
Steric Clash in the SET Domain of Histone Methyltransferase NSD1 as a Cause of Sotos Syndrome and Its Genetic Heterogeneity in a Brazilian Cohort

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genomic DNA Extraction
2.3. NSD1 Mutation Analysis
2.4. Reverse Transcription-quantitative PCR (RT-qPCR)
2.5. Protein Modeling and Bioinformatics Analysis
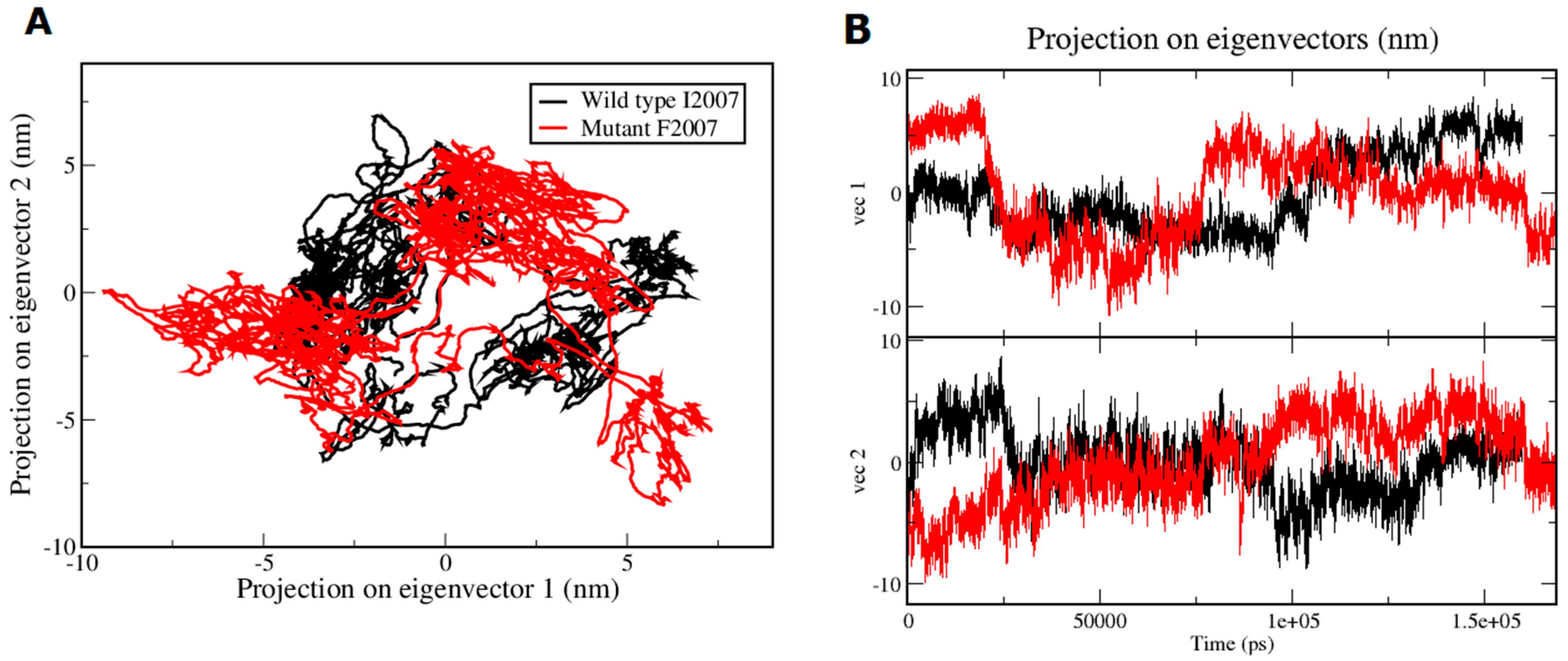
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cecconi, M.; Forzano, F.; Milani, D.; Cavani, S.; Baldo, C.; Selicorni, A.; Pantaleoni, C.; Silengo, M.; Ferrero, G.B.; Scarano, G.; et al. Mutation analysis of the NSD1 gene in a group of 59 patients with congenital overgrowth. Am. J. Med. Genet. A 2005, 134, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.R.; Hughes, H.E. Sotos syndrome: A study of the diagnostic criteria and natural history. J. Med. Genet. 1994, 31, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Sotos, J.F.; Dodge, P.R.; Muirhead, D.; Crawford, J.D.; Talbot, N.B. Cerebral Gigantism in Childhood. A Syndrome of Excessively Rapid Growth and Acromegalic Features and a Nonprogressive Neurologic Disorder. N. Engl. J. Med. 1964, 271, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Douglas, J.; Coleman, K.; Baujat, G.; Cole, T.R.; Das, S.; Horn, D.; Hughes, H.E.; Temple, I.K.; Faravelli, F.; et al. Genotype-phenotype associations in Sotos syndrome: An analysis of 266 individuals with NSD1 aberrations. Am. J. Hum. Genet. 2005, 77, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Saugier-Veber, P.; Bonnet, C.; Afenjar, A.; Drouin-Garraud, V.; Coubes, C.; Fehrenbach, S.; Holder-Espinasse, M.; Roume, J.; Malan, V.; Portnoi, M.F.; et al. Heterogeneity of NSD1 alterations in 116 patients with Sotos syndrome. Hum. Mutat. 2007, 28, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.J.; Ward, R.E.; Moore, E.; Fremion, A.S.; Wappner, R.S. Overgrowth, congenital hypotonia, nystagmus, strabismus, and mental retardation: Variant of dominantly inherited Sotos sequence? Am. J. Med. Genet. 1988, 29, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Zonana, J.; Sotos, J.F.; Romshe, C.A.; Fisher, D.A.; Elders, M.J.; Rimoin, D.L. Dominant inheritance of cerebral gigantism. J. Pediatr. 1977, 91, 251–256. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Rahman, N. Sotos syndrome. Eur. J. Hum. Genet. 2007, 15, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, K.; Kimura, J.; Matsuo, M.; Kurosawa, K.; Masuno, M.; Niikawa, N.; Kuroki, Y. Sotos syndrome associated with a de novo balanced reciprocal translocation t(5;8)(q35;q24.1). Am. J. Med. Genet. 2002, 107, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, N.; Imaizumi, K.; Harada, N.; Masuno, M.; Kondoh, T.; Nagai, T.; Ohashi, H.; Naritomi, K.; Tsukahara, M.; Makita, Y.; et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 2002, 30, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Hanks, S.; Temple, I.K.; Davies, S.; Murray, A.; Upadhyaya, M.; Tomkins, S.; Hughes, H.E.; Cole, T.R.P.; Rahman, N. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am. J. Hum. Genet. 2003, 72, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Faravelli, F. NSD1 mutations in Sotos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2005, 137C, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, D.J.; Raca, G.; Welch, K.; Dempsey, M.; Anderes, E.; Ostrovnaya, I.; Alkhateeb, A.; Kamimura, J.; Matsumoto, N.; Schaeffer, G.B.; et al. NSD1 analysis for Sotos syndrome: Insights and perspectives from the clinical laboratory. Genet. Med. 2005, 7, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, N.; Harada, N.; Shimokawa, O.; Miyake, N.; Kawame, H.; Uetake, K.; Makita, Y.; Kondoh, T.; Ogata, T.; Hasegawa, T.; et al. Fifty microdeletions among 112 cases of Sotos syndrome: Low copy repeats possibly mediate the common deletion. Hum. Mutat. 2003, 22, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Matsumoto, N.; Kurotaki, N.; Harada, N.; Niikawa, N.; Ogata, T.; Imaizumi, K.; Kurosawa, K.; Kondoh, T.; Ohashi, H.; et al. Sotos syndrome and haploinsufficiency of NSD1: Clinical features of intragenic mutations and submicroscopic deletions. J. Med. Genet. 2003, 40, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Z.; Khan, S.I.; Horton, J.R.; Tamaru, H.; Selker, E.U.; Cheng, X. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell 2003, 12, 177–185. [Google Scholar] [CrossRef]
- Zhang, X.; Tamaru, H.; Khan, S.I.; Horton, J.R.; Keefe, L.J.; Selker, E.U.; Cheng, X. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell 2002, 111, 117–127. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Kuznetsova, E.; Hajian, T.; Wu, H.; Dombrovski, L.; Li, Y.; Gräslund, S.; Arrowsmith, C.H.; Schapira, M.; Vedadi, M. A Basic Post-SET Extension of NSDs Is Essential for Nucleosome Binding In Vitro. J. Biomol. Screen. 2014, 19, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Pasillas, M.P.; Shah, M.; Kamps, M.P. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Hum. Mutat. 2011, 32, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Stec, I.; Nagl, S.B.; van Ommen, G.J.; den Dunnen, J.T. The PWWP domain: A potential protein-protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett. 2000, 473, 1–5. [Google Scholar] [CrossRef]
- Wang, X.; Yeh, S.; Wu, G.; Hsu, C.; Wang, L.; Chiang, T.; Yang, Y.; Guo, Y.; Chang, C. Identification and characterization of a novel androgen receptor coregulator ARA267-alpha in prostate cancer cells. J. Biol. Chem. 2001, 276, 40417–40423. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, N.; Harada, N.; Yoshiura, K.; Sugano, S.; Niikawa, N.; Matsumoto, N. Molecular characterization of NSD1, a human homologue of the mouse Nsd1 gene. Gene 2001, 279, 197–204. [Google Scholar] [CrossRef]
- Huang, N.; vom Baur, E.; Garnier, J.M.; Lerouge, T.; Vonesch, J.L.; Lutz, Y.; Chambon, P.; Losson, R. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J. 1998, 17, 3398–3412. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Ropero, S.; Setien, F.; Fraga, M.F.; Lapunzina, P.; Losson, R.; Alaminos, M.; Cheung, N.K.; Rahman, N.; Esteller, M. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl. Acad. Sci. USA 2009, 106, 21830–21835. [Google Scholar] [CrossRef] [PubMed]
- Rayasam, G.V.; Wendling, O.; Angrand, P.O.; Mark, M.; Niederreither, K.; Song, L.; Lerouge, T.; Hager, G.L.; Chambon, P.; Losson, R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003, 22, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.; Kant, S.; Karperien, M.; van Beers, L.; Tjon, J.; Vink, G.R.; van Tol, D.; Dauwerse, H.; le Cessie, S.; Beemer, F.A.; et al. Genotype-phenotype correlation in patients suspected of having Sotos syndrome. Horm Res. 2004, 62, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Rio, M.; Clech, L.; Amiel, J.; Faivre, L.; Lyonnet, S.; le Merrer, M.; Odent, S.; Lacombe, D.; Edery, P.; Brauner, R.; et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J. Med. Genet. 2003, 40, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Duno, M.; Skovby, F.; Schwartz, M. Leukocyte cDNA analysis of NSD1 derived from confirmed Sotos syndrome patients. Ann. Hum. Genet. 2007, 71, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.E.; Turnpenny, P.; McConnell, V.P. Phenotypic variability in a three-generation Northern Irish family with Sotos syndrome. Clin. Dysmorphol. 2011, 20, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Labonne, J.D.; Chung, M.J.; Jones, J.R.; Anand, P.; Wenzel, W.; Iacoboni, D.; Layman, L.C.; Kim, H.G. Concomitant partial exon skipping by a unique missense mutation of RPS6KA3 causes Coffin-Lowry syndrome. Gene 2016, 575, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Kutzner, C.; van der Spoel, D.; Fechner, M.; Lindahl, E.; Schmitt, U.W.; de Groot, B.L.; Grubmüller, H. Speeding up parallel GROMACS on high-latency networks. J. Comput. Chem. 2007, 28, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins 2007, 66, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Poudel, S.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016, 44, D336–D342. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012, 40, D302–D305. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Kotilainen, J.; Pohjola, P.; Pirinen, S.; Arte, S.; Nieminen, P. Premolar hypodontia is a common feature in Sotos syndrome with a mutation in the NSD1 gene. Am. J. Med. Genet. A 2009, 149A, 2409–2414. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.M.; Hau, E.W.; Lo, I.F.; Chan, D.H.; Lam, S.T. Spectrum of NSD1 gene mutations in southern Chinese patients with Sotos syndrome. Chin. Med. J. (Engl.) 2005, 118, 1499–1506. [Google Scholar] [PubMed]
- Nicita, F.; Tarani, L.; Spalice, A.; Grasso, M.; Papetti, L.; Cecconi, M.; di Biasi, C.; Ursitti, F.; Iannetti, P. Novel missense mutation (L1917P) involving sac-domain of NSD1 gene in a patient with Sotos syndrome. J. Genet. 2011, 90, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Li, Y.; Chen, Z.; Wang, M.; Reinberg, D.; Xu, R.M. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J. Biol. Chem. 2011, 286, 8361–8368. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting the Effects of Amino Acid Substitutions on Protein Function. Annu. Rev. Genom. Hum. Genet. 2006, 7, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Accounting for Human Polymorphisms Predicted to Affect Protein Function. Genome Res. 2002, 12, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [PubMed]
- Morishita, M.; di Luccio, E. Structural insights into the regulation and the recognition of histone marks by the SET domain of NSD1. Biochem. Biophys. Res. Commun. 2011, 412, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Kozbial, P.Z.; Mushegian, A.R. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 2005, 5. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.A.; McCarthy, J.G. The structure of two N-methyltransferases from the caffeine biosynthetic pathway. Plant Physiol. 2007, 144, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Kudithipudi, S.; Lungu, C.; Rathert, P.; Happel, N.; Jeltsch, A. Substrate specificity analysis and novel substrates of the protein lysine methyltransferase NSD1. Chem. Biol. 2014, 21, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, S.; Gillessen-Kaesbach, G.; Meinecke, P.; Albrecht, B.; Neumann, L.M.; Hesse, V.; Palanduz, S.; Balg, S.; Majewski, F.; Fuchs, S.; et al. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur. J. Hum. Genet. 2003, 11, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Fagali, C.; Kok, F.; Nicola, P.; Kim, C.; Bertola, D.; Albano, L.; Koiffmann, C.P. MLPA analysis in 30 Sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur. J. Med. Genet. 2009, 52, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Rahman, N. Clinical features of NSD1-positive Sotos syndrome. Clin. Dysmorphol. 2004, 13, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, S.R.; Smith, E.; Shilatifard, A. Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007, 14, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA 2010, 107, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Caballero, O.L.; Levy, S.; Stevenson, B.J.; Iseli, C.; de Souza, S.J.; Galante, P.A.; Busam, D.; Leversha, M.A.; Chadalavada, K.; et al. Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc. Natl. Acad. Sci. USA 2009, 106, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002, 12, 198–209. [Google Scholar] [CrossRef]
- Xiao, B.; Wilson, J.R.; Gamblin, S.J. SET domains and histone methylation. Curr. Opin. Struct. Biol. 2003, 13, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.; Houtz, R.L.; Hurley, J.H. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, Y. Catalytic mechanism and product specificity of the histone lysine methyltransferase SET7/9: An ab initio QM/MM-FE study with multiple initial structures. J. Am. Chem. Soc. 2006, 128, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, G.M.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Stec, I.; Wright, T.J.; van Ommen, G.J.; de Boer, P.A.; van Haeringen, A.; Moorman, A.F.; Altherr, M.R.; den Dunnen, J.T. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet. 1998, 7, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Tonon, G.; Wong, K.K.; Maulik, G.; Brennan, C.; Feng, B.; Zhang, Y.; Khatry, D.B.; Protopopov, A.; You, M.J.; Aguirre, A.J.; et al. High-resolution genomic profiles of human lung cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 9625–9630. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Smidt, M.; Banning, M.J.; Oudakker, A.R.; Van Esch, H.; de Brouwer, A.P.; Nillesen, W.; Sistermans, E.A.; Hamel, B.C.; de Bruijn, D.; et al. Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J. Med. Genet. 2005, 42, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.T.; Hood, R.L.; Zhan, S.H.; Bulman, D.E.; Fejes, A.P.; Moore, R.; Mungall, A.J.; Eydoux, P.; Babul-Hirji, R.; An, J.; et al. Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 2012, 90, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Coleman, K.; Tatton-Brown, K.; Hughes, H.E.; Temple, I.K.; Cole, T.R.; Rahman, N. Childhood Overgrowth Collaboration. Evaluation of NSD2 and NSD3 in overgrowth syndromes. Eur. J. Hum. Genet. 2005, 13, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Ginter, D.N.; Scott, C.I. Cerebral gigantism. Birth Defects Orig. Artic. Ser. 1975, 11, 415–422. [Google Scholar] [PubMed]
- Li, M.; Shuman, C.; Fei, Y.L.; Cutiongco, E.; Bender, H.A.; Stevens, C.; Wilkins-Haug, L.; Day-Salvatore, D.; Yong, S.L.; Geraghty, M.T.; et al. GPC3 mutation analysis in a spectrum of patients with overgrowth expands the phenotype of Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. 2001, 102, 161–168. [Google Scholar] [CrossRef]
- Baujat, G.; Rio, M.; Rossignol, S.; Sanlaville, D.; Lyonnet, S.; le Merrer, M.; Munnich, A.; Gicquel, C.; Cormier-Daire, V.; Colleaux, L. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am. J. Hum. Genet. 2004, 74, 715–720. [Google Scholar] [CrossRef] [PubMed]
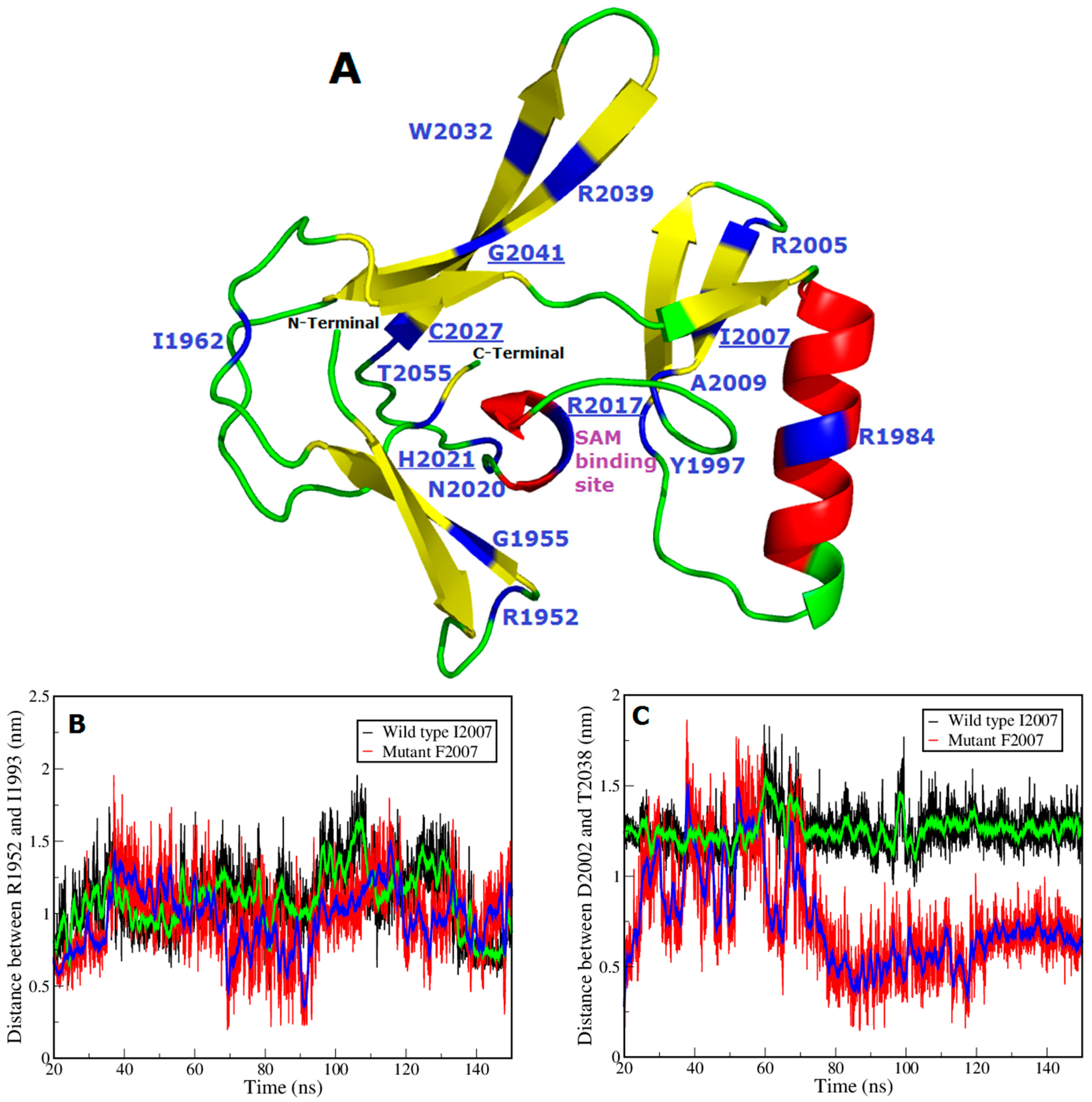

{kind=link}
{kind=link}
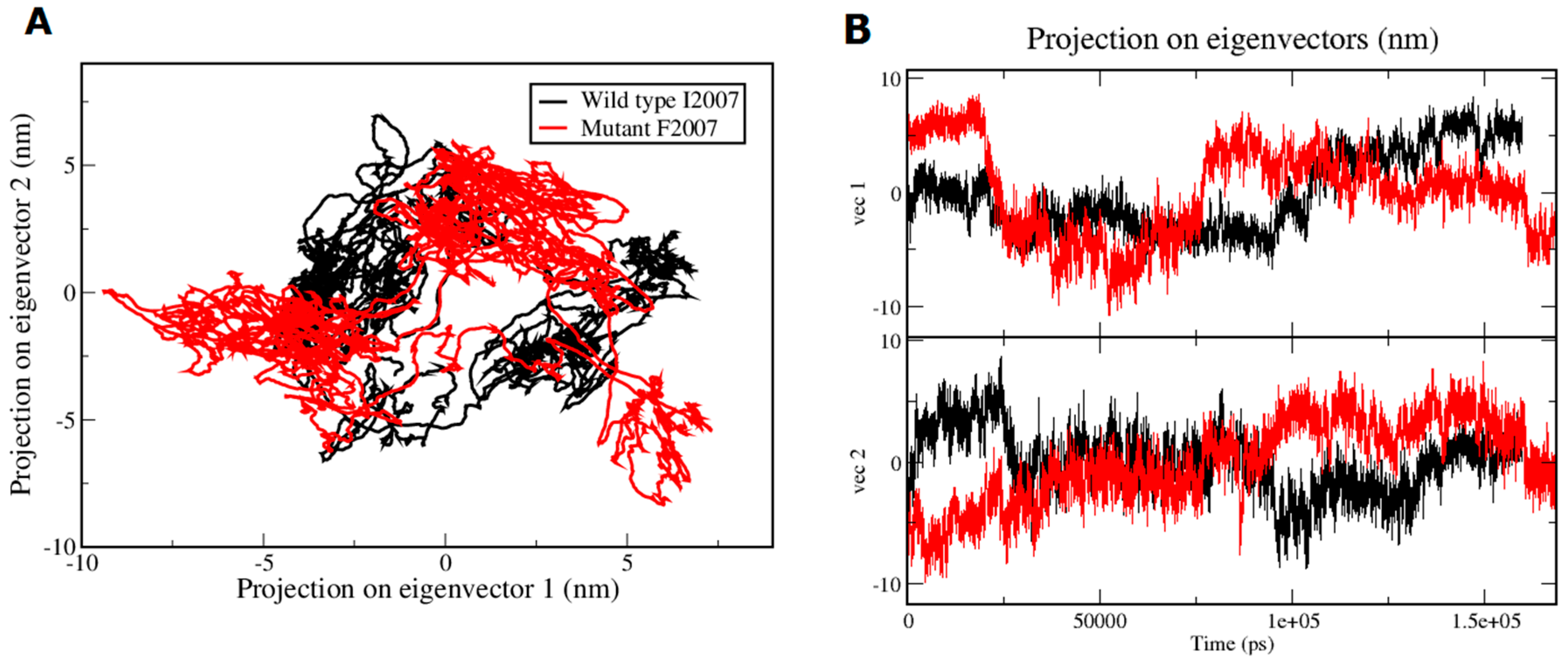
{kind=link}
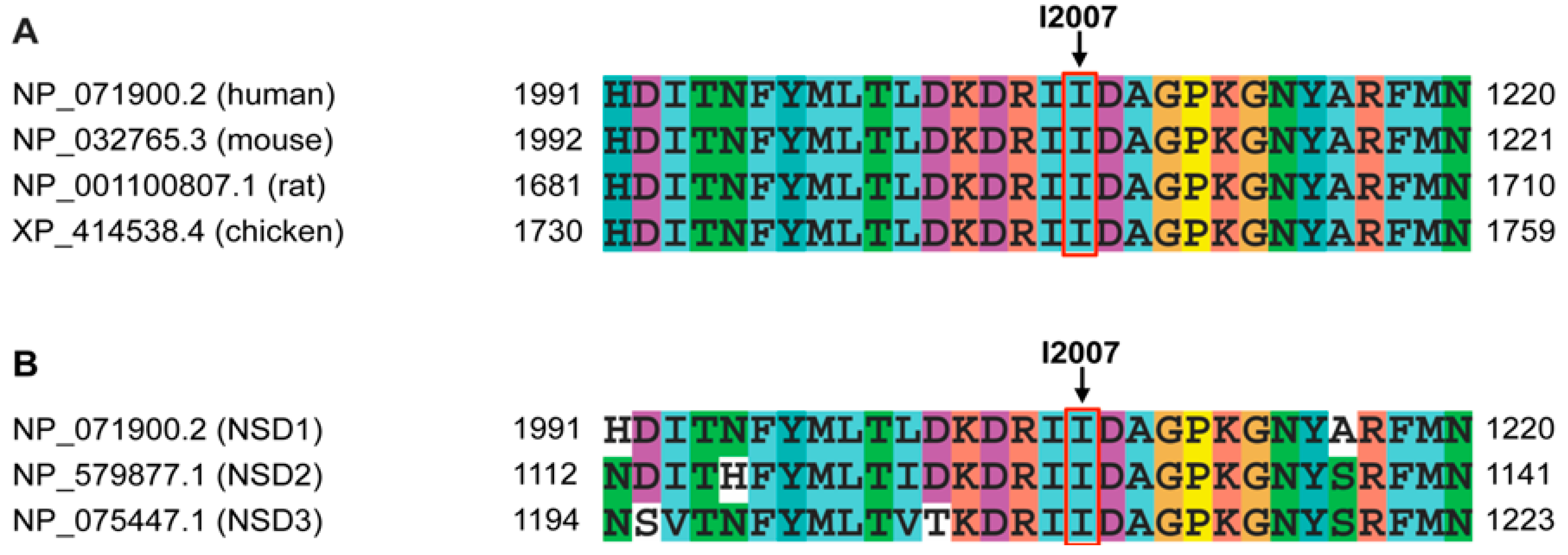
{kind=link}
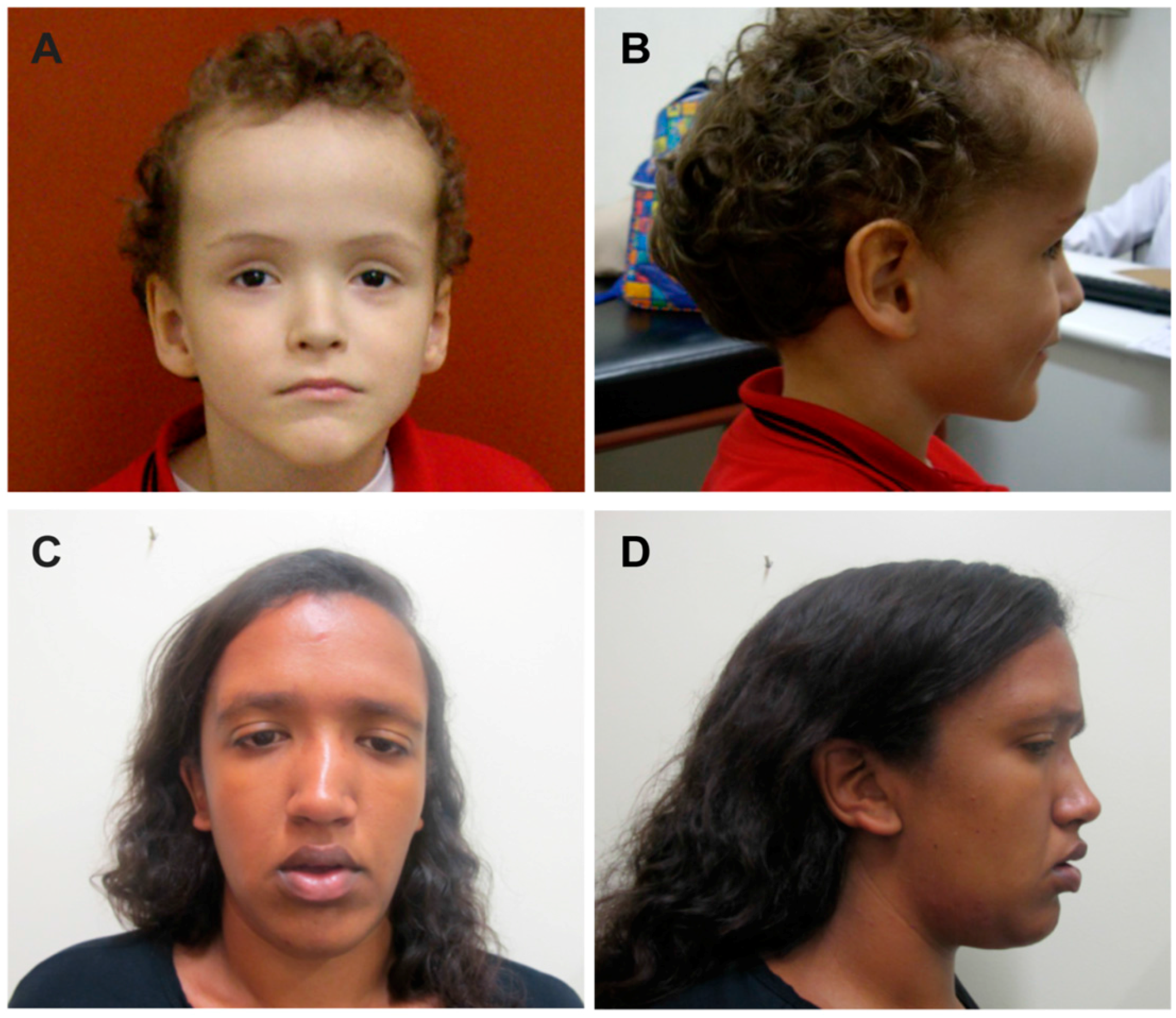
{kind=link}
{kind=link}
| Domains | Amino acid Positions |
|---|---|
| PWWP1 | 323–388 |
| NLS1 | 512–529 |
| NLS2 | 1157–1174 |
| NLS3 | 1471–1488 |
| PHD1 | 1543–1589 |
| PHD2 | 1590–1639 |
| PHD3 | 1640–1693 |
| PHD4 | 1707–1751 |
| PWWP2 | 1756–1818 |
| AWS | 1890–1940 |
| SET | 1942–2065 |
| Post-SET | 2066–2082 |
| PHD5 | 2120–2160 |
| Patient | DGDP186 (Male) | DGDP 291 (Female) | DGDP306 (Female) |
|---|---|---|---|
| Age at diagnosis | 9 months | 2 years 7 months | 1 year 5 months |
| Current age | 10 years | 23 year | 5 years |
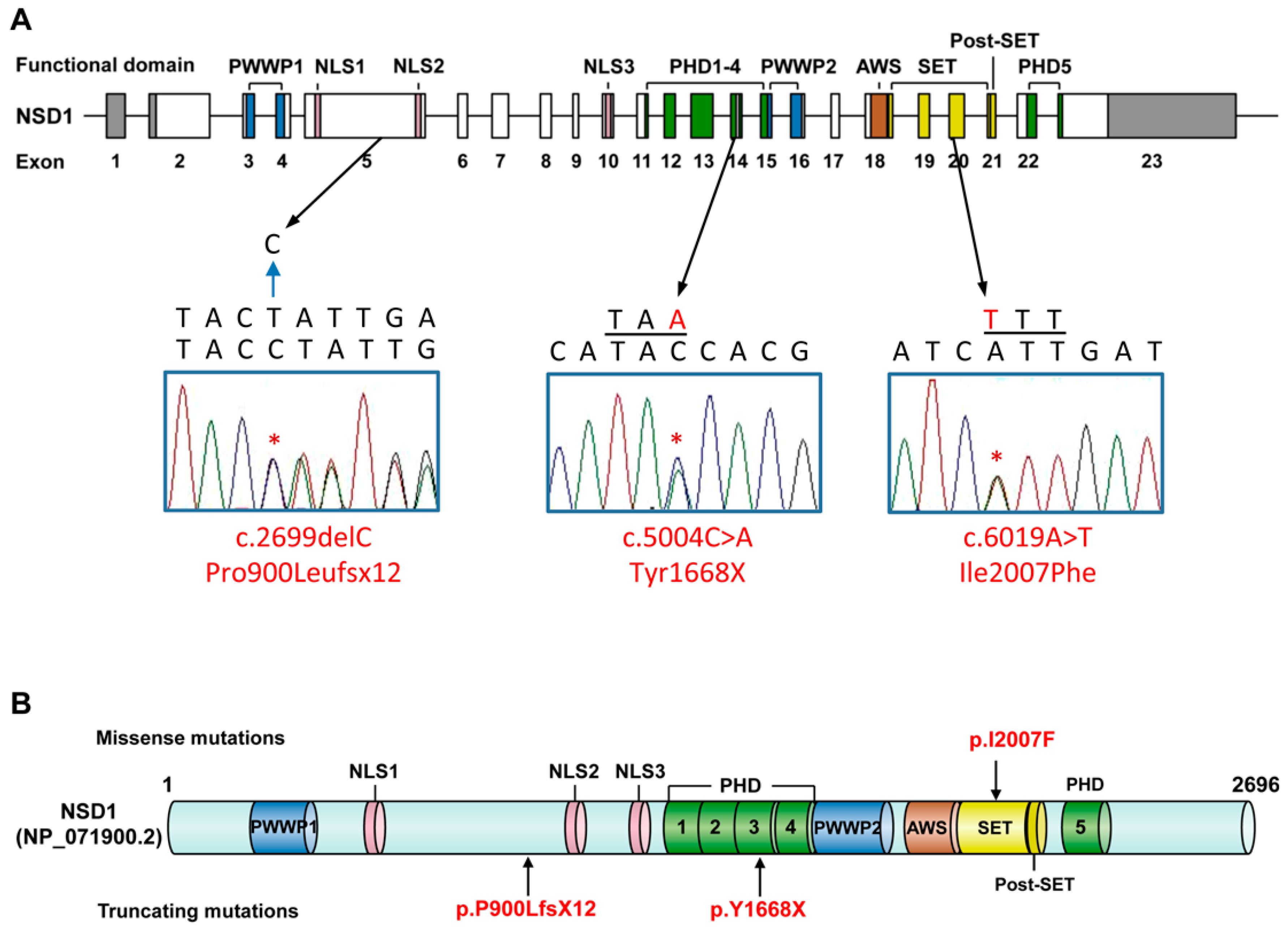
| Nucleotide change (NM_022455.4) | c.5004C>A | c.6019A>T | c.2699delC |
| Exon | 14 | 20 | 5 |
| Amino acid change (NP_071900.2) | p.Y1668X | p.I2007F | p.P900LfsX12 |
| Familial analysis | de novo | Both mother and brother with no variant, father unavailable | de novo |
| Brazilian control analysis | not found in 600 controls | not found in 600 controls | not found in 600 controls |
| Developmental Milestone | |||
| Prenatal overgrowth | − | − | − |
| Excessive growth velocity | + | + | + |
| Advanced bone age | − | + | + |
| Large hands and feet | + | + | + |
| Developmental delay | Mild | Mild | Mild |
| Lack of fine motor control | − | − | − |
| Walking at | 22 months | 24 months | 23 months |
| Speaking | Delayed | Delayed | Delayed |
| Delayed learning ability | − | + | N/A |
| Facial Dysmorphism | |||
| Macrocephaly | + | + | + |
| Prominent forehead | + | + | + |
| High-arched palate | + | − | + |
| Pointed chin | + | + | + |
| Other Features | |||
| Hypotonia | + | + | + |
| Seizures | − | − | + |
| Scoliosis | + | + | − |
| Enlarged ventricles | + | N/A | + |
| Artrial septal defect | − | + | − |
| Pulmonary stenosis | − | + | − |
| Patient | DGDP168 | DGDP171 | DGDP173 | DGDP174 | DGDP176 | DGDP180 | DGDP183 | DGDP305 |
|---|---|---|---|---|---|---|---|---|
| Age at diagnosis | 7 years | 14 years 3 months | 8 years 3 months | 9 months | 1 years 6 months | 1 years 9 months | 7 years | 2 years 8 months |
| Current age | 25 years | 27 years | 18 years | 13 years | 20 years | 15 years | 18 years | 7 years |
| Nucleotide change (NM_022455.4) | 3004_3005 delAA | 5750T>A | 4740delA | 6050G>A | 5892+1G>T | 2954_2955 delCT | 1894C>T | 5965C>T |
| Exon or intron | 5 | 18 | 12 | 20 | Intron 19 | 5 | 5 | 19 |
| Amino acid change (NP_071900.2) | p.K1002EfsX8 | p.L1917P | p.K1580NfsX62 | p.R2017Q | Exon 19 skipping | p.S985CfsX25 | p.R632X | p.Q1989X |
| Developmental Milestone | ||||||||
| Prenatal overgrowth | + | + | + | − | + | − | − | + |
| Excessive growth velocity | + | + | + | − | + | + | + | + |
| Advanced bone age | − | + | + | + | + | + | − | + |
| Large hands and feet | + | + | + | + | + | + | + | + |
| Developmental delay | Mild | Mild | Mild | Mild | Mild | Mild | Mild | Mild |
| Lack of fine motor control | − | + | + | + | + | + | − | + |
| Walking at | 15 months | 24 months | 18 months | 20 months | 17 months | 20 months | Delayed | 20 months |
| Speaking | Delayed | Delayed | Normal | Delayed | Delayed | Delayed | Delayed | Normal |
| Delayed learning ability | + | + | + | + | + | + | + | + |
| Facial Dysmorphism | ||||||||
| Macrocephaly | + | + | + | − | + | + | + | + |
| Prominent forehead | + | + | + | + | + | + | + | + |
| High-arched palate | − | + | + | + | + | + | + | + |
| Pointed chin | + | + | + | + | + | + | + | + |
| Other features | ||||||||
| Hypotonia | + | + | + | + | + | + | + | + |
| Seizures | − | − | − | − | − | − | + | − |
| Scoliosis | − | − | − | − | + | + | − | + |
| Enlarged ventricles | + | + | + | + | + | − | − | − |
| AA Position | Polyphen2 (Predicted Impact) | PolyPhen2 (PSIC Score) | SPPIDER | SIFT Predicted Impact (Tolerance Index) | DDG Value Kcal/Mol | I-Mutant 2.0 Predicted Impact | PANTHER (subPSEC) |
|---|---|---|---|---|---|---|---|
| p.R1952W | Probably damaging | 0.999 | Non-interacting | Intolerant (0.00) | 0.18 | Increase Stability | −3.04478 |
| p.G1955D | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −1.47 | Decrease Stability | −7.27103 |
| p.I1962T | Possibly damaging | 0.934 | Non-interacting | Intolerant (0.00) | −0.95 | Decrease Stability | −2.82147 |
| p.R1984G | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −0.93 | Decrease Stability | −6.53802 |
| p.R1984Q | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −0.53 | Decrease Stability | −6.59809 |
| p.Y1997H | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −0.95 | Decrease Stability | −3.31488 |
| p.Y1997S | Probably damaging | 0.999 | Non-interacting | Intolerant (0.00) | −0.70 | Decrease Stability | −3.43065 |
| p.Y1997C | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −1.22 | Decrease Stability | −4.51444 |
| p.R2005Q | Probably damaging | 1.000 | Non-interacting | Intolerant (0.01) | −1.13 | Decrease Stability | −1.95697 |
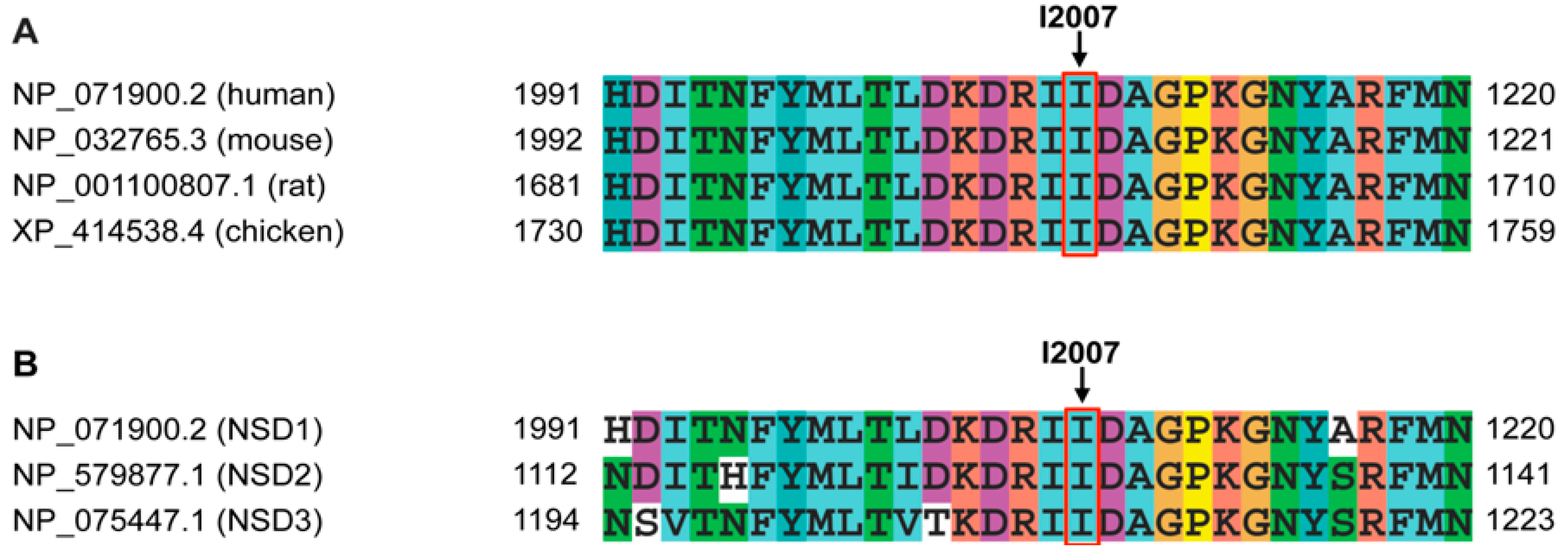
| p.I2007F | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −1.65 | Decrease Stability | −2.97211 |
| p.A2009V | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −1.25 | Decrease Stability | −2.74529 |
| p.R2017Q | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −0.27 | Decrease Stability | −6.4321 |
| p.R2017W | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | 0.02 | Increase Stability | −8.09732 |
| p.N2020S | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −0.81 | Decrease Stability | −6.32023 |
| p.H2021R | Probably damaging | 1.000 | Non-interacting | Intolerant (0.01) | 0.16 | Increase Stability | −9.24832 |
| p.C2027R | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | 0.31 | Increase Stability | −3.94723 |
| p.C2027Y | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −0.52 | Decrease Stability | −4.23261 |
| p.W2032L | Probably damaging | 0.999 | Non-interacting | Intolerant (0.001) | −1.03 | Decrease Stability | −6.15612 |
| p.R2039C | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −1.58 | Decrease Stability | −3.90311 |
| p.G2041D | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | −2.27 | Decrease Stability | −3.7109 |
| p.T2055I | Probably damaging | 0.999 | Non-interacting | Intolerant (0.00) | −0.97 | Decrease Stability | −3.05772 |
| p.Y2058C | Probably damaging | 1.000 | Non-interacting | Intolerant (0.00) | 0.27 | Increase Stability | −7.73506 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, K.; Anand, P.; Lee, J.A.; Jones, J.R.; Kim, C.A.; Bertola, D.R.; Labonne, J.D.J.; Layman, L.C.; Wenzel, W.; Kim, H.-G. Steric Clash in the SET Domain of Histone Methyltransferase NSD1 as a Cause of Sotos Syndrome and Its Genetic Heterogeneity in a Brazilian Cohort. Genes 2016, 7, 96. https://doi.org/10.3390/genes7110096
Ha K, Anand P, Lee JA, Jones JR, Kim CA, Bertola DR, Labonne JDJ, Layman LC, Wenzel W, Kim H-G. Steric Clash in the SET Domain of Histone Methyltransferase NSD1 as a Cause of Sotos Syndrome and Its Genetic Heterogeneity in a Brazilian Cohort. Genes. 2016; 7(11):96. https://doi.org/10.3390/genes7110096
Chicago/Turabian StyleHa, Kyungsoo, Priya Anand, Jennifer A. Lee, Julie R. Jones, Chong Ae Kim, Debora Romeo Bertola, Jonathan D. J. Labonne, Lawrence C. Layman, Wolfgang Wenzel, and Hyung-Goo Kim. 2016. "Steric Clash in the SET Domain of Histone Methyltransferase NSD1 as a Cause of Sotos Syndrome and Its Genetic Heterogeneity in a Brazilian Cohort" Genes 7, no. 11: 96. https://doi.org/10.3390/genes7110096
APA StyleHa, K., Anand, P., Lee, J. A., Jones, J. R., Kim, C. A., Bertola, D. R., Labonne, J. D. J., Layman, L. C., Wenzel, W., & Kim, H.-G. (2016). Steric Clash in the SET Domain of Histone Methyltransferase NSD1 as a Cause of Sotos Syndrome and Its Genetic Heterogeneity in a Brazilian Cohort. Genes, 7(11), 96. https://doi.org/10.3390/genes7110096