
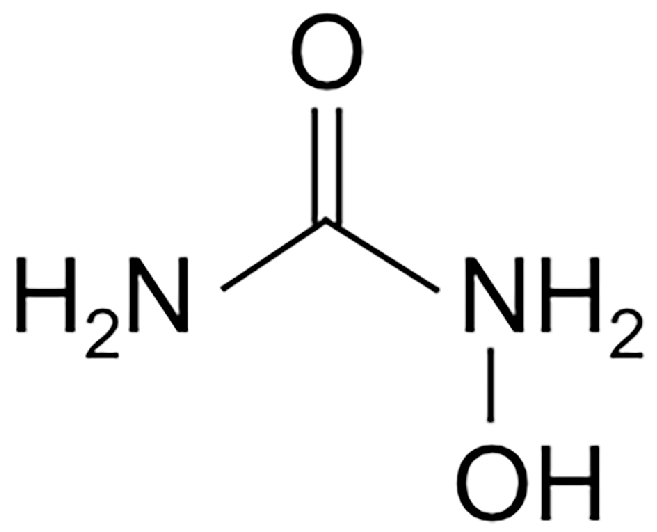
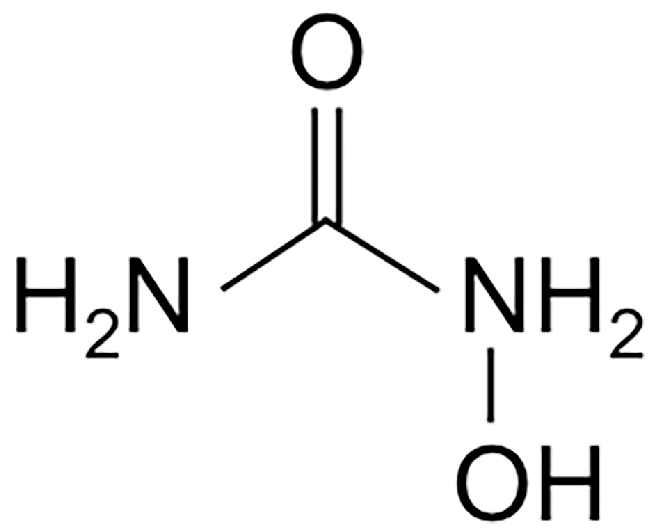
The Cell Killing Mechanisms of Hydroxyurea

Abstract
:1. Introduction
2. Inhibition of RNR and Other Potential Metalloenzymes
3. S Phase Arrest, DNA Damage and the Checkpoint Response
4. Accumulation of Reactive Oxygen Species (ROS)
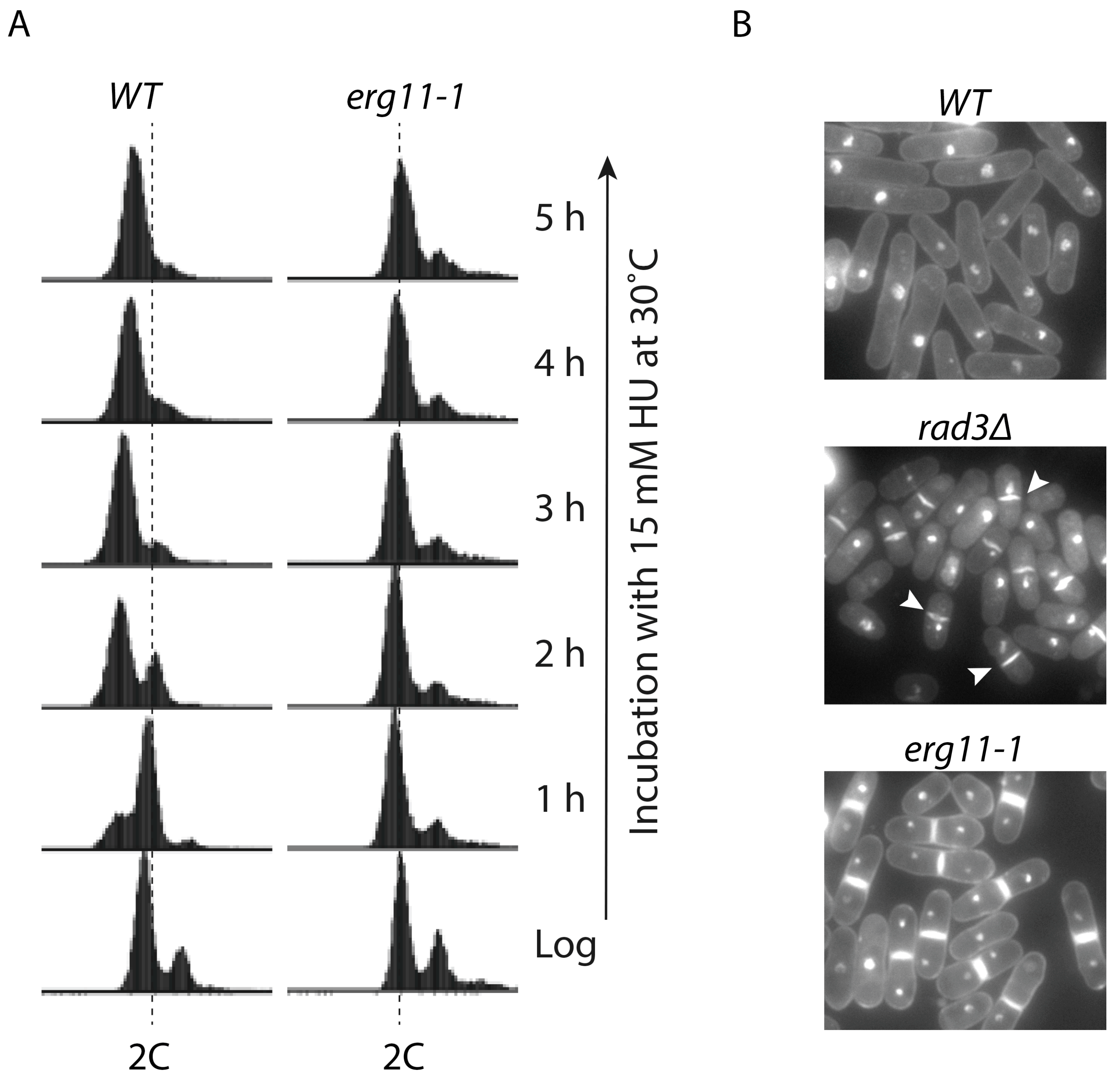
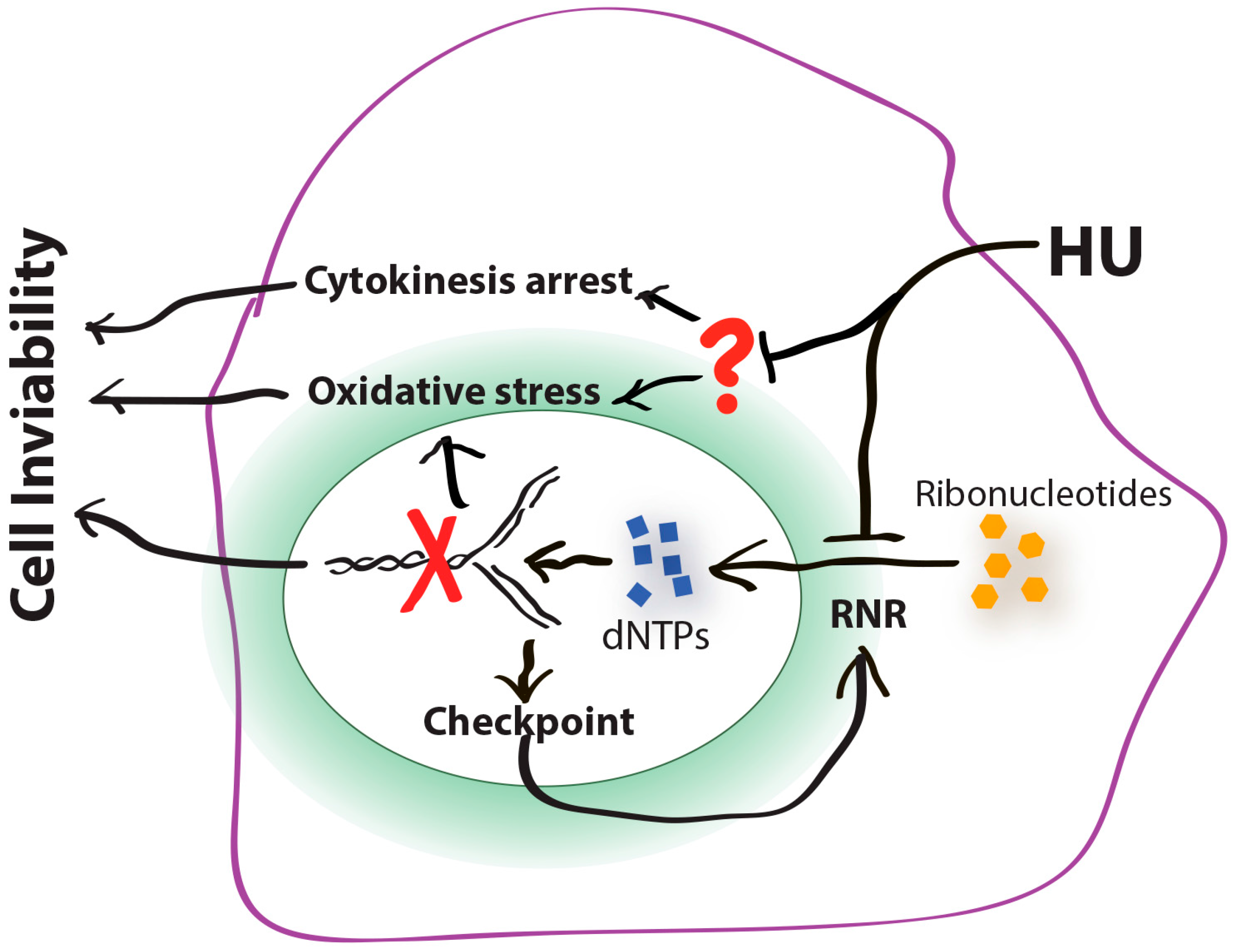
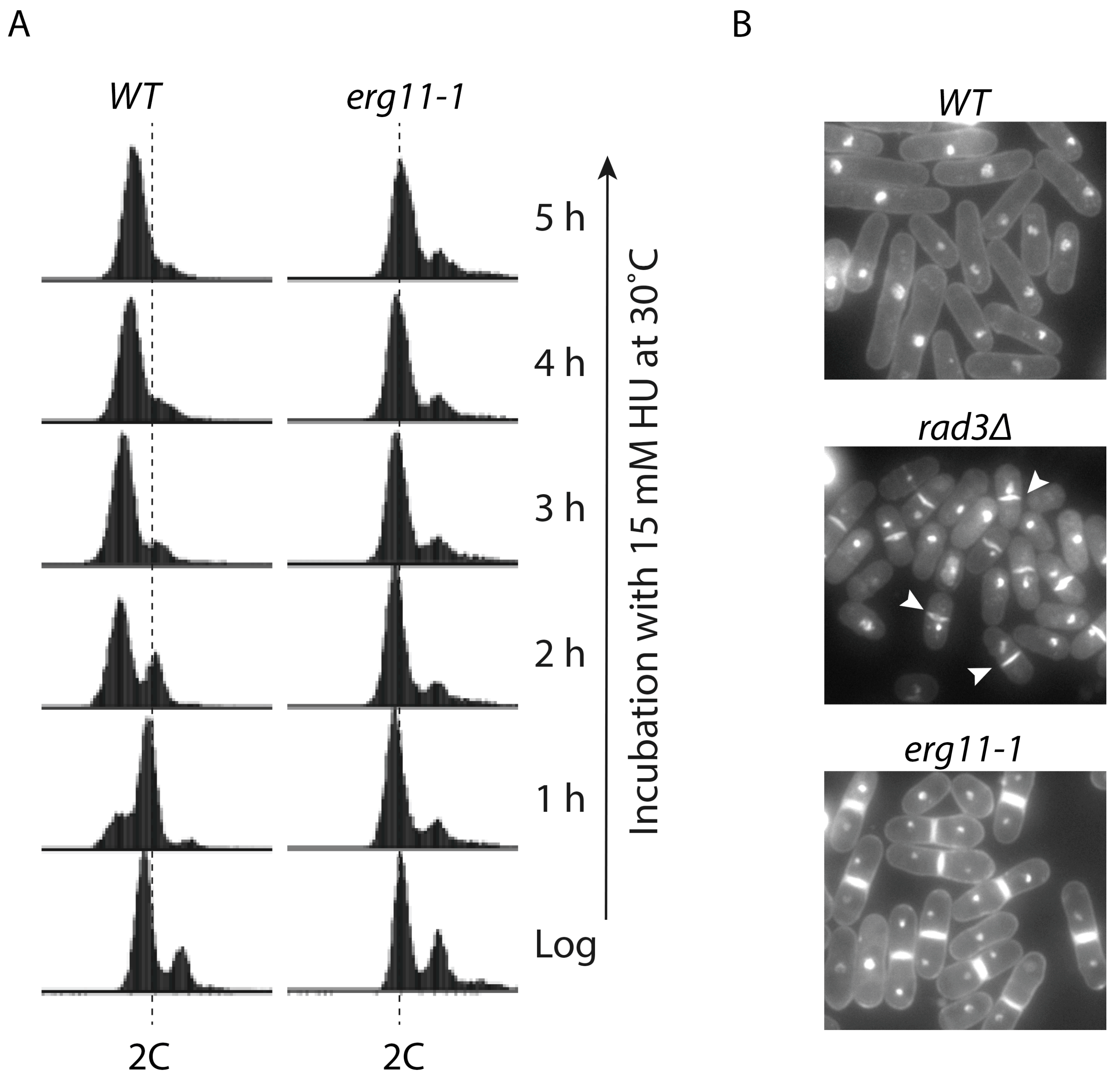
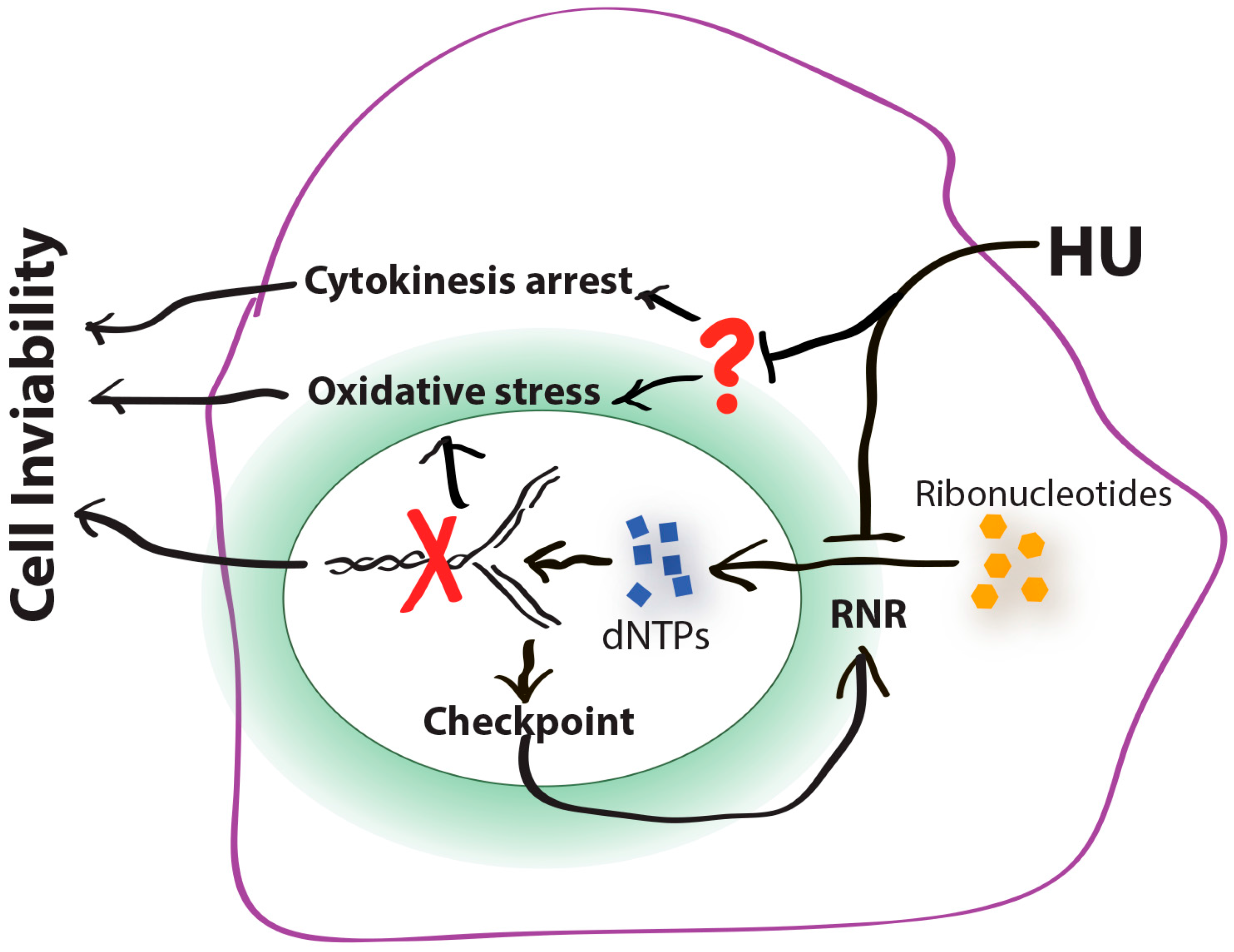
5. Cytokinesis Arrest and the Potentially Unidentified Cellular Target(s) of HU
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dresler, W.F.C.; Stein, R. Ueber Den Hydroxylharnstoff. Eur. J. Org. Chem. 1869, 150, 242–252. [Google Scholar] [CrossRef]
- Rosenthal, F.; Wislicki, L.; Kollek, L. Über die Beziehungen von Schwersten Blutgiften zu Abbauprodukten des Eiweisses. Klin. Wochenschr. 1928, 7, 972–977. [Google Scholar] [CrossRef]
- Stock, C.C.; Clarke, D.A.; Philips, F.S.; Barclay, R.K.; Myron, S.A. Sarcoma 180 Screening Data. Cancer Res. 1960, 20 Pt 2, 193–381. [Google Scholar] [PubMed]
- Stearns, B.; Losee, K.A.; Bernstein, J. Hydroxyurea. A New Type of Potential Antitumor Agent. J. Med. Chem. 1963, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, H.S.; Gutter, B.; Becker, Y. Studies on the Developmental Cycle of Chlamydia Trachomatis: Selective Inhibition by Hydroxyurea. J. Bacteriol. 1973, 115, 682–690. [Google Scholar] [PubMed]
- Feiner, R.R.; Coward, J.E.; Rosenkranz, H.S. Effect of Hydroxyurea on Staphylococcus Epidermidis and Micrococcus Lysodeikticus: Thickening of the Cell Wall. Antimicrob. Agents Chemother. 1973, 3, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Gale, G.R.; Kendall, S.M.; Mclain, H.H.; Dubois, S. Effect of Hydroxyurea on Pseudomonas Aeruginosa. Cancer Res. 1964, 24, 1012–1020. [Google Scholar] [PubMed]
- Nozaki, A.; Numata, K.; Morimoto, M.; Kondo, M.; Sugimori, K.; Morita, S.; Miyajima, E.; Ikeda, M.; Kato, N.; Maeda, S.; et al. Hydroxyurea Suppresses Hcv Replication in Humans: A Phase I Trial of Oral Hydroxyurea in Chronic Hepatitis C Patients. Antivir. Ther. 2010, 15, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Lori, F.; Malykh, A.; Cara, A.; Sun, D.; Weinstein, J.N.; Lisziewicz, J.; Gallo, R.C. Hydroxyurea as an Inhibitor of Human Immunodeficiency Virus-Type 1 Replication. Science 1994, 266, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Leavell, U.W., Jr.; Yarbro, J.W. Hydroxyurea. A New Treatment for Psoriasis. Arch. Dermatol. 1970, 102, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Yarbro, J.W. Hydroxyurea in the Treatment of Refractory Psoriasis. Lancet 1969, 2, 846–847. [Google Scholar] [CrossRef]
- Donehower, R.C. An Overview of the Clinical Experience with Hydroxyurea. Semin. Oncol. 1992, 19, 11–19. [Google Scholar] [PubMed]
- Spivak, J.L.; Hasselbalch, H. Hydroxycarbamide: A User’s Guide for Chronic Myeloproliferative Disorders. Expert Rev. Anti-Cancer Ther. 2011, 11, 403–414. [Google Scholar] [CrossRef] [PubMed]
- 19th WHO Model List of Essential Medicines (April 2015). http://www.who.int/medicines/publications/essentialmedicines/EML2015_8-May-15.pdf.
- Reichard, P.; Ehrenberg, A. Ribonucleotide Reductase—A Radical Enzyme. Science 1983, 221, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Akerblom, L.; Ehrenberg, A.; Graslund, A.; Lankinen, H.; Reichard, P.; Thelander, L. Overproduction of the Free Radical of Ribonucleotide Reductase in Hydroxyurea-Resistant Mouse Fibroblast 3t6 Cells. Proc. Natl. Acad. Sci. USA 1981, 78, 2159–2163. [Google Scholar] [CrossRef] [PubMed]
- Timson, J. Hydroxyurea. Mutat. Res. 1975, 32, 115–132. [Google Scholar] [CrossRef]
- Desesso, J.M. Cell Death and Free Radicals: A Mechanism for Hydroxyurea Teratogenesis. Med. Hypotheses 1979, 5, 937–951. [Google Scholar] [CrossRef]
- Davies, B.W.; Kohanski, M.A.; Simmons, L.A.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Hydroxyurea Induces Hydroxyl Radical-Mediated Cell Death in Escherichia coli. Mol. Cell 2009, 36, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Nakayashiki, T.; Mori, H. Genome-Wide Screening with Hydroxyurea Reveals a Link between Nonessential Ribosomal Proteins and Reactive Oxygen Species Production. J. Bacteriol. 2013, 195, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Singh, A.; Alter, G.M. Hydroxyurea Induces Cytokinesis Arrest in Cells Expressing a Mutated Sterol-14alpha-Demethylase in the Ergosterol Biosynthesis Pathway. Genetics 2016, in press. [Google Scholar]
- Marchetti, M.A.; Weinberger, M.; Murakami, Y.; Burhans, W.C.; Huberman, J.A. Production of Reactive Oxygen Species in Response to Replication Stress and Inappropriate Mitosis in Fission Yeast. J. Cell Sci. 2006, 119, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.E.; Facca, C.; Fatmi, Z.; Baille, D.; Benakli, S.; Vernis, L. DNA Replication Inhibitor Hydroxyurea Alters Fe-S Centers by Producing Reactive Oxygen Species in Vivo. Sci. Rep. 2016, 6, 29361. [Google Scholar] [CrossRef] [PubMed]
- Dubacq, C.; Chevalier, A.; Courbeyrette, R.; Petat, C.; Gidrol, X.; Mann, C. Role of the Iron Mobilization and Oxidative Stress Regulons in the Genomic Response of Yeast to Hydroxyurea. Mol. Genet. Genom. 2006, 275, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Cokic, V.P.; Smith, R.D.; Beleslin-Cokic, B.B.; Njoroge, J.M.; Miller, J.L.; Gladwin, M.T.; Schechter, A.N. Hydroxyurea Induces Fetal Hemoglobin by the Nitric Oxide-Dependent Activation of Soluble Guanylyl Cyclase. J. Clin. Investig. 2003, 111, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Adragna, N.C.; Fonseca, P.; Lauf, P.K. Hydroxyurea Affects Cell Morphology, Cation Transport, and Red Blood Cell Adhesion in Cultured Vascular Endothelial Cells. Blood 1994, 83, 553–560. [Google Scholar] [PubMed]
- Fraser, D.I.; Liu, K.T.; Reid, B.J.; Hawkins, E.; Sevier, A.; Pyle, M.; Robinson, J.W.; Ouellette, P.H.; Ballantyne, J.S. Widespread Natural Occurrence of Hydroxyurea in Animals. PLoS ONE 2015, 10, E0142890. [Google Scholar] [CrossRef] [PubMed]
- Kettani, T.; Cotton, F.; Gulbis, B.; Ferster, A.; Kumps, A. Plasma Hydroxyurea Determined by Gas Chromatography-Mass Spectrometry. J. Chromatogr. B 2009, 877, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Krakoff, I.H.; Brown, N.C.; Reichard, P. Inhibition of Ribonucleoside Diphosphate Reductase by Hydroxyurea. Cancer Res. 1968, 28, 1559–1565. [Google Scholar] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide Reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Stubbe, J.; Van Der Donk, W.A. Ribonucleotide Reductases: Radical Enzymes with Suicidal Tendencies. Chem. Biol. 1995, 2, 793–801. [Google Scholar] [CrossRef]
- Uhlin, U.; Eklund, H. Structure of Ribonucleotide Reductase Protein R1. Nature 1994, 370, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M. Ribonucleotide Reductases and Radical Reactions. Cell. Mol. Life Sci. 1998, 54, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Zimanyi, C.M.; Chen, P.Y.; Kang, G.; Funk, M.A.; Drennan, C.L. Molecular Basis for Allosteric Specificity Regulation in Class Ia Ribonucleotide Reductase From Escherichia coli. Elife 2016, 5, E07141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Elia, A.E.; Naylor, M.L.; Dephoure, N.; Ballif, B.A.; Goel, G.; Xu, Q.; Ng, A.; Chou, D.M.; Xavier, R.J.; et al. Profiling DNA Damage-Induced Phosphorylation in Budding Yeast Reveals Diverse Signaling Networks. Proc. Natl. Acad. Sci. USA 2016, 113, E3667–E3675. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.A.; Zhou, C.; Elia, A.E.; Murray, J.M.; Carr, A.M.; Elledge, S.J.; Rhind, N. Identification of S-Phase DNA Damage-Response Targets in Fission Yeast Reveals Conservation of Damage-Response Networks. Proc. Natl. Acad. Sci. USA 2016, 113, E3676–E3685. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.; Georgieva, B.; Domkin, V.; Zhao, X.; Rothstein, R.; Thelander, L. Survival of DNA Damage in Yeast Directly Depends on Increased Dntp Levels Allowed by Relaxed Feedback Inhibition of Ribonucleotide Reductase. Cell 2003, 112, 391–401. [Google Scholar] [CrossRef]
- Elledge, S.J.; Zhou, Z.; Allen, J.B. Ribonucleotide Reductase: Regulation, Regulation, Regulation. Trends Biochem. Sci. 1992, 17, 119–123. [Google Scholar] [CrossRef]
- Nyholm, S.; Thelander, L.; Graslund, A. Reduction and Loss of the Iron Center in the Reaction of The Small Subunit of Mouse Ribonucleotide Reductase with Hydroxyurea. Biochemistry 1993, 32, 11569–11574. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, G.; Thelander, L.; Graslund, A. Epr Stopped-Flow Studies of the Reaction of the Tyrosyl Radical of Protein R2 from Ribonucleotide Reductase with Hydroxyurea. Biochem. Biophys. Res. Commun. 1992, 188, 879–887. [Google Scholar] [CrossRef]
- Shao, J.; Zhou, B.; Zhu, L.; Bilio, A.J.; Su, L.; Yuan, Y.C.; Ren, S.; Lien, E.J.; Shih, J.; Yen, Y. Determination of the Potency and Subunit-Selectivity of Ribonucleotide Reductase Inhibitors with a Recombinant-Holoenzyme-Based in Vitro Assay. Biochem. Pharmacol. 2005, 69, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, B.; Chen, X.; Jiang, H.; Shao, J.; Yen, Y. Inhibitory Mechanisms of Heterocyclic Carboxaldehyde Thiosemicabazones for Two Forms of Human Ribonucleotide Reductase. Biochem. Pharmacol. 2009, 78, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Larsen, I.K.; Sjoberg, B.M.; Thelander, L. Characterization of the Active Site of Ribonucleotide Reductase of Escherichia coli, Bacteriophage T4 and Mammalian Cells by Inhibition Studies with Hydroxyurea Analogues. Eur. J. Biochem. 1982, 125, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Gerez, C.; Elleingand, E.; Kauppi, B.; Eklund, H.; Fontecave, M. Reactivity of the Tyrosyl Radical of Escherichia coli Ribonucleotide Reductase—Control by the Protein. Eur. J. Biochem. 1997, 249, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Zhou, B.; Chu, B.; Yen, Y. Ribonucleotide Reductase Inhibitors and Future Drug Design. Curr. Cancer Drug Targets 2006, 6, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Sjoberg, B.M.; Eklund, H. Three-Dimensional Structure of the Free Radical Protein of Ribonucleotide Reductase. Nature 1990, 345, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Ormo, M.; Regnstrom, K.; Wang, Z.; Que, L., Jr.; Sahlin, M.; Sjoberg, B.M. Residues Important for Radical Stability in Ribonucleotide Reductase from Escherichia coli. J. Biol. Chem. 1995, 270, 6570–6576. [Google Scholar] [PubMed]
- Sneeden, J.L.; Loeb, L.A. Mutations in the R2 Subunit of Ribonucleotide Reductase That Confer Resistance to Hydroxyurea. J. Biol. Chem. 2004, 279, 40723–40728. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, G.P.; Bowdon, B.J.; Adamson, D.J.; Vail, M.H. Comparison of the Effects of Several Inhibitors of the Synthesis of Nucleic Acids Upon the Viability and Progression through the Cell Cycle of Cultured H. Ep. No. 2 Cells. Cancer Res. 1972, 32, 2661–2669. [Google Scholar] [PubMed]
- Fontecave, M.; Lepoivre, M.; Elleingand, E.; Gerez, C.; Guittet, O. Resveratrol, a Remarkable Inhibitor of Ribonucleotide Reductase. FEBS Lett. 1998, 421, 277–279. [Google Scholar] [CrossRef]
- Karlsson, M.; Sahlin, M.; Sjoberg, B.M. Escherichia coli Ribonucleotide Reductase. Radical Susceptibility to Hydroxyurea Is Dependent on the Regulatory State of the Enzyme. J. Biol. Chem. 1992, 267, 12622–12626. [Google Scholar] [PubMed]
- Juul, T.; Malolepszy, A.; Dybkaer, K.; Kidmose, R.; Rasmussen, J.T.; Ersen, G.R.; Johnsen, H.E.; Jorgensen, J.E.; Andersen, S.U. The in Vivo Toxicity of Hydroxyurea Depends on Its Direct Target Catalase. J. Biol. Chem. 2010, 285, 21411–21415. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Supuran, C.T. Hydroxyurea Is a Carbonic Anhydrase Inhibitor. Bioorg. Med. Chem. 2003, 11, 2241–2246. [Google Scholar] [CrossRef]
- Campestre, C.; Agamennone, M.; Tortorella, P.; Preziuso, S.; Biasone, A.; Gavuzzo, E.; Pochetti, G.; Mazza, F.; Hiller, O.; Tschesche, H.; et al. N-Hydroxyurea as Zinc Binding Group in Matrix Metalloproteinase Inhibition: Mode of Binding in a Complex with Mmp-8. Bioorg. Med. Chem. Lett. 2006, 16, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Temperini, C.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. N-Hydroxyurea—A Versatile Zinc Binding Function in the Design of Metalloenzyme Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4316–4320. [Google Scholar] [CrossRef] [PubMed]
- Day, J.A.; Cohen, S.M. Investigating the Selectivity of Metalloenzyme Inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.L.; Pfleger, C.M.; Kirschner, M.W.; Thelander, L. Mouse Ribonucleotide Reductase R2 Protein: A New Target for Anaphase-Promoting Complex-Cdh1-Mediated Proteolysis. Proc. Natl. Acad. Sci. USA 2003, 100, 3925–3929. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.L.; Bjorklund, S.; Thelander, L. S Phase-Specific Transcription of the Mouse Ribonucleotide Reductase R2 Gene Requires Both a Proximal Repressive E2f-Binding Site and an Upstream Promoter Activating Region. J. Biol. Chem. 2004, 279, 10796–10807. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, S.; Skog, S.; Tribukait, B.; Thelander, L. S-Phase-Specific Expression of Mammalian Ribonucleotide Reductase R1 and R2 Subunit Mrnas. Biochemistry 1990, 29, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, Y.; Eriksson, S.; Jildevik, I.; Skog, S.; Thelander, L.; Tribukait, B. Cell Cycle-Dependent Expression of Mammalian Ribonucleotide Reductase. Differential Regulation of the Two Subunits. J. Biol. Chem. 1985, 260, 9114–9116. [Google Scholar] [PubMed]
- Eriksson, S.; Graslund, A.; Skog, S.; Thelander, L.; Tribukait, B. Cell Cycle-Dependent Regulation of Mammalian Ribonucleotide Reductase. The S Phase-Correlated Increase in Subunit M2 Is Regulated by de Novo Protein Synthesis. J. Biol. Chem. 1984, 259, 11695–11700. [Google Scholar] [PubMed]
- Zhao, X.; Muller, E.G.; Rothstein, R. A Suppressor of Two Essential Checkpoint Genes Identifies a Novel Protein That Negatively Affects Dntp Pools. Mol. Cell 1998, 2, 329–340. [Google Scholar] [CrossRef]
- Nestoras, K.; Mohammed, A.H.; Schreurs, A.S.; Fleck, O.; Watson, A.T.; Poitelea, M.; O’shea, C.; Chahwan, C.; Holmberg, C.; Kragelund, B.B.; et al. Regulation of Ribonucleotide Reductase by Spd1 Involves Multiple Mechanisms. Genes Dev. 2010, 24, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, P.; Dahl, L.; Chilkova, O.; Domkin, V.; Thelander, L. The Schizosaccharomyces pombe Replication Inhibitor Spd1 Regulates Ribonucleotide Reductase Activity and Dntps by Binding to the Large Cdc22 Subunit. J. Biol. Chem. 2006, 281, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Zhang, Z.; An, X.; Bucci, B.; Perlstein, D.L.; Stubbe, J.; Huang, M. Subcellular Localization of Yeast Ribonucleotide Reductase Regulated by the DNA Replication and Damage Checkpoint Pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 6628–6633. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.D.; Wang, J.; Stubbe, J.; Elledge, S.J. Dif1 Is a DNA-Damage-Regulated Facilitator of Nuclear Import for Ribonucleotide Reductase. Mol. Cell 2008, 32, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.D.; Elledge, S.J. Control of Ribonucleotide Reductase Localization through an Anchoring Mechanism Involving Wtm1. Genes Dev. 2006, 20, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Mckethan, B.L.; Spiro, S. Cooperative and Allosterically Controlled Nucleotide Binding Regulates the DNA Binding Activity of Nrdr. Mol. Microbiol. 2013, 90, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Herrick, J.; Sclavi, B. Ribonucleotide Reductase snd the Regulation of DNA Replication: An Old Story and an Ancient Heritage. Mol. Microbiol. 2007, 63, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Furuya, K.; Carr, A.M. DNA Checkpoints in Fission Yeast. J. Cell Sci. 2003, 116, 3847–3848. [Google Scholar] [CrossRef] [PubMed]
- Alvino, G.M.; Collingwood, D.; Murphy, J.M.; Delrow, J.; Brewer, B.J.; Raghuraman, M.K. Replication in Hydroxyurea: It’s a Matter of Time. Mol. Cell. Biol. 2007, 27, 6396–6406. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F. Checkpoint-Dependent Inhibition of DNA Replication Initiation by Sld3 and Dbf4 Phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mosqueda, J.; Maas, N.L.; Jonsson, Z.O.; Defazio-Eli, L.G.; Wohlschlegel, J.; Toczyski, D.P. Damage-Induced Phosphorylation of Sld3 Is Important to Block Late Origin Firing. Nature 2010, 467, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication Stress-Induced Genome Instability: The Dark Side of Replication Maintenance by Homologous Recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Funahashi, S.; Uemura, T.; Yanagida, M. Isolation and Characterization of Schizosaccharomyces pombe Cut Mutants That Block Nuclear Division But Not Cytokinesis. EMBO J. 1986, 5, 2973–2979. [Google Scholar] [PubMed]
- Saka, Y.; Yanagida, M. Fission Yeast Cut5+, Required for S Phase Onset and M Phase Restraint, Is Identical to the Radiation-Damage Repair Gene Rad4+. Cell 1993, 74, 383–393. [Google Scholar] [CrossRef]
- Sinclair, W.K. Hydroxyurea: Differential Lethal Effects on Cultured Mammalian Cells during the Cell Cycle. Science 1965, 150, 1729–1731. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, W.K. Hydroxyurea: Effects on Chinese Hamster Cells Grown in Culture. Cancer Res. 1967, 27, 297–308. [Google Scholar] [PubMed]
- Barranco, S.C.; Novak, J.K. Survival Responses of Dividing and Nondividing Mammalian Cells after Treatment with Hydroxyurea, Arabinosylcytosine, or Adriamycin. Cancer Res. 1974, 34, 1616–1618. [Google Scholar] [PubMed]
- Timson, J. Hydroxyurea: Comparison of Cytotoxic and Antimitotic Activities against Human Lymphocytes in Vitro. Br. J. Cancer 1969, 23, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.K.; Sinclair, W.K. Cytological Effects on Chinese Hamster Cells of Synchronizing Concentrations of Hydroxyurea. J. Cell. Physiol. 1968, 72, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Przybyszewski, W.M.; Malec, J. Protection against Hydroxyurea-Induced Cytotoxic Effects in L5178y Cells by Free Radical Scavengers. Cancer Lett. 1982, 17, 223–228. [Google Scholar] [CrossRef]
- Li, J.C.; Kaminskas, E. Progressive Formation of DNA Lesions in Cultured Ehrlich Ascites Tumor Cells Treated with Hydroxyurea. Cancer Res. 1987, 47, 2755–2758. [Google Scholar] [PubMed]
- Veale, D.; Cantwell, B.M.; Kerr, N.; Upfold, A.; Harris, A.L. Phase 1 Study of High-Dose Hydroxyurea in Lung Cancer. Cancer Chemother. Pharmacol. 1988, 21, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Mohler, W.C. Cytotoxicity of Hydroxyurea (Nsc-32065) Reversible by Pyrimidine Deoxyribosides in a Mammalian Cell Line Grown in Vitro. Cancer Chemother. Rep. 1964, 34, 1–6. [Google Scholar] [PubMed]
- Philips, F.S.; Sternberg, S.S.; Schwartz, H.S.; Cronin, A.P.; Sodergren, J.E.; Vidal, P.M. Hydroxyurea. I. Acute Cell Death in Proliferating Tissues in Rats. Cancer Res. 1967, 27, 61–75. [Google Scholar] [PubMed]
- Farber, E.; Baserga, R. Differential Effects of Hydroxyurea on Survival of Proliferating Cells In Vivo. Cancer Res. 1969, 29, 136–139. [Google Scholar] [PubMed]
- Scott, W.J.; Ritter, E.J.; Wilson, J.G. DNA Synthesis Inhibition and Cell Death Associated with Hydroxyurea Teratogenesis in Rat Embryos. Dev. Biol. 1971, 26, 306–315. [Google Scholar] [CrossRef]
- Hennings, H.; Devik, F. Comparison of Cytotoxicity of Hydroxyurea in Normal and Rapidly Proliferating Epidermis and Small Intestine in Mice. Cancer Res. 1971, 31, 277–282. [Google Scholar] [PubMed]
- Coyle, M.B.; Strauss, B. Cell Killing and the Accumulation of Breaks in the DNA of Hep-2 Cells Incubated in the Presence of Hydroxyurea. Cancer Res. 1970, 30, 2314–2319. [Google Scholar] [PubMed]
- Walker, I.G.; Yatscoff, R.W.; Sridhar, R. Hydroxyurea: Induction of Breaks in Template Strands of Replicating DNA. Biochem. Biophys. Res. Commun. 1977, 77, 403–408. [Google Scholar] [CrossRef]
- Massafi, K.; Carr, H.S.; Rosenkranz, H.S. Hydroxyurea and Cell Death. Isr. J. Med. Sci. 1972, 8, 559–561. [Google Scholar] [PubMed]
- Sinha, N.K.; Snustad, D.P. Mechanism of Inhibition of Deoxyribonucleic acid Synthesis in Escherichia coli by Hydroxyurea. J. Bacteriol. 1972, 112, 1321–1324. [Google Scholar] [PubMed]
- Jacobs, S.J.; Rosenkranz, H.S. Detection of a Reactive Intermediate in the Reaction between DNA and Hydroxyurea. Cancer Res. 1970, 30, 1084–1094. [Google Scholar] [PubMed]
- Rosenkranz, H.S.; Rosenkranz, S. Degradation of DNA by Carbamoyloxyurea—An Oxidation Product of Hydroxyurea. Biochim. Biophys. Acta 1969, 195, 266–267. [Google Scholar] [CrossRef]
- Sakano, K.; Oikawa, S.; Hasegawa, K.; Kawanishi, S. Hydroxyurea Induces Site-Specific DNA Damage via Formation of Hydrogen Peroxide and Nitric Oxide. Jpn. J. Cancer Res. 2001, 92, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Pathways of Oxidative Damage. Annu. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Przybyszewski, W.M.; Kasperczyk, J. [Radical Mechanism of Hydroxyurea Side Toxicity]. Postepy Hig. Med. Dosw. 2006, 60, 516–526. [Google Scholar]
- Desesso, J.M. Amelioration of Teratogenesis. I. Modification of Hydroxyurea-Induced Teratogenesis by the Antioxidant Propyl Gallate. Teratology 1981, 24, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Godoy, V.G.; Jarosz, D.F.; Walker, F.L.; Simmons, L.A.; Walker, G.C. Y-Family DNA Polymerases Respond to DNA Damage-Independent Inhibition of Replication Fork Progression. EMBO J. 2006, 25, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A.; Chin, S.M.; Linn, S. Toxic DNA Damage by Hydrogen Peroxide through the Fenton Reaction in Vivo and in Vitro. Science 1988, 240, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Seaver, L.C.; Imlay, J.A. Alkyl Hydroperoxide Reductase Is the Primary Scavenger of Endogenous Hydrogen Peroxide in Escherichia coli. J. Bacteriol. 2001, 183, 7173–7181. [Google Scholar] [CrossRef] [PubMed]
- Foti, J.J.; Devadoss, B.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Oxidation of the Guanine Nucleotide Pool Underlies Cell Death by Bactericidal Antibiotics. Science 2012, 336, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. DNA Damage-Induced Reactive Oxygen Species (Ros) Stress Response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008, 45, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, A.; Pflieger, D.; Barrault, M.B.; Vinh, J.; Toledano, M.B. A Thiol Peroxidase Is an H2O2 Receptor and Redox-Transducer in Gene Activation. Cell 2002, 111, 471–481. [Google Scholar] [CrossRef]
- Tang, H.M.; Pan, K.; Kong, K.Y.; Hu, L.; Chan, L.C.; Siu, K.L.; Sun, H.; Wong, C.M.; Jin, D.Y. Loss of Apd1 in Yeast Confers Hydroxyurea Sensitivity Suppressed by Yap1p Transcription Factor. Sci. Rep. 2015, 5, 7897. [Google Scholar] [CrossRef] [PubMed]
- Han, T.X.; Xu, X.Y.; Zhang, M.J.; Peng, X.; Du, L.L. Global Fitness Profiling of Fission Yeast Deletion Strains by Barcode Sequencing. Genome Biol. 2010, 11, R60. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Shimanuki, M.; Yanagida, M. Fission Yeast Genes That Confer Resistance to Staurosporine Encode an Ap-1-Like Transcription Factor and a Protein Kinase Related to the Mammalian Erk1/Map2 and Budding Yeast Fus3 and Kss1 Kinases. Genes Dev. 1991, 5, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Benko, Z.; Fenyvesvolgyi, C.; Pesti, M.; Sipiczki, M. The Transcription Factor Pap1/Caf3 Plays a Central Role in the Determination of Caffeine Resistance in Schizosaccharomyces pombe. Mol. Genet. Genom. 2004, 271, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Turi, T.G.; Webster, P.; Rose, J.K. Brefeldin a Sensitivity and Resistance in Schizosaccharomyces pombe. Isolation of Multiple Genes Conferring Resistance. J. Biol. Chem. 1994, 269, 24229–24236. [Google Scholar] [PubMed]
- Belfield, C.; Queenan, C.; Rao, H.; Kitamura, K.; Walworth, N.C. The Oxidative Stress Responsive Transcription Factor Pap1 Confers DNA Damage Resistance on Checkpoint-Deficient Fission Yeast Cells. PLoS ONE 2014, 9, E89936. [Google Scholar] [CrossRef] [PubMed]
- Boronat, S.; Domenech, A.; Paulo, E.; Calvo, I.A.; Garcia-Santamarina, S.; Garcia, P.; Encinar Del Dedo, J.; Barcons, A.; Serrano, E.; Carmona, M.; et al. Thiol-Based H2O2 Signalling in Microbial Systems. Redox Biol. 2014, 2, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A.; Linn, S. DNA Damage and Oxygen Radical Toxicity. Science 1988, 240, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Netz, D.J.; Stith, C.M.; Stumpfig, M.; Kopf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.; Pierik, A.J. Eukaryotic DNA Polymerases Require an Iron-Sulfur Cluster for the Formation of Active Complexes. Nat. Chem. Biol. 2012, 8, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lyver, E.R.; Nakamaru-Ogiso, E.; Yoon, H.; Amutha, B.; Lee, D.W.; Bi, E.; Ohnishi, T.; Daldal, F.; Pain, D.; et al. Dre2, a Conserved Eukaryotic Fe/S Cluster Protein, Functions in Cytosolic Fe/S Protein Biogenesis. Mol. Cell. Biol. 2008, 28, 5569–5582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, H.; Zhang, C.; An, X.; Liu, L.; Stubbe, J.; Huang, M. Conserved Electron Donor Complex Dre2-Tah18 Is Required for Ribonucleotide Reductase Metallocofactor Assembly and DNA Synthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E1695–E1704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Wu, X.; An, X.; Stubbe, J.; Huang, M. Investigation of in Vivo Diferric Tyrosyl Radical Formation in Saccharomyces cerevisiae Rnr2 Protein: Requirement of Rnr4 and Contribution of Grx3/4 and Dre2 Proteins. J. Biol. Chem. 2011, 286, 41499–41509. [Google Scholar] [CrossRef] [PubMed]
- Vernis, L.; Facca, C.; Delagoutte, E.; Soler, N.; Chanet, R.; Guiard, B.; Faye, G.; Baldacci, G. A Newly Identified Essential Complex, Dre2-Tah18, Controls Mitochondria Integrity and Cell Death after Oxidative Stress in Yeast. PLoS ONE 2009, 4, E4376. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Jiang, W.; Krebs, C.; Stubbe, J. Yfae, a Ferredoxin Involved in Diferric-Tyrosyl Radical Maintenance in Escherichia coli Ribonucleotide Reductase. Biochemistry 2007, 46, 11577–11588. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Imlay, J.A. Hydrogen Peroxide Inactivates the Escherichia coli Isc Iron-Sulphur Assembly System, and Oxyr Induces the Suf System to Compensate. Mol. Microbiol. 2010, 78, 1448–1467. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Imlay, J.A. Micromolar Intracellular Hydrogen Peroxide Disrupts Metabolism by Damaging Iron-Sulfur Enzymes. J. Biol. Chem. 2007, 282, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Flint, D.H.; Tuminello, J.F.; Emptage, M.H. The Inactivation of Fe-S Cluster Containing Hydro-Lyases by Superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [PubMed]
- Klinge, S.; Hirst, J.; Maman, J.D.; Krude, T.; Pellegrini, L. An Iron-Sulfur Domain of the Eukaryotic Primase Is Essential for RNA Primer Synthesis. Nat. Struct. Mol. Biol. 2007, 14, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Weiner, B.E.; Huang, H.; Dattilo, B.M.; Nilges, M.J.; Fanning, E.; Chazin, W.J. An Iron-Sulfur Cluster in the C-Terminal Domain of the P58 Subunit of Human DNA Primase. J. Biol. Chem. 2007, 282, 33444–33451. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Alvim Kamei, C.L.; Cools, T.; Vanderauwera, S.; Takahashi, N.; Okushima, Y.; Eekhout, T.; Yoshiyama, K.O.; Larkin, J.; Van Den Daele, H.; et al. The Arabidopsis Siamese-Related Cyclin-Dependent Kinase Inhibitors Smr5 and Smr7 Regulate the DNA Damage Checkpoint in Response to Reactive Oxygen Species. Plant Cell 2014, 26, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.D.; Kitchen, L.E.; Au, W.C.; Babic, C.M.; Basrai, M.A. Loss of Sod1 and Lys7 Sensitizes Saccharomyces cerevisiae to Hydroxyurea and DNA Damage Agents and Downregulates Mec1 Pathway Effectors. Mol. Cell. Biol. 2005, 25, 10273–10285. [Google Scholar] [CrossRef] [PubMed]
- Mcculley, A.; Haarer, B.; Viggiano, S.; Karchin, J.; Feng, W. Chemical Suppression of Defects in Mitotic Spindle Assembly, Redox Control, and Sterol Biosynthesis by Hydroxyurea. Genes Genomes Genet. 2014, 4, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Yin, D.; Dawood, D.H.; Liu, X.; Chen, Y.; Ma, Z. Functional Characterization of Fgerg3 and Fgerg5 Associated with Ergosterol Biosynthesis, Vegetative Differentiation and Virulence of Fusarium Graminearum. Fungal Genet. Biol. 2014, 68, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Gould, K.L.; Simanis, V. The Control of Septum Formation in Fission Yeast. Genes Dev. 1997, 11, 2939–2951. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.A.; Morabito, L.; Holloway, S.L. A Novel Yeast Mutant That Is Defective in Regulation of the Anaphase-Promoting Complex by the Spindle Damage Checkpoint. Mol. Genet. Genom. 2003, 270, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Gruneberg, U.; Nigg, E.A. Regulation of Cell Division: Stop the Sin! Trends Cell Biol. 2003, 13, 159–162. [Google Scholar] [CrossRef]
- Yanagida, M. Gene Products Required for Chromosome Separation. J. Cell Sci. Suppl. 1989, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Jwa, M.; Song, K. Byr4, a Dosage-Dependent Regulator of Cytokinesis in S. Pombe, Interacts with a Possible Small Gtpase Pathway Including Spg1 and Cdc16. Mol. Cells 1998, 8, 240–245. [Google Scholar] [PubMed]
- Lee, I.J.; Coffman, V.C.; Wu, J.Q. Contractile-Ring Assembly in Fission Yeast Cytokinesis: Recent Advances and New Perspectives. Cytoskeleton 2012, 69, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Bathe, M.; Chang, F. Cytokinesis and the Contractile Ring in Fission Yeast: Towards a Systems-Level Understanding. Trends Microbiol. 2010, 18, 38–45. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Potential Targets | Discovery Methods | Organisms | Biological Functions | Ref. |
|---|---|---|---|---|
| Catalase | Genetics | A. thaliana | Decomposition of H2O2 | [52] |
| Carbonic anhydrase | in vitro | ? | Interconversion of CO2 and H2O to H2CO3 | [53] |
| Matrix metalloproteinases | in vitro | ? | Cleavage of the peptide bond | [54] |
| Unknown yet | Genetics | S. pombe | Cytokinesis | [21] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.; Xu, Y.-J. The Cell Killing Mechanisms of Hydroxyurea. Genes 2016, 7, 99. https://doi.org/10.3390/genes7110099
Singh A, Xu Y-J. The Cell Killing Mechanisms of Hydroxyurea. Genes. 2016; 7(11):99. https://doi.org/10.3390/genes7110099
Chicago/Turabian StyleSingh, Amanpreet, and Yong-Jie Xu. 2016. "The Cell Killing Mechanisms of Hydroxyurea" Genes 7, no. 11: 99. https://doi.org/10.3390/genes7110099
APA StyleSingh, A., & Xu, Y.-J. (2016). The Cell Killing Mechanisms of Hydroxyurea. Genes, 7(11), 99. https://doi.org/10.3390/genes7110099