
Selection and Verification of Candidate Reference Genes for Mature MicroRNA Expression by Quantitative RT-PCR in the Tea Plant (Camellia sinensis)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Experimental Stress Treatments
2.2. Selection of Candidate Reference Genes
2.3. Primer Design for Reverse Transcription of ncRNAs
2.4. RNA extraction, cDNA synthesisand qRT-PCR protocol for ncRNAs
2.5. Evaluation of Expression Stability of Candidate Reference Genes
2.6. Evaluation of Utility of the Candidate Reference Genes
3. Results and Discussion
3.1. Verification of Amplification Specificity and Efficiency of the Primer Sets
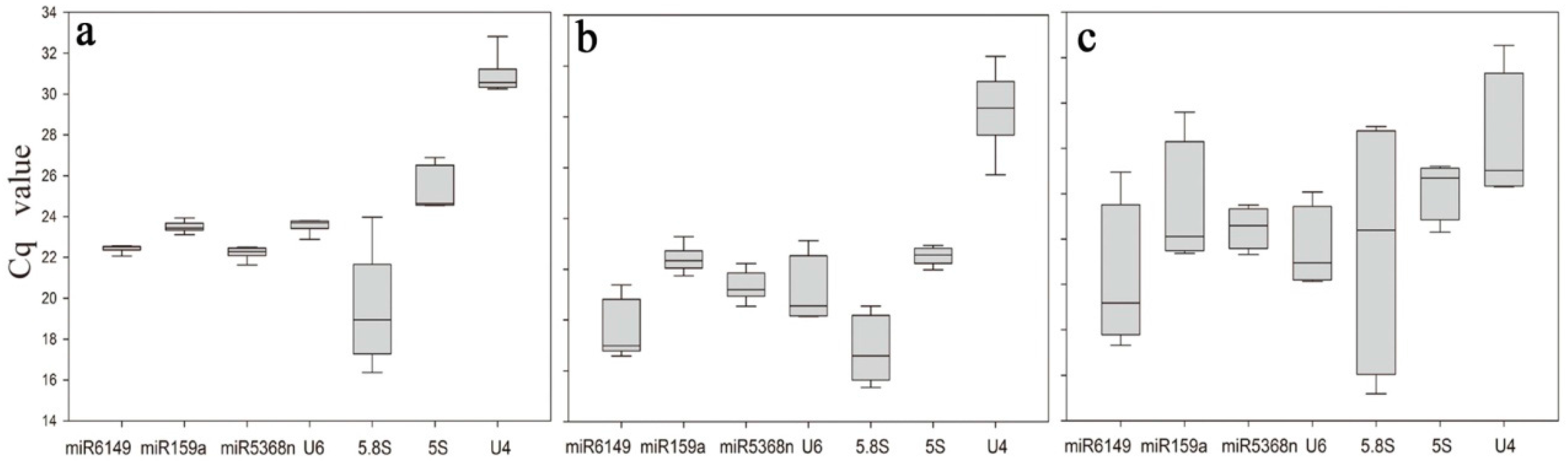
3.2. Description of Expression Profiling of Candidate Reference Genes
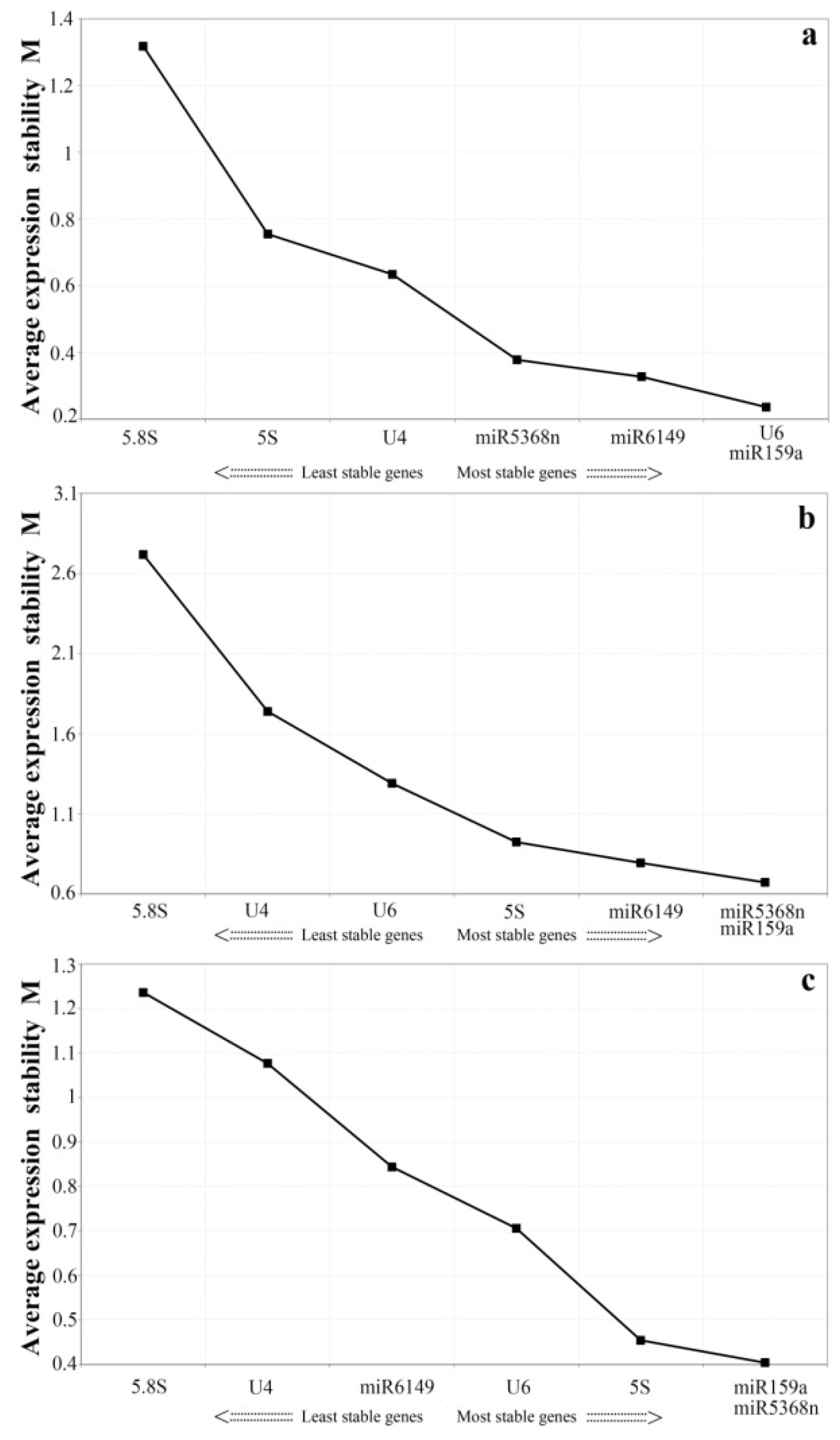
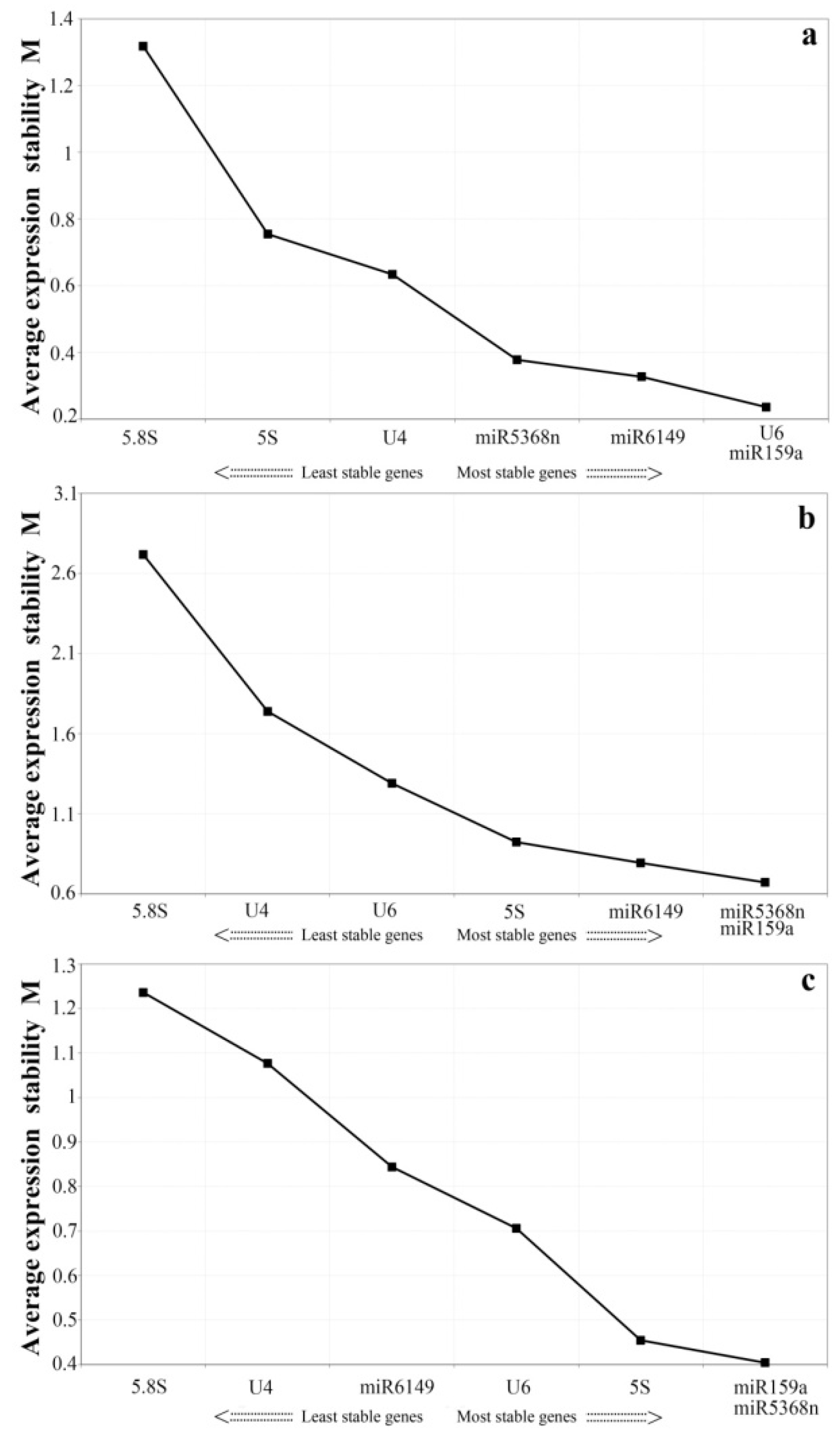
3.3. Expression Stability Analysis of All Candidate Reference Genes by GeNorm and NormFinder
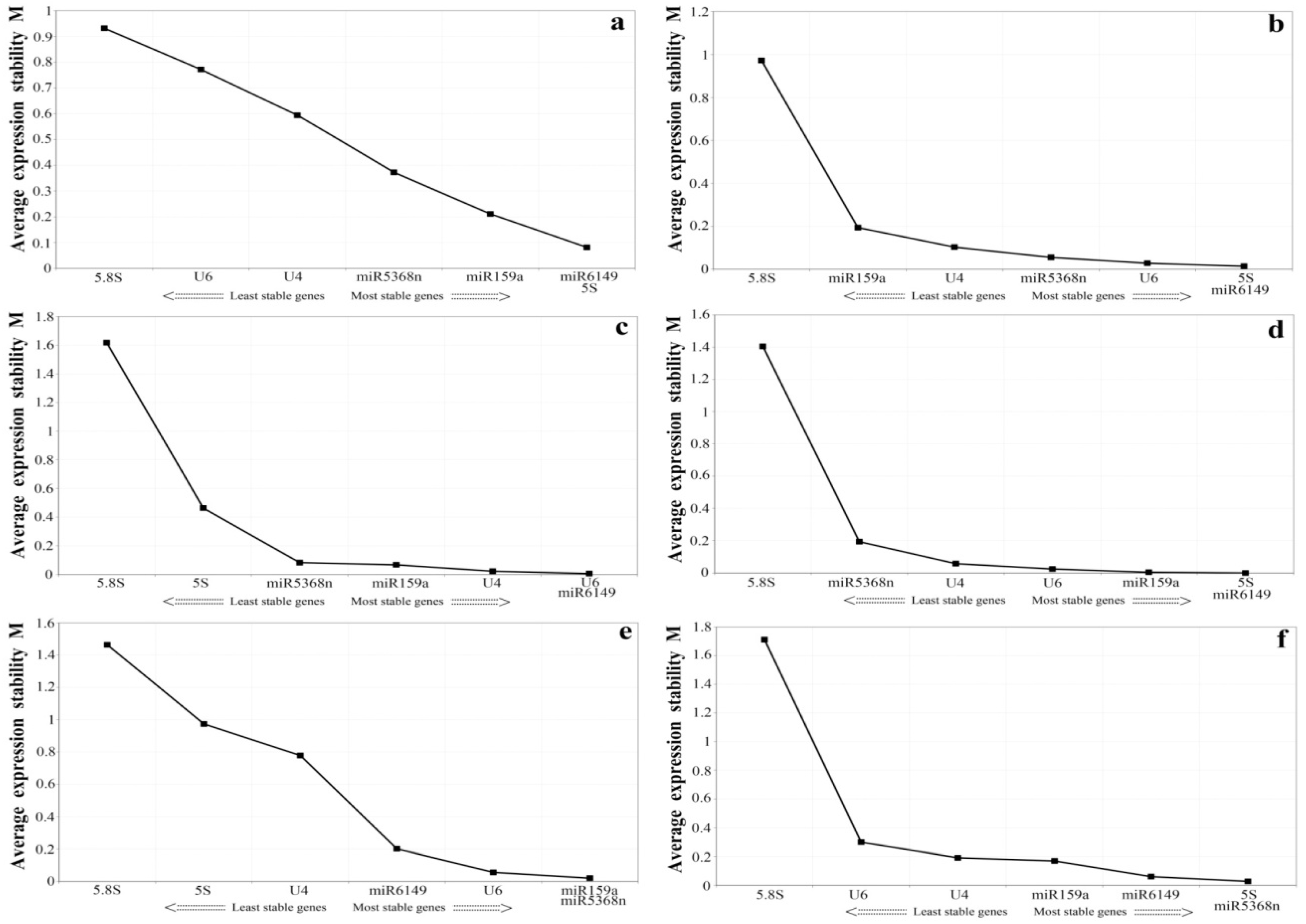
3.4. Expression Stability Assessment of Candidate Reference Genes under Experimental Stresses and among the Bud to the Second Leaf
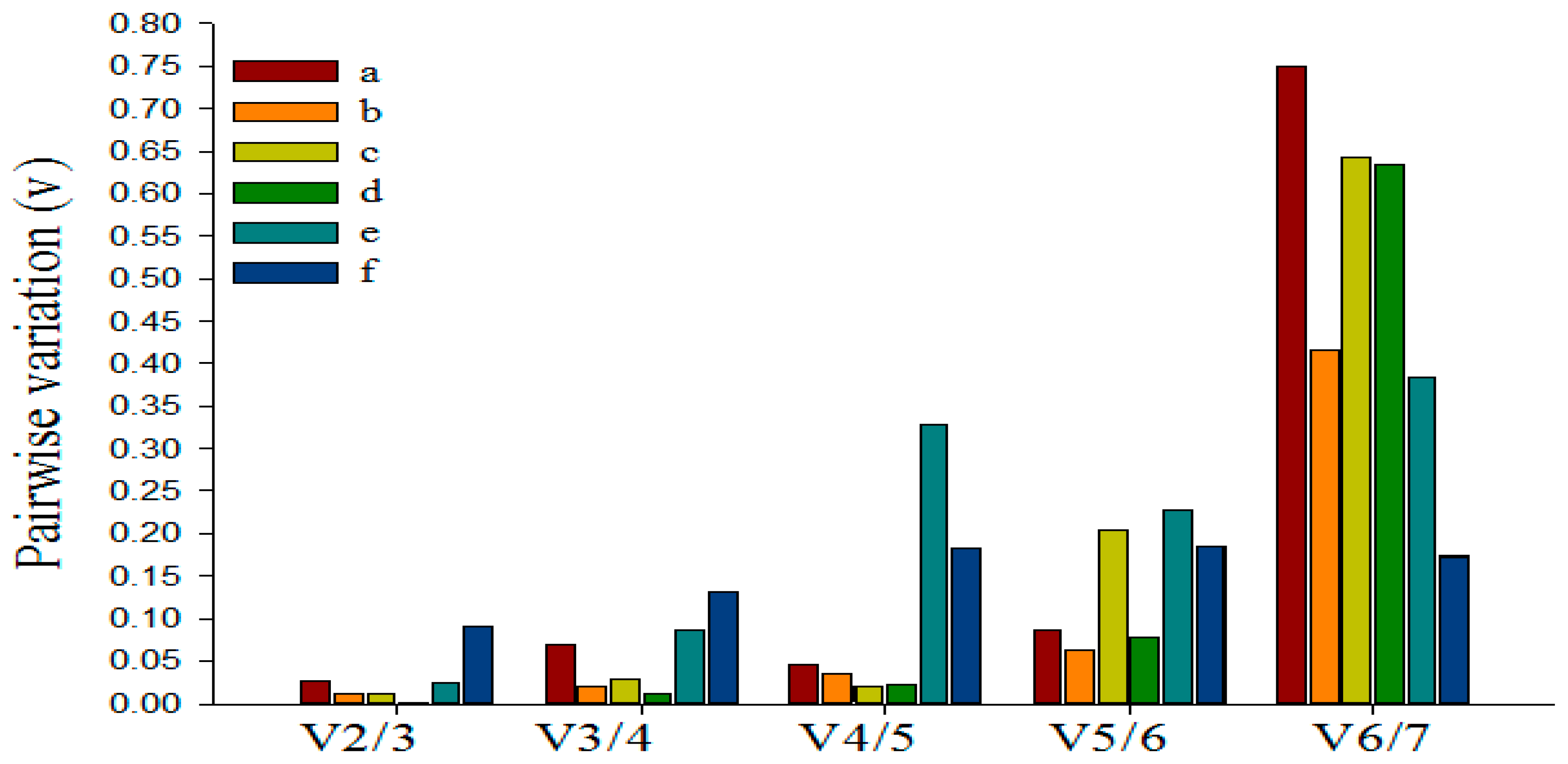
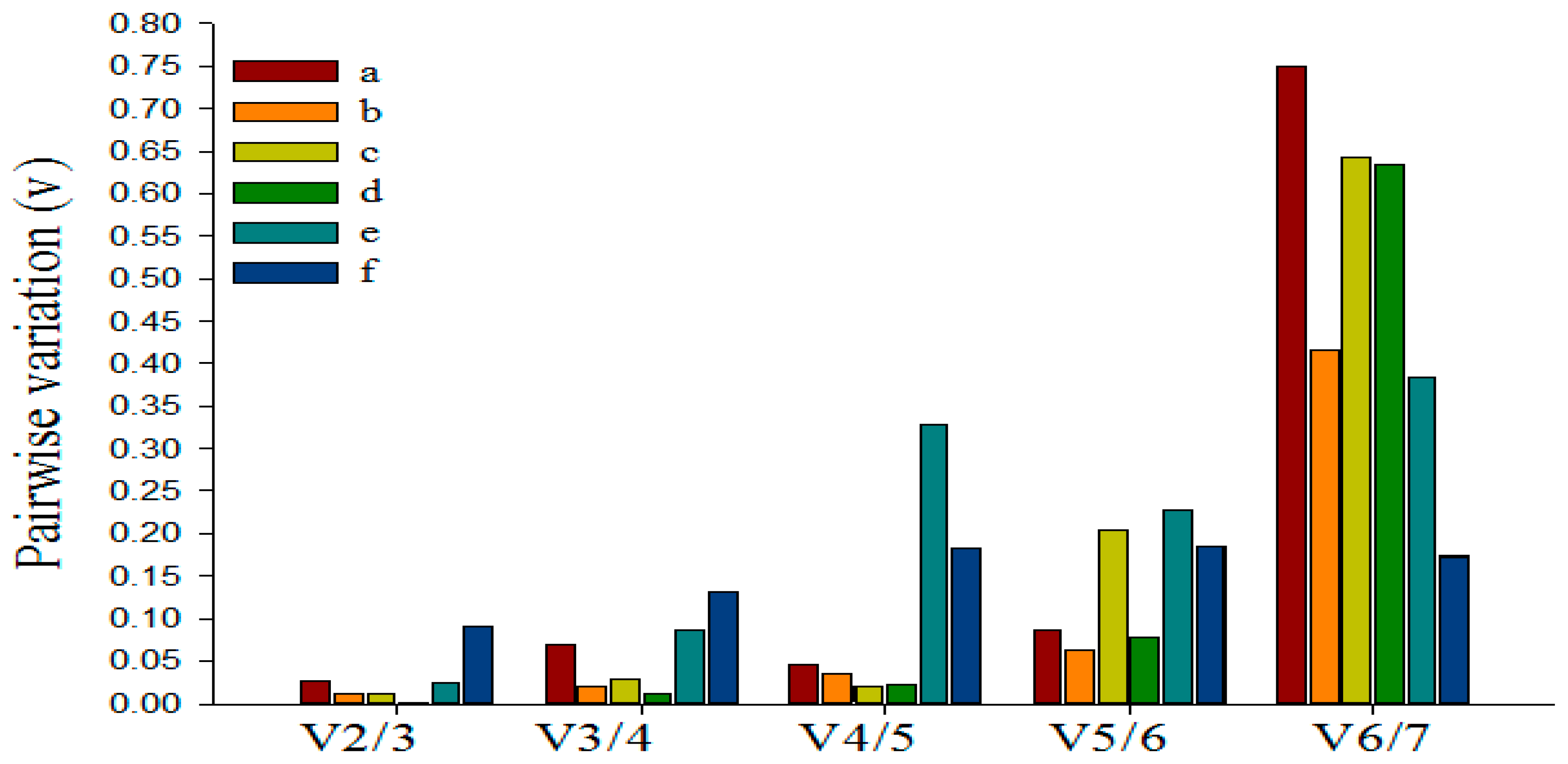
3.5. Determining the Minimum Number of Reference Genes Using GeNorm
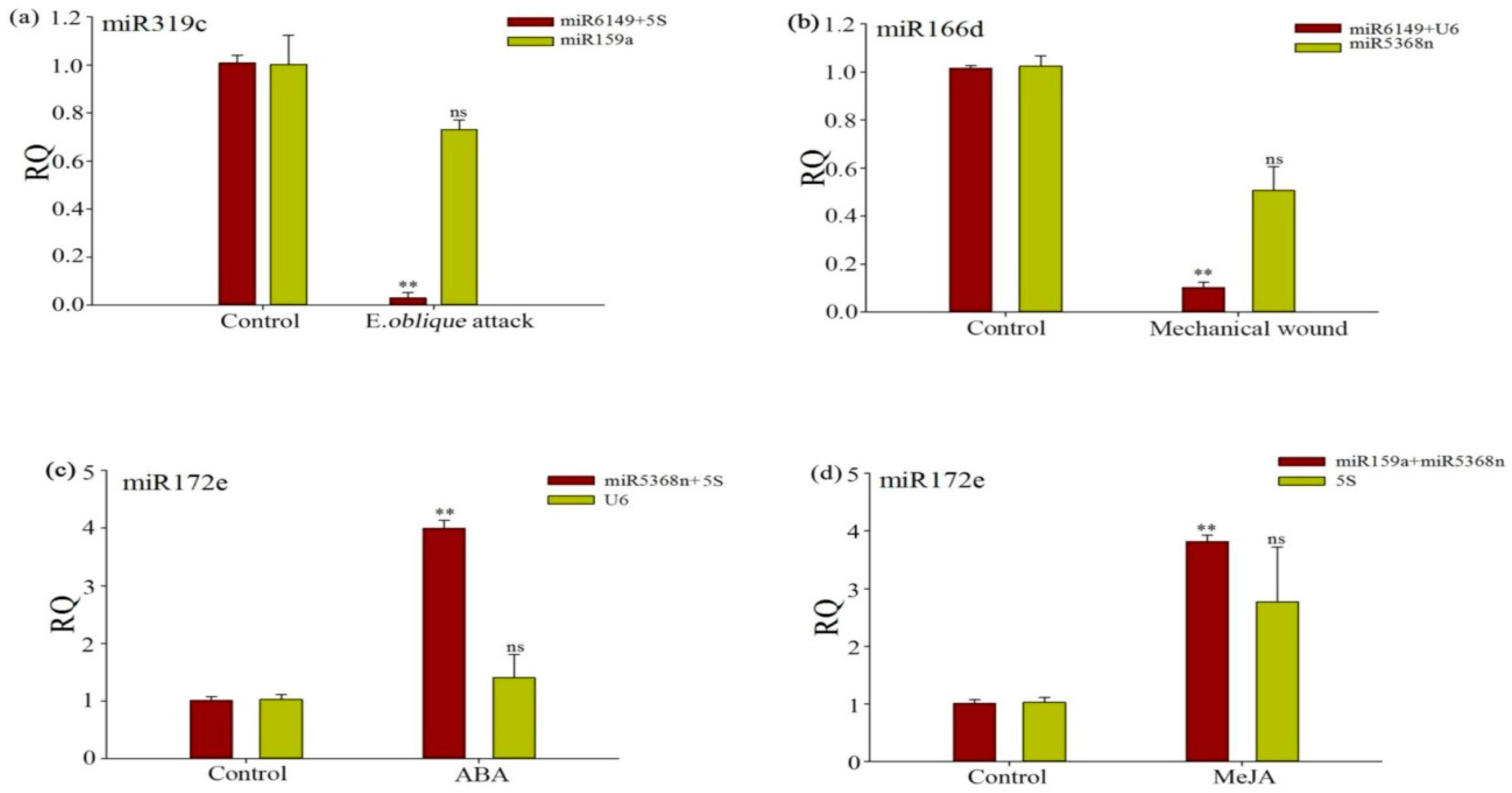
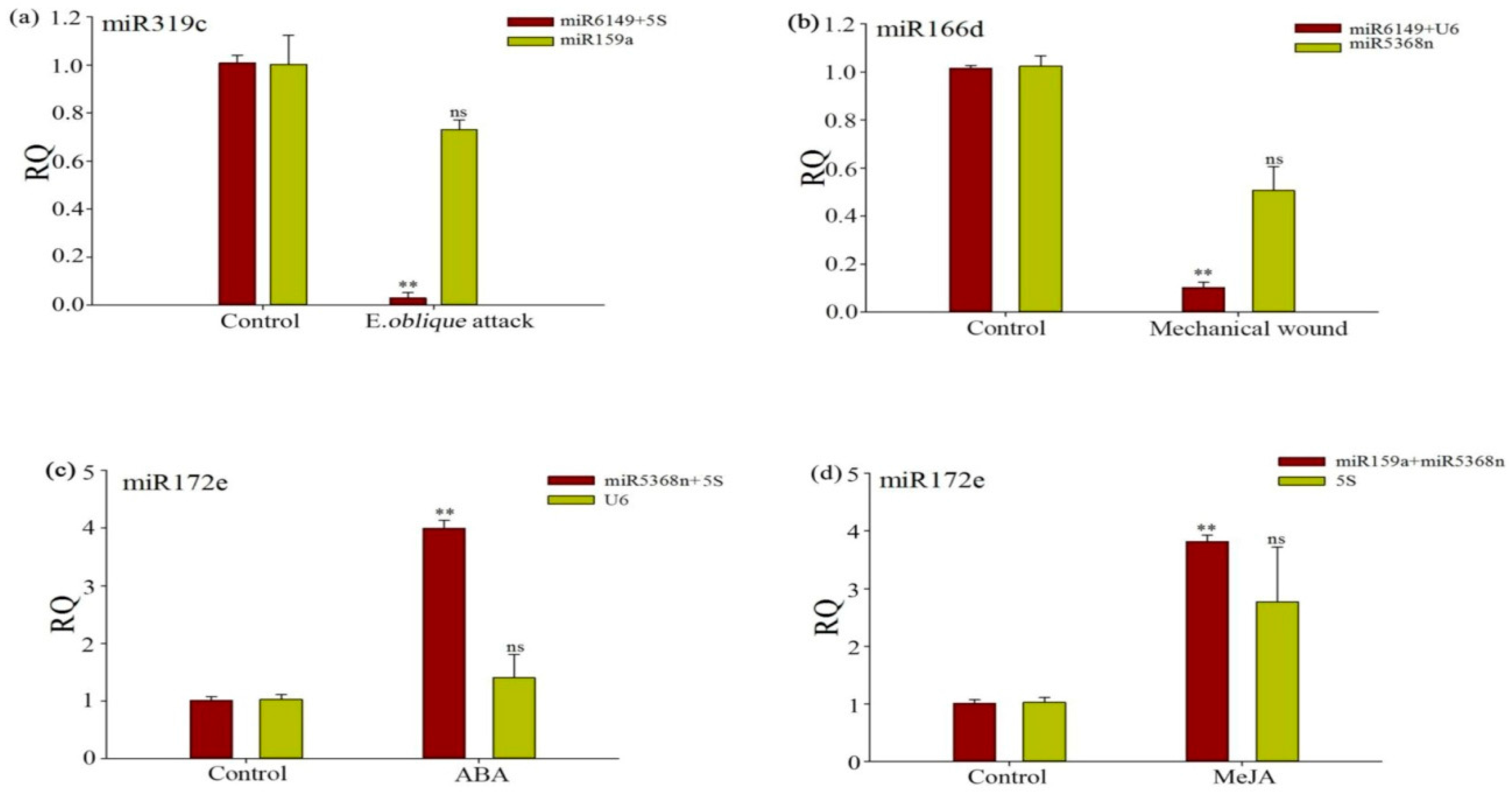
3.6. Evaluation of the Validity of a Selected Reference Gene Normalizing a Target Gene under Different Experimental Conditions
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5.8S | 5.8S ribosomal RNA |
| 5S | 5S ribosomal RNA |
| U6 | U6 small nucleolar RNA |
| U4 | U4 small nucleolar RNA |
| ABA | Abscisic acid |
| miRNA | microRNA |
| ncRNA | non-coding RNA |
| MeJA | Methyl Jasmonate |
| qRT-PCR | quantitative reverse transcription-polymerase chain reaction |
References
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell. Dev. Biol. 2009, 25, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Shukla, L.I.; Chinnusamy, V.; Sunkar, R. The role of microRNAs and other endogenous small RNAs in plant stress responses. Biochim. Biophys. Acta 2008, 1779, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Yan-Du, L.; Qin-Hua, G.; Xiao-Yuan, C.; Song, Q. Roles of microRNA in plant defense and virus offense interaction. Plant. Cell. Rep. 2008, 27, 1571–1579. [Google Scholar]
- Kulcheski, F.R.; Oliveira, L.F.D.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimarães, F.C. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom. 2011, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qin, Y.; Duan, H.; Yin, W.; Xia, X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot. 2011, 62, 3765–3779. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Huang, X.; Zhang, Q.; Wei, G.; Wang, X.; Kang, Z. Selection of suitable inner reference genes for relative quantification expression of microRNA in wheat. Plant. Physiol. Biochem. 2012, 51, 116–122. [Google Scholar]
- Jin, S.J.; Simon, V.A.; Feddersen, R.M.; Rakhshan, F.; Schultz, D.A.; Zschunke, M.A.; Lingle, W.L.; Kolbert, C.P.; Jin, J. Quantitative miRNA expression analysis using fluidigm microfluidics dynamic arrays. BMC Genom. 2011, 12, S44. [Google Scholar] [CrossRef]
- Shen, J.; Xie, K.; Xiong, L. Global expression profiling of rice microRNAs by one-tube stem-loop reverse transcription quantitative PCR revealed important roles of microRNAs in abiotic stress responses. Mol. Genet. Genom. MGG 2010, 284, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tan, B.; Guo, W.W. Estimating transgene copy number in precocious trifoliate orange by TaqMan real-time PCR. Plant. Cell. Tissue Organ. Cult. 2012, 109, 363–371. [Google Scholar] [CrossRef]
- Artico, S.; Nardeli, S.M.; Brilhante, O.; Grossi-De-Sa, M.F.; Alves-Ferreira, M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant. Biol. 2010, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Klein, D. Quantification using real-time PCR technology: Applications and limitations. Trends Mol. Med. 2002, 8, 257–260. [Google Scholar] [CrossRef]
- Bustin, S.A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J. Mol. Endocrinol. 2002, 29, 4021–4022. [Google Scholar] [CrossRef]
- Lin, Y.L.; Lai, Z.X. Reference gene selection for qPCR analysis during somatic embryogenesis in longan tree. Plant Sci. 2010, 178, 359–365. [Google Scholar] [CrossRef]
- Derveaux, S.; Vandesompele, J.; Hellemans, J. How to do successful gene expression analysis using real-time PCR. Methods 2010, 50, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Keertan, D.; Huggett, J.F.; Bustin, S.A.; Johnson, M.A.; Graham, R.; Alimuddin, Z. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 2004, 37, 112–119. [Google Scholar]
- Kulcheski, F.R.; Nepomuceno, A.L.; Abdelnoor, R.V.; Margis, R. The use of microRNAs as reference genes for quantitative PCR in soybean. Anal. Biochem. 2010, 406, 185–92. [Google Scholar] [CrossRef] [PubMed]
- Hai-Ying, Z.; Jian-Wen, C.; Cai-Qin, L.; Lei, C.; Jian-Yang, W.; Jian-Ye, C.; Wang-Jin, L.; Jian-Guo, L. Selection of reliable reference genes for expression studies by reverse transcription quantitative real-time PCR in litchi under different experimental conditions. Plant. Cell. Rep. 2011, 30, 641–653. [Google Scholar]
- Schmittgen, T.D.; Zakrajsek, B.A. Effect of experimental treatment on housekeeping gene expression: Validation by real-time, quantitative RT-PCR. J. Biochem. Biophys. Methods 2000, 46, 69–81. [Google Scholar] [CrossRef]
- Zhong, H.; Simons, J.W. Direct comparison of GAPDH, β-actin, cyclophilin, and 28S rRNA as internal standards for quantifying RNA levels under hypoxia. Biochem. Biophys. Res. Commun. 1999, 259, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nijhawan, A.; Tyagi, A.K.; Khurana, J.P. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2006, 345, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhong, H.-Y.; Kuang, J.-F.; Li, J.-G.; Lu, W.-J.; Chen, J.-Y. Validation of reference genes for RT-qPCR studies of gene expression in banana fruit under different experimental conditions. Planta 2011, 234, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Maroufi, A.; Bockstaele, E.V.; Loose, M.D. Validation of reference genes for gene expression analysis in chicory (Cichorium intybus) using quantitative real-time PCR. BMC Mol. Biol. 2010, 11, 397. [Google Scholar] [CrossRef] [PubMed]
- Kou, S.-J.; Wu, X.-M.; Liu, Z.; Liu, Y.-L.; Xu, Q.; Guo, W.-W. Selection and validation of suitable reference genes for miRNA expression normalization by quantitative RT-PCR in citrus somatic embryogenic and adult tissues. Plant. Cell Rep. 2012, 31, 2151–2163. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kumar, S. Differential display-mediated identification of three drought-responsive expressed sequence tags in tea (Camellia sinensis (L.) O. Kuntze). J. Biosci. 2005, 30, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Zaveri, N.T. Green tea and its polyphenolic catechins: Medicinal uses in cancer and noncancer applications. Life Sci. 2006, 78, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Graham, H.N. Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 1992, 21, 334–350. [Google Scholar] [CrossRef]
- Juneja, L.R.; Chu, D.-C.; Okubo, T.; Nagato, Y.; Yokogoshi, H. L-theanine—A unique amino acid of green tea and its relaxation effect in humans. Trends Food Sci. Technol. 1999, 10, 199–204. [Google Scholar] [CrossRef]
- Deng, W.-W.; Zhang, M.; Wu, J.-Q.; Jiang, Z.-Z.; Tang, L.; Li, Y.-Y.; Wei, C.-L.; Jiang, C.-J.; Wan, X.-C. Molecular cloning, functional analysis of three cinnamyl alcohol dehydrogenase (CAD) genes in the leaves of tea plant, Camellia sinensis. J. Plant Physiol. 2013, 170, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, C.-J.; Li, Y.-Y.; Wei, C.-L.; Deng, W.-W. CsICE1 and CsCBF1: Two transcription factors involved in cold responses in Camellia sinensis. Plant. Cell Rep. 2012, 31, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Luo, Y. Identification of microRNAs and their targets in tea (Camellia sinensis). J. Zhejiang Univ. SC B 2013, 14, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Prabu, G.; Mandal, A. Computational identification of miRNAs and their target genes from expressed sequence tags of tea (Camellia sinensis). Genom. Proteom. Bioinform. 2010, 8, 113–121. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, X.; Chen, X.; Song, C.; Zou, Z.; Wang, Y.; Wang, M.; Fang, W.; Li, X. Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol. 2014, 14, 271. [Google Scholar] [CrossRef] [PubMed]
- Mohanpuria, P.; Yadav, S.K. Characterization of novel small RNAs from tea (Camellia sinensis L.). Mol. Biol. Rep. 2012, 39, 3977–3986. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, A.; Chandran, V.; Gajjeraman, P. Differential expression of microRNAs in dormant bud of tea (Camellia sinensis (L.) O. Kuntze). Plant. Cell Rep. 2014, 33, 1053–1069. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, S. Primer premier 5. Biotech. Softw. Internet Rep.: Comput. Softw. J. Sci. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, L.; Liu, X.; Cui, D.; Chen, T.; Zhang, H.; Jiang, C.; Xu, C.; Li, P.; Li, S. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi-Gasic, E.; Wu, R.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant. Methods 2007, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Gao, Z.; Wang, F.; Zhou, J.; Zhang, Z. Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol. Biol. 2009, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immunity 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Silveira, É.D.; Alves-Ferreira, M.; Guimarães, L.A.; da Silva, F.R.; Carneiro, V.T. Selection of reference genes for quantitative real-time PCR expression studies in the apomictic and sexual grass Brachiaria brizantha. BMC Plant. Biol. 2009, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Cohen, C.D.; Henger, A.; Irrgang, S.; Schlöndorff, D.; Kretzler, M. Validation of endogenous controls for gene expression analysis in microdissected human renal biopsies. Kidney Int. 2003, 64, 356–360. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ncRNA | Template/Primer Sequences |
|---|---|
| miRNA6149 | Seq: ATACGCACCTGAATCGGTAG RT: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGAT ACGACCTACCG F:GCGCGATACGCACCTGAAT |
| miRNA159a | Seq: CTTGGATTGAAGGGAGCTCC RT: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATA CGACGGAGCT F: CGCGCTTGGATTGAAGGG |
| miRNA5368n | Seq: GAGATACCACTCTGGAAGAGC RT: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGAT ACGACGCTCTT F: GCGGAGATACCACTCTGG |
| miRNA319c | Seq: TTGGACTGAAGGGAGCTCC RT: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGAT ACGACGGAGCT F: GCGCTTGGACTGAAGGG |
| miRNA166d | Seq: TCGGACCAGGCTTCATTCCCCAG RT: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGAT ACGACCTGGGG F: GCTCGGACCAGGCTTCATT |
| miRNA172e | Seq: GGAATCTTGATGATGCTGCAT RT: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGAT ACGACATGCAG F: GCGCGGGAATCTTGATGATG |
| Universal reverse primer for miRNA qRT-PCR & reverse transcription | R: GTGCAGGGTCCGAGGTATTC |
| U6 | Seq:GTCCCTTCGGGGACATCCGATAAAATTGGAACGATACAGAGAAGATTAGCATGGCCCCTGCGCAAGGATGACACGCACAAATCGAGAAATGGTCCAAATTTTTTT F: CGGGGACATCCGATAAAATTG R: GGACCATTTCTCGATTTGTGC |
| U4 | Seq:GCAATGACGCAGCTAGTGAGGTAATAACCGAGGCGCGTCAATTGCTGGTTGAAAACTATTTCCAAACCCCCTCATTGGCCTGGGTTCAGCCCGGGCCTCTGAGAATTTCTGGAAGGGCTCCCTTTG F: TGAGGTAATAACCGAGGCGC R: CAGAAATTCTCAGAGGCCCG |
| 5S -rRNA | Seq:CGGATGCGATCATACCAGCACCAATGCACTGGATCCCATCAGAACTATGCAGTTAAGCATGCTTGGGAGAGAATAGAGCTGGGGTGGGTGACCCCCTAGGAAGTCCTTGTGTTG F: GCGATCATACCAGCACCAATG R: CAACACAAGGACTTCCTAGGG |
| 5.8S-rRNA | Seq:TAAACGACTCTCGGCAACGGATATCTCGGCTCTCGCATCGATGAAGAACGTAGCGAAATGCGATACTTGGTGTGAATTGCAGAATCCCGCGAACCATCGAGTCTTTGAACGCAAGTTGCGCCCGAAGCCATTAGGTTGAGGGCACGCCTGCCTGGGCGTCTCAC F: CTCGGCAACGGATATCTCG R: GCCCTCAACCTAATGGCTTC |
| Rank | The Bud to theFifth Leaf | Tissues | All Treatment Samples | |||
|---|---|---|---|---|---|---|
| Gene | Stability | Gene | Stability | Gene | Stability | |
| 1 | 5SrRNA | 0.091 | 5SrRNA | 0.4 | U6 | 0.099 |
| 2 | miR5368n | 0.312 | miR5368n | 0.465 | miR159a | 0.193 |
| 3 | miR159a | 0.379 | miR159a | 0.525 | miR5368n | 0.211 |
| 4 | U6 | 0.48 | U6 | 0.574 | miR6149 | 0.261 |
| 5 | U4 | 0.579 | U4 | 0.728 | U4 | 0.341 |
| 6 | miR6149 | 0.721 | miR6149 | 0.921 | 5SrRNA | 0.403 |
| 7 | 5.8SrRNA | 0.835 | 5.8SrRNA | 2.789 | 5.8SrRNA | 1.019 |
| Rank | The Bud to the Second Leaf | E. oblique Attack | Mechanical Wounding | SA | MeJA | ABA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Stab. | Gene | Stab. | Gene | Stab. | Gene | Stab. | Gene | Stab. | Gene | Stab. | |
| 1 | miR6149 | 0.028 | miR5368n | 0.002 | U6 | 0.002 | U6 | 0.02 | miR159a | 0.007 | miR159a | 0.005 |
| 2 | 5S rRNA | 0.028 | miR6149 | 0.002 | U4 | 0.002 | miR5368n | 0.123 | miR5368n | 0.007 | U4 | 0.005 |
| 3 | miR159a | 0.113 | 5S rRNA | 0.005 | miR6149 | 0.002 | miR6149 | 0.179 | U6 | 0.022 | U6 | 0.118 |
| 4 | miR5368n | 0.404 | U4 | 0.051 | miR159a | 0.01 | 5S rRNA | 0.179 | miR6149 | 0.108 | miR5368n | 0.217 |
| 5 | U4 | 0.684 | U6 | 0.064 | miR5368n | 0.01 | miR159a | 0.194 | U4 | 1.14 | miR6149 | 0.379 |
| 6 | U6 | 0.744 | miR159a | 0.452 | 5S rRNA | 1.207 | U4 | 0.339 | 5S rRNA | 1.181 | 5S rRNA | 0.461 |
| 7 | 5.8S rRNA | 0.79 | 5.8S rRNA | 2.025 | 5.8S rRNA | 3.093 | 5.8S rRNA | 3.085 | 5.8S rRNA | 1.801 | 5.8S rRNA | 3.626 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.; Zhang, X.; Shi, C.; Wang, S.; Wu, A.; Wei, C. Selection and Verification of Candidate Reference Genes for Mature MicroRNA Expression by Quantitative RT-PCR in the Tea Plant (Camellia sinensis). Genes 2016, 7, 25. https://doi.org/10.3390/genes7060025
Song H, Zhang X, Shi C, Wang S, Wu A, Wei C. Selection and Verification of Candidate Reference Genes for Mature MicroRNA Expression by Quantitative RT-PCR in the Tea Plant (Camellia sinensis). Genes. 2016; 7(6):25. https://doi.org/10.3390/genes7060025
Chicago/Turabian StyleSong, Hui, Xiao Zhang, Cong Shi, Shuangshuang Wang, Ailin Wu, and Chaoling Wei. 2016. "Selection and Verification of Candidate Reference Genes for Mature MicroRNA Expression by Quantitative RT-PCR in the Tea Plant (Camellia sinensis)" Genes 7, no. 6: 25. https://doi.org/10.3390/genes7060025
APA StyleSong, H., Zhang, X., Shi, C., Wang, S., Wu, A., & Wei, C. (2016). Selection and Verification of Candidate Reference Genes for Mature MicroRNA Expression by Quantitative RT-PCR in the Tea Plant (Camellia sinensis). Genes, 7(6), 25. https://doi.org/10.3390/genes7060025