- Furojo, Y.; Tomiyama, K. Distribution and microhabitat of coexisting two freshwater snail species, Semisulcospira libertine (Gould, Prosobranchia: Pleuroceridae) and Clithon retropictus (Martens, Prosobranchia: Neritidae). Jpn. J. Malacol. Venus 2000, 59, 245–260. [Google Scholar]
- Noseworthy, R.G.; Lee, H.-J.; Choi, K.-S. The occurrence of Clithon retropictus (v. Martens, 1879) (Gastropoda: Neritidae) in an unusual habitat, Northern Jeju Island, Republic of Korea. Ocean Sci. J. 2013, 48, 259–262. [Google Scholar] [CrossRef]
- Miyajima, H.; Wada, K. Spatial distribution in relation to life history in the neritid gastropod Clithon retropictus in the Kanzaki River Estuary, Osaka, Japan. Plankton Benthos Res. 2014, 9, 207–216. [Google Scholar] [CrossRef]
- Kwon, O.K.; Park, G.M.; Lee, J.S. Colored Shells of Korea; Academic Publishing Co.: Seoul, Korea, 1993; p. 445. [Google Scholar]
- Kwon, O.G.; Min, D.K.; Lee, J.R.; Lee, J.S.; Je, J.G.; Choe, B.L. Korean Mollusks with Color Illustrations; Hangul Graphics: Busan, Korea, 2001; p. 332. (In Korean) [Google Scholar]
- Min, D.K. Mollusks in Korea; Hanguel Graphics: Busan, Korea, 2004; p. 113. (In Korean) [Google Scholar]
- Noseworthy, R.G.; Ju, S.J.; Choi, K.S. The occurrence of Clithon retropictus (von Martens in Kobelt, 1879, Gastropoda: Neritidae) in Jeju Island, Republic of Korea. Korean J. Malacol. 2012, 28, 81–90. [Google Scholar] [CrossRef]
- Malmqvist, B.; Rundle, S. Threats to the running water ecosystems of the world. Environ. Conserv. 2002, 29, 78–107. [Google Scholar] [CrossRef]
- Primm, S.L.; Dollar, L.; Bass, O.L., Jr. The genetic rescue of Florida panther. Animal Conserv. 2006, 9, 115–122. [Google Scholar]
- Ellstrand, N.C.; Biggs, D.; Kaus, A.; Lubinsky, P.; McDade, L.A.; Preston, K.; Prince, L.M.; Regan, H.M.; Rorive, V.; Ryder, O.A.; et al. Got hybridization? A multidisciplinary approach for informing science policy. BioScience 2010, 60, 384–388. [Google Scholar] [CrossRef]
- Wang, X.W.; Zhao, Q.Y.; Luan, J.B.; Yan, G.H.; Liu, S.S. Analysis of a native whitefly transcriptome and its sequence divergence with two invasive whitefly species. BMC Genomics 2012. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, P.; Lu, Y.; Kumar, N.; Creasy, T.; Daugherty, S.; Chibucos, M.C.; Orvis, J.; Shetty, A.; Ott, S.; Flowers, M.; et al. Rapid transcriptome analysis of an invasive pest, the brown marmorated stink bug Halyomorpha halys. BMC Genomics 2014. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.F.; Sherman, C.D.H. De novo assembly and characterization of the invasive Northern Pacific Seastar transcriptome. PLoS ONE 2015, 10, e0142003. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Patnaik, B.B.; Hwang, H.-J.; Park, S.Y.; Wang, T.H.; Park, E.B.; Chung, J.M.; Song, D.K.; Patnaik, H.H.; Lee, J.B.; et al. De novo transcriptome generation and annotation for two Korean endemic land snails, Aegista chejuensis and Aegista quelpartensis, using Illumina paired-end sequencing technology. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, B.B.; Wang, T.H.; Kang, S.W.; Hwang, H.-J.; Park, S.Y.; Park, E.B.; Chung, J.M.; Song, D.K.; Kim, C.; Kim, S.; et al. Sequencing, de novo assembly, and annotation of the transcriptome of the endangered freshwater pearl bivalve, Cristaria plicata, provides novel insights into functional genes and marker discovery. PLoS ONE 2016, 11, e0148622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, S.; Duan, J.; Wang, J.; Chen, S.; Cheng, Z.; Zhang, Q.; Liang, X.; Li, Y. De novo assembly and characterization of the transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogea L.). BMC Genomics 2012. [Google Scholar] [CrossRef]
- Castellanos-Martinez, S.; Arteta, D.; Catarino, S.; Gestal, C. De novo transcriptome sequencing of the Octopus vulgaris hemocytes using Illumina RNA-Seq technology: Response to the infection by the gastrointestinal parasite Aggregata octopiana. PLoS ONE 2014, 9, e107873. [Google Scholar]
- Blankenberg, D.; Gordon, A.; von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A. The Galaxy Team. Manipulation of FASTQ data with Galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices Clustering Tool (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulorious, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLASTX+ architectures and structure. BMC Bioinform. 2009. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucl. Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopaedia of Genes and Genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version including eukaryotes. BMC Bioinform. 2003. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.-C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer 3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008. [Google Scholar] [CrossRef] [PubMed]
- Prentis, P.J.; Pavasovic, A. The Anadara trapezia transcriptome: A resource for molluscan physiological genomics. Mar. Genomics 2014, 18, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hui, J.H.L.; Chan, T.F.; Chu, K.H. De novo transcriptome sequencing of the snail Echinolittorina malaccana: Identification of genes responsive to thermal stress and development of genetic markers for population studies. Mar. Biotechnol. 2014, 16, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, Y.; Xu, G.; Xie, L.; Hu, X.; Bao, Z.; Zhang, R. Characterization of the Zhikong scallop (Chlamys farreri) mantle transcriptome and identification of biomineralization-related genes. Mar. Biotechnol. 2013, 15, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Weng, S.; Chen, Y.; Yu, X.; Lu, L.; Zhang, H.; He, J.; Xu, X. Analysis of Litopenaeus vannamei transcriptome using the next-generation DNA sequencing technique. PLoS ONE 2012, 7, e47442. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.H.; Ryu, T.; Seridi, L.; Ghosheh, Y.; Bougouffa, S.; Qian, P.-Y.; Ravasi, T. Transcriptome analysis elucidates key developmental components of bryozoan lophophore development. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, B.B.; Hwang, H.-J.; Kang, S.W.; Park, S.Y.; Wang, T.H.; Park, E.B.; Chung, J.M.; Song, D.K.; Kim, C.; Kim, S.; et al. Transcriptome characterization of non-model endangered lycaenids, Protantigius superans and Spindasis takanosis, using Illumina Hi-Seq 2500 sequencing. Int. J. Mol. Sci. 2015, 16, 29948–29970. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Park, S.Y.; Patnaik, B.B.; Hwang, H.J.; Kim, C.; Kim, S.; Lee, J.S.; Han, Y.S.; Lee, Y.S. Construction of PANM Database (Protostome DB) for rapid annotation of NGS data in Mollusks. Korean J. Malacol. 2015, 31, 243–247. [Google Scholar] [CrossRef]
- Mittapalli, O.; Bai, X.; Mamidala, P.; Rajarapu, S.P.; Bonello, P.; Herms, D.A. Tissue specific transcriptomics of the exotic invasive insect pest Emerald Ash Borer (Agrilus planipennis). PLoS ONE 2010, 5, e13708. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wang, B.; Pan, L.; Ye, Y.; He, M.; Han, S.; Zheng, S.; Wang, X.; Lin, Y. Comprehensive structural annotation of Pichia pastoris transcriptome and the response to various carbon sources using deep-paired RNA sequencing. BMC Genomics 2012. [Google Scholar] [CrossRef] [PubMed]
- Simakov, O.; Marletaz, F.; Cho, S.J.; Edsinger-Gonzales, E.; Havlak, P.; Hellsten, U.; Kuo, D.H.; Larsson, T.; Lv, J.; Arendt, D.; et al. Insights into bilateria evolution from three spiralian genomes. Nature 2013, 493, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Heyland, A.; Vue, Z.; Voolstra, C.R.; Medina, M.; Moroz, L.L. Developmental transcriptome of Aplysia californica. J. Exp. Zool. B Mol. Dev. Ecol. 2011, 316, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Consea, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucl. Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Du Plessis, L.; Skunca, N.; Dessimoz, C. The what, where, how and why of gene ontology—A primer for bioinformaticians. Brief Bioinform. 2011, 12, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Lovering, R.C.; Camon, E.B.; Blake, J.A.; Diehl, A.D. Access to immunology through the Gene Ontology. Immunology 2008, 125, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Wood, V.; Dolinski, K.; Draghici, S. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 2008, 9, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Barell, D.; Dimmer, E.; Huntley, R.P.; Binns, D.; O’Donovan, C.; Apweiler, R. The GOA database in 2009—An integrated Gene Ontology Annotation resource. Nucl. Acids Res. 2009, 37, D396–D403. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopaedia of Genes and Genomes. Nucl. Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Annadurai, R.S.; Neethiraj, R.; Jayakumar, V.; Damodaran, A.C.; Rao, S.N.; Katta, M.A.V.S.K.; Gopinathan, S.; Sarma, S.P.; Senthilkumar, V.; Niranjan, V.; et al. De novo transcriptome assembly (NGS) of Curcuma longa L. rhizome reveals novel transcripts related to anticancer and antimalarial terpenoids. PLoS ONE 2013, 8, e56217. [Google Scholar]
- Lv, J.; Liu, P.; Gao, B.; Wang, Y.; Wang, Z.; Chen, P.; Li, J. Transcriptome analysis of the Portunus trituberculatus: De novo assembly, growth related gene identification and marker discovery. PLoS ONE 2014, 9, e94055. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ma, C.; Li, S.; Jiang, W.; Li, X.; Liu, Y.; Ma, L. Transcriptome analysis of the Mud Crab (Scylla paramamosain) by 454 deep sequencing: Assembly, annotation and marker discovery. PLoS ONE 2014, 9, e102668. [Google Scholar] [CrossRef] [PubMed]
- Khush, R.S.; Leulier, F.; Lemaitre, B. Drosophila immunity: Two paths to NF-ĸB. Trends Immunol. 2001, 22, 260–264. [Google Scholar] [CrossRef]
- Brayer, K.J.; Segal, D.J. Keep your fingers off my DNA: Protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys. 2008, 50, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Pairett, A.N.; Serb, J.M. De novo assembly and characterization of two transcriptomes reveal multiple light-mediated functions in the Scallop eye (Bivalvia: Pectinidae). PLoS ONE 2013, 8, e69852. [Google Scholar] [CrossRef] [PubMed]
- Albertin, C.B.; Simakov, O.; Mitros, T.; Wang, Z.Y.; Pungor, J.R.; Edsinger-Gonzalez, E.; Brenner, S.; Ragsdale, C.W.; Rokhsar, D.S. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 2015, 524, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Huang, M.; Zhang, H.; Song, L. The immune role of C-type lectins in molluscs. ISJ 2011, 8, 241–246. [Google Scholar]
- Wang, X.; Li, J.; Li, Y. Isolation and characterization of Microsatellite markers for an endemic tree in East Asia, Quercus variabilis (Fagaceae). Appl. Plant Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Genetics and conservation biology. Comptes Rendus 2003, 326, S22–S29. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptome Summary | |
| Number of raw read sequences | 246,743,798 |
| Number of raw read bases | 31,089,718,548 |
| Number of clean read sequences | 241,696,750 |
| Number of clean read bases | 30,038,741,871 |
| Mean length of contig (bp) | 124.3 |
| N50 length of contig (bp) | 126 |
| GC % of contig | 50.93 |
| High-quality reads (%) | 97.95 (sequences), 96.62 (bases) |
| Contig Information | |
| Total number of contigs | 503,882 |
| Number of bases | 330,800,003 |
| Mean length of contig (bp) | 656.5 |
| N50 length of contig (bp) | 953 |
| GC % of contig | 46.80 |
| Largest contig (bp) | 22,900 |
| No. of large contigs (≥500 bp) | 164,129 |
| Unigene Information | |
| Total number of unigenes | 282,838 |
| Number of bases | 208,418,920 |
| Mean length of unigene (bp) | 736.9 |
| N50 length of unigene (bp) | 1201 |
| GC % of unigene | 46.61 |
| Length ranges (bp) | 110–22,900 |
| Database | All | ≤300 bp | 300–1000 bp | ≥1000 bp |
|---|---|---|---|---|
| PANM | 94,634 | 17,610 | 43,881 | 33,143 |
| Unigene | 78,825 | 16,601 | 38,056 | 24,168 |
| COG | 43,306 | 5916 | 15,559 | 21,831 |
| GO | 17,162 | 3702 | 7915 | 5545 |
| KEGG | 896 | 138 | 240 | 518 |
| All Database | 125,616 | 26,229 | 61,447 | 37,940 |
| Domain | Short Name | Description | Number of Unigenes |
|---|---|---|---|
| IPR015880 | Znf_C2H2-like | Zinc finger, C2H2-like domain | 1141 |
| IPR002110 | Ankyrin_rpt | Ankyrin repeat | 862 |
| IPR027417 | P_loop_NTPase | P-loop containing nucleoside triphosphate hydrolase domain | 566 |
| IPR013783 | Ig-like_fold | Immunoglobulin-like fold domain | 475 |
| IPR020683 | Ankyrin_rpt-contain_dom | Ankyrin repeat-containing domain | 360 |
| IPR000477 | RT_dom | Reverse transcriptase domain | 338 |
| IPR003599 | Ig_sub | Immunoglobulin subtype domain | 307 |
| IPR000504 | RRM_dom | RNA recognition motif domain | 271 |
| IPR002290 | Ser/Thr_dual-sp_kinase | Serine/threonine/dual specificity protein kinase, catalytic domain | 257 |
| IPR000742 | EGF-like_dom | EGF-like domain | 244 |
| IPR011989 | ARM-like | Armadillo-like helical domain | 206 |
| IPR005135 | Endonuclease/exonuclease/ phosphatase | Endonuclease/exonuclease/phosphatase domain | 202 |
| IPR000276 | GPCR_rhodopsn | G protein-coupled receptor, rhodopsin-like family | 198 |
| IPR001680 | WD40_repeat | WD40 repeat | 187 |
| IPR003598 | Ig_sub2 | Immunoglobulin subtype 2 domain | 181 |
| IPR001841 | Znf_RING | Zinc finger, RING-type domain | 176 |
| IPR002048 | EF_hand_dom | EF-hand domain | 161 |
| IPR001881 | EGF-like_Ca-bd_dom | EGF-like calcium-binding domain | 160 |
| IPR001304 | C-type_lectin | C-type lectin domain | 159 |
| IPR002035 | VWF_A | von Willebrand factor, type A domain | 158 |
| SSR parameters | Numbers identified |
|---|---|
| Total number of sequences examined | 48,973 |
| Total size of examined sequences (bp) | 106,535,022 |
| Total number of identified SSRs | 49,280 |
| Number of SSR containing sequences | 25,365 |
| Number of sequences containing more than 1 SSR | 12,546 |
| Number of SSRs present in compound formation | 11,278 |
| Unit size | Number of SSRs |
| 2 | 33,718 |
| 3 | 8373 |
| 4 | 5756 |
| 5 | 1180 |
| 6 | 253 |
| Repeats | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | ≥21 | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Di | 0 | 0 | 7713 | 5048 | 3566 | 2579 | 2119 | 2543 | 2180 | 676 | 811 | 710 | 614 | 556 | 504 | 479 | 435 | 3185 | 33,718 |
| Tri | 0 | 4165 | 1899 | 940 | 729 | 123 | 139 | 74 | 51 | 43 | 26 | 28 | 35 | 28 | 20 | 10 | 23 | 40 | 8373 |
| Tetra | 3461 | 1263 | 680 | 91 | 77 | 51 | 29 | 15 | 28 | 15 | 6 | 6 | 8 | 6 | 3 | 4 | 0 | 13 | 5756 |
| Penta | 740 | 220 | 39 | 34 | 25 | 27 | 27 | 17 | 13 | 11 | 7 | 11 | 3 | 2 | 0 | 0 | 0 | 4 | 1180 |
| Hexa | 222 | 18 | 9 | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 253 |
| Total | 4423 | 5666 | 10,340 | 6114 | 4398 | 2782 | 2314 | 2649 | 2272 | 745 | 850 | 755 | 660 | 592 | 527 | 493 | 458 | 3242 | 49,280 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.Y.; Patnaik, B.B.; Kang, S.W.; Hwang, H.-J.; Chung, J.M.; Song, D.K.; Sang, M.K.; Patnaik, H.H.; Lee, J.B.; Noh, M.Y.; et al. Transcriptomic Analysis of the Endangered Neritid Species Clithon retropictus: De Novo Assembly, Functional Annotation, and Marker Discovery. Genes 2016, 7, 35. https://doi.org/10.3390/genes7070035
Park SY, Patnaik BB, Kang SW, Hwang H-J, Chung JM, Song DK, Sang MK, Patnaik HH, Lee JB, Noh MY, et al. Transcriptomic Analysis of the Endangered Neritid Species Clithon retropictus: De Novo Assembly, Functional Annotation, and Marker Discovery. Genes. 2016; 7(7):35. https://doi.org/10.3390/genes7070035
Chicago/Turabian StylePark, So Young, Bharat Bhusan Patnaik, Se Won Kang, Hee-Ju Hwang, Jong Min Chung, Dae Kwon Song, Min Kyu Sang, Hongray Howrelia Patnaik, Jae Bong Lee, Mi Young Noh, and et al. 2016. "Transcriptomic Analysis of the Endangered Neritid Species Clithon retropictus: De Novo Assembly, Functional Annotation, and Marker Discovery" Genes 7, no. 7: 35. https://doi.org/10.3390/genes7070035
APA StylePark, S. Y., Patnaik, B. B., Kang, S. W., Hwang, H.-J., Chung, J. M., Song, D. K., Sang, M. K., Patnaik, H. H., Lee, J. B., Noh, M. Y., Kim, C., Kim, S., Park, H. S., Lee, J. S., Han, Y. S., & Lee, Y. S. (2016). Transcriptomic Analysis of the Endangered Neritid Species Clithon retropictus: De Novo Assembly, Functional Annotation, and Marker Discovery. Genes, 7(7), 35. https://doi.org/10.3390/genes7070035