The SaeRS Two‐Component System of Staphylococcus aureus


Abstract
:1. Introduction
2. Components of the SaeRS TCS
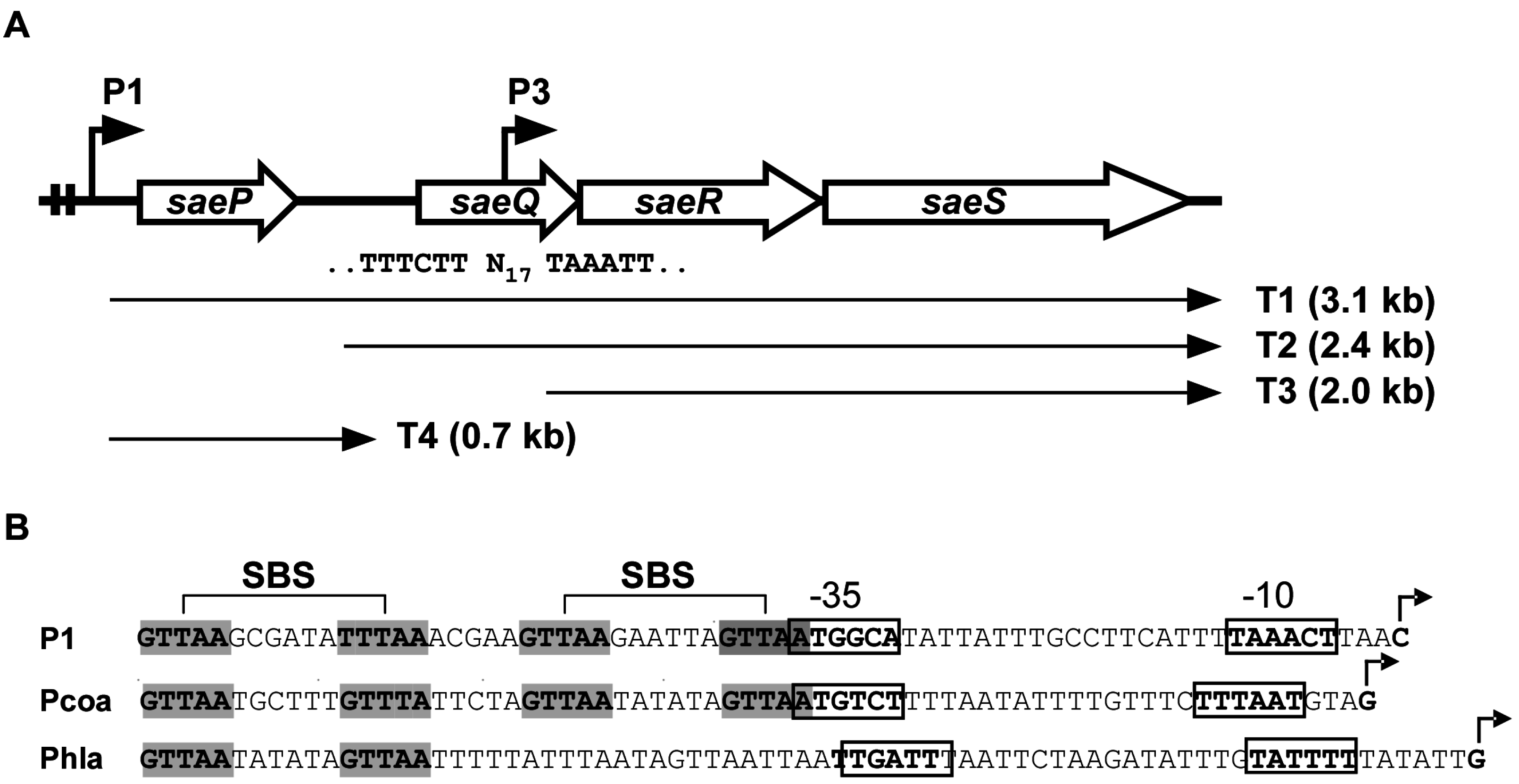
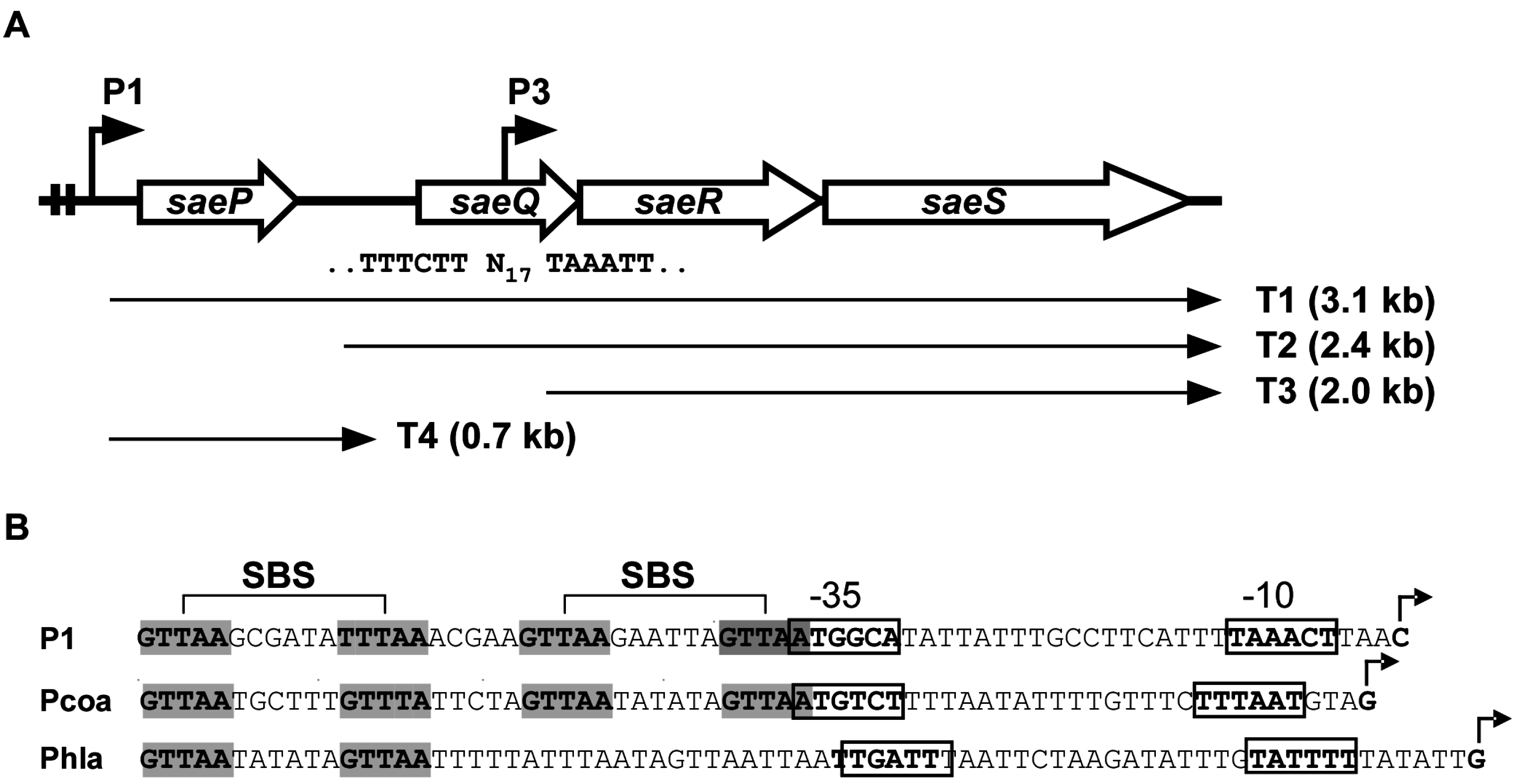
2.1. Structure of the sae Operon
2.1.1. P3 Promoter (saeP3)
2.1.2. P1 Promoter (saeP1)
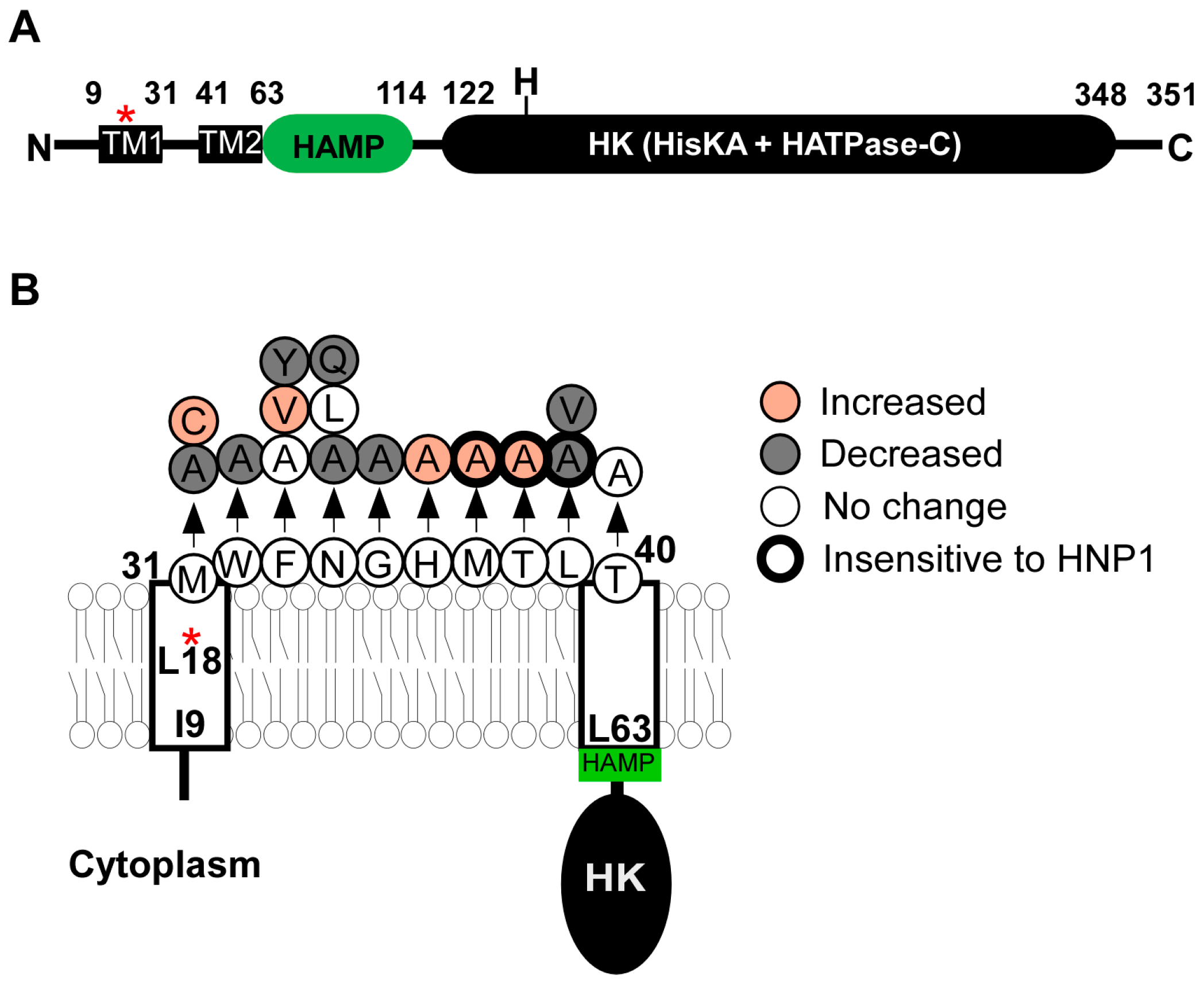
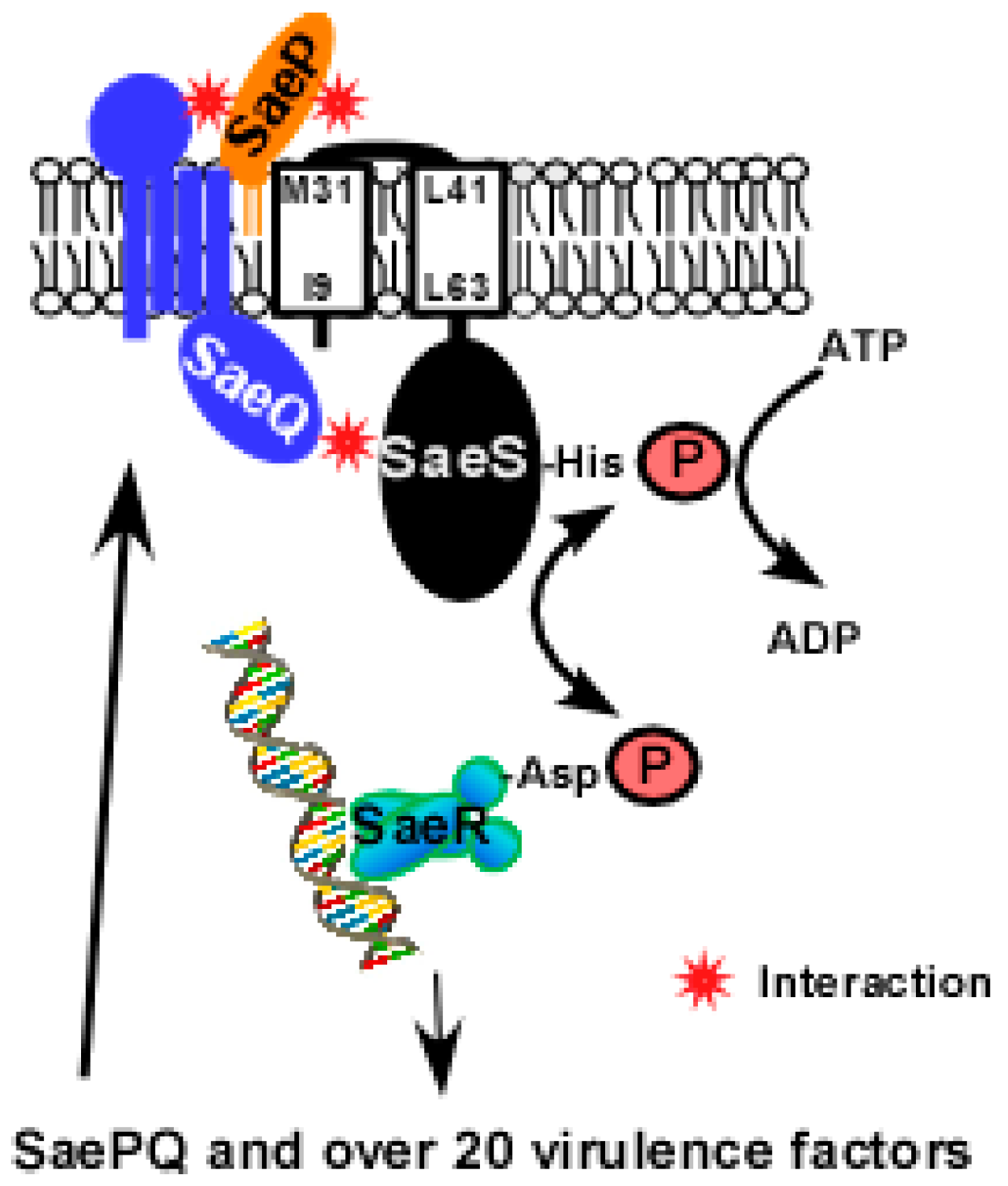
2.2. SaeS
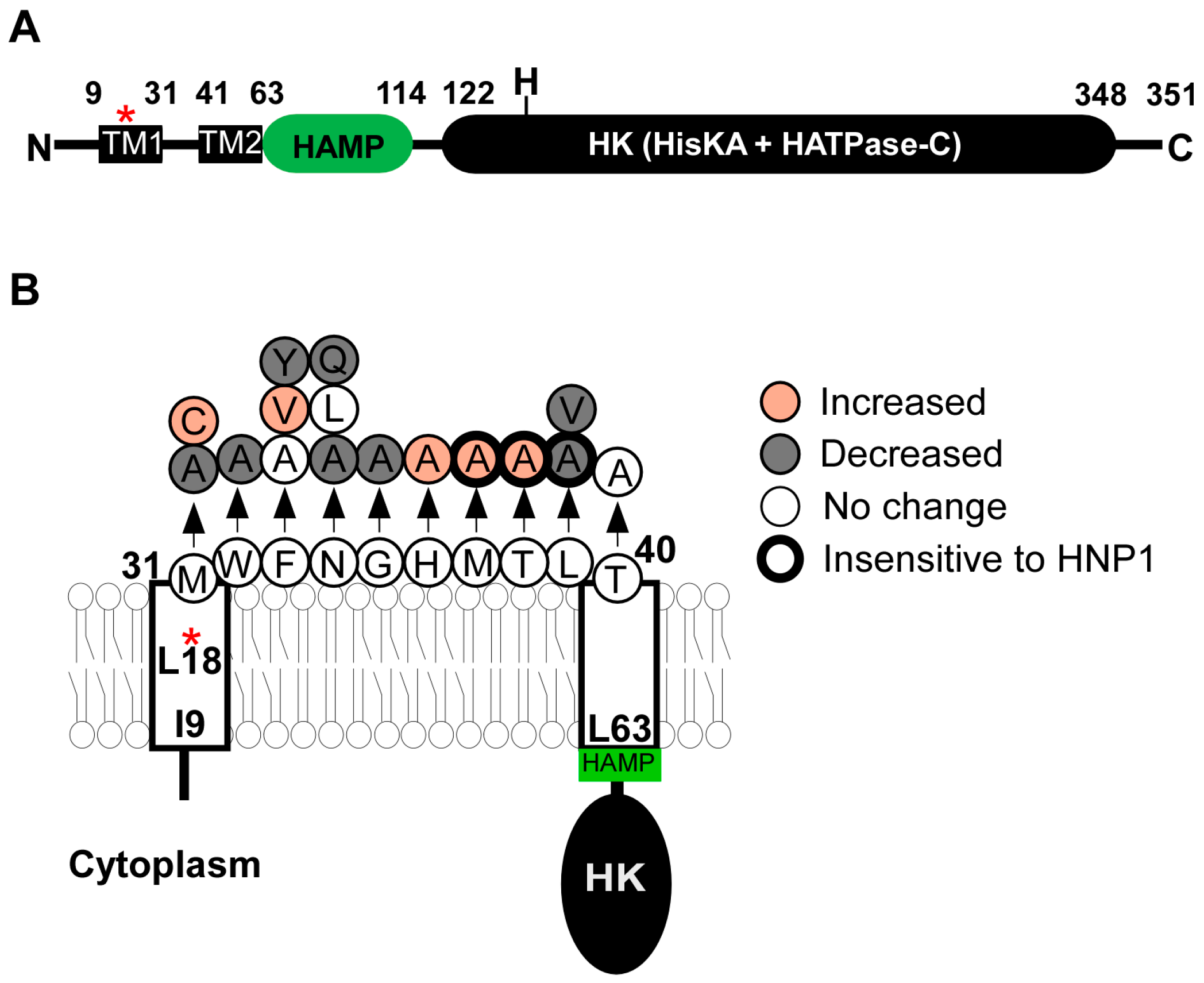
2.2.1. Transmembrane Domain
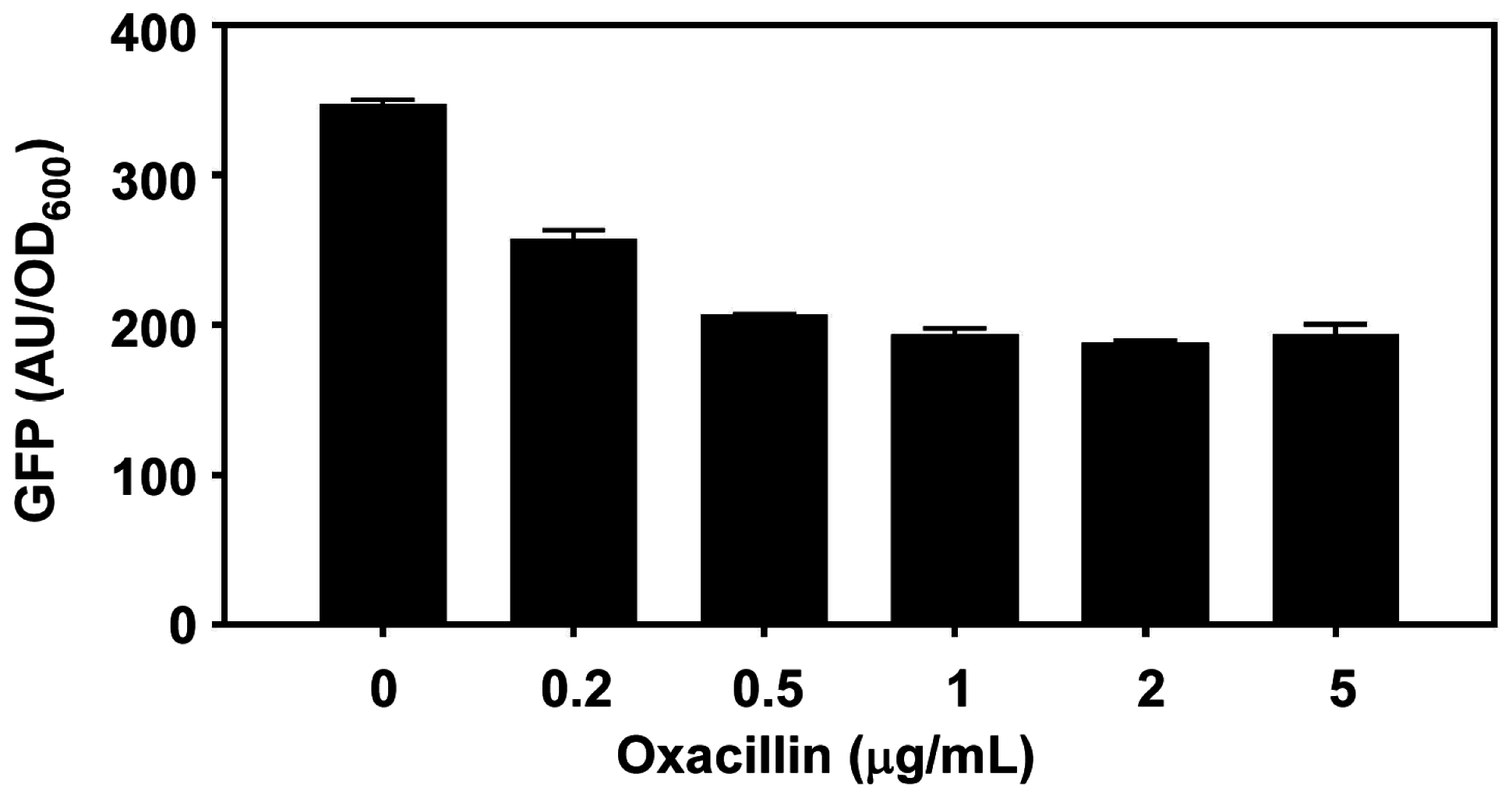
2.2.2. Growth-Phase Dependent Activation
2.3. SaeS Variants
2.3.1. SaeSP (SaeS L18P)
2.3.2. SaeSSK and SaeSSKT
2.4. SaeR
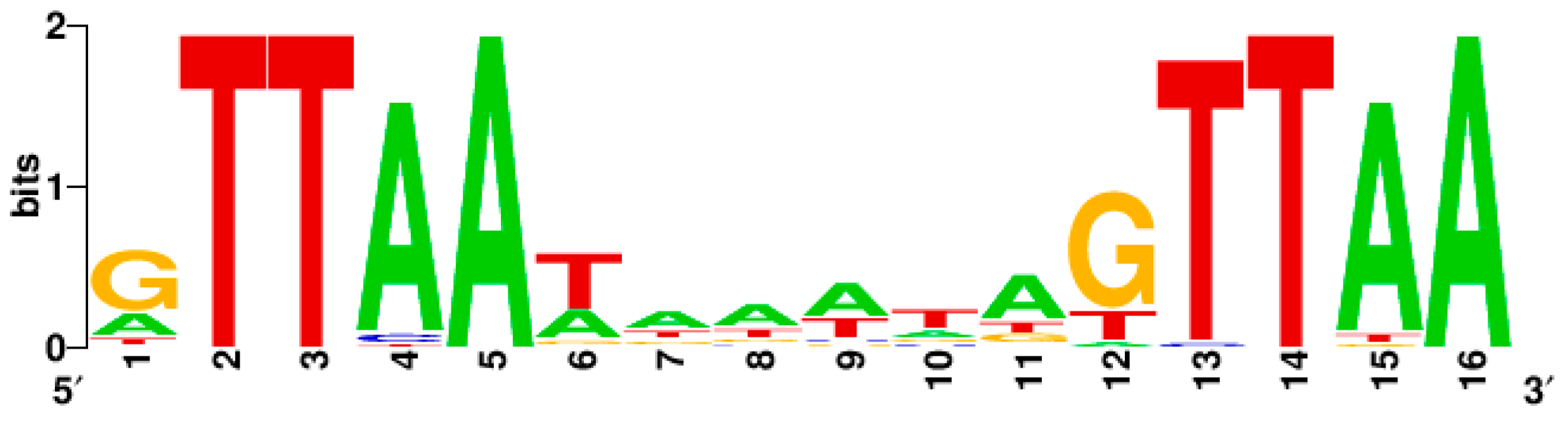
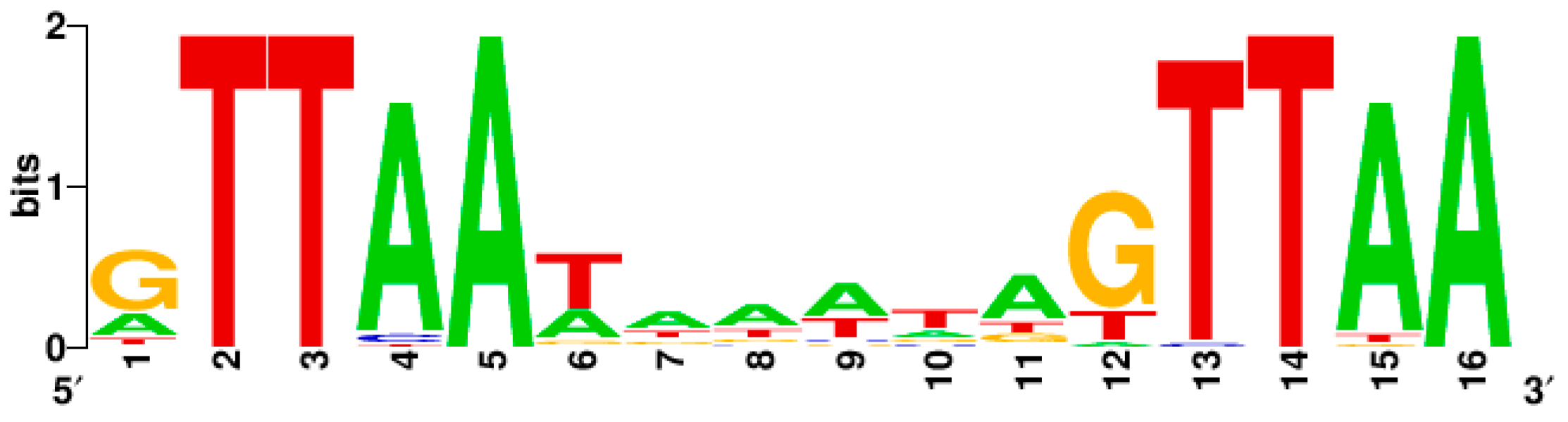
SaeR Binding Sequence (SBS)
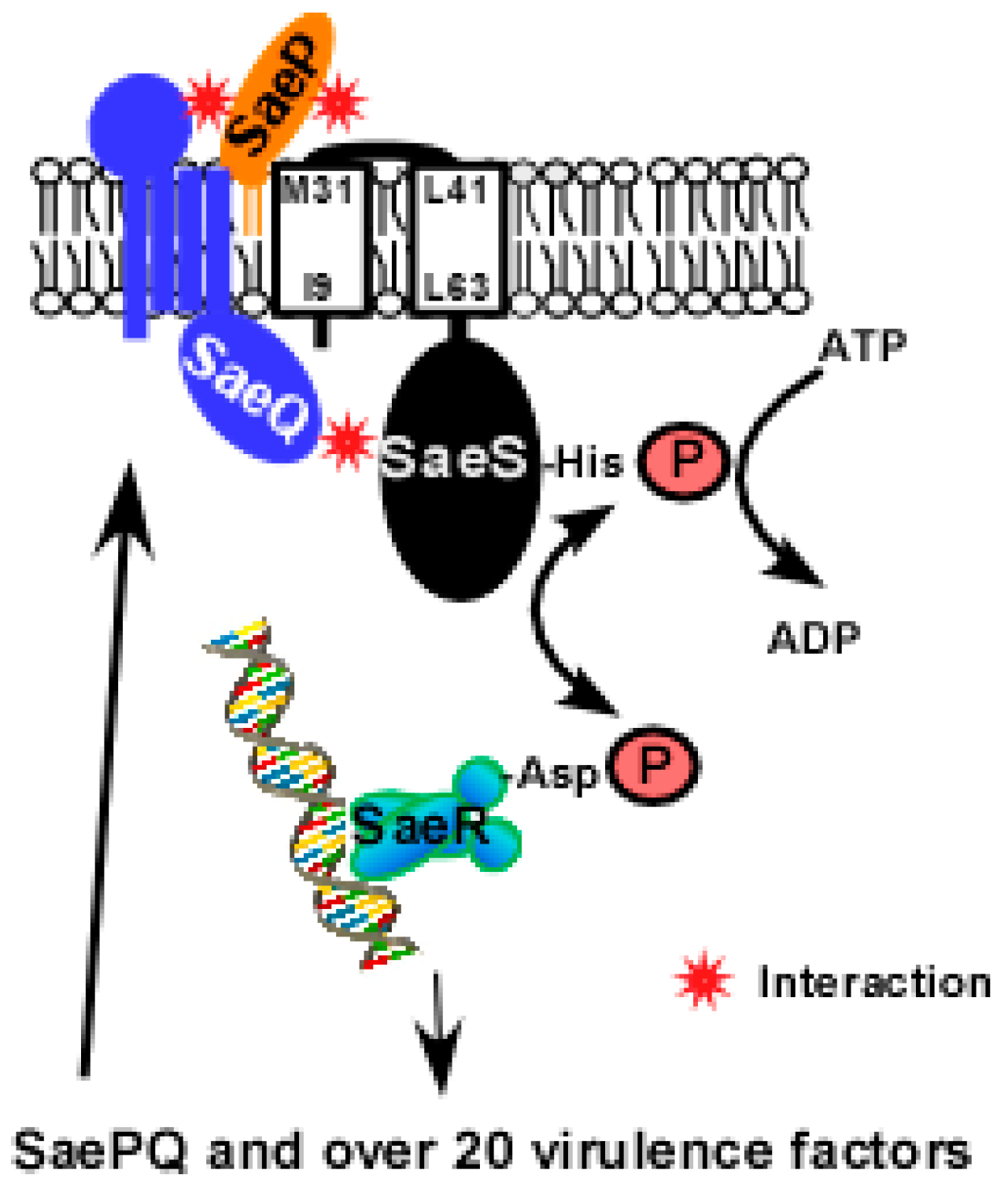
2.5. SaeP and SaeQ
3. Signals
3.1. Activation Signals
3.1.1. Human Neutrophil Peptide 1, 2, and 3 (HNP1-3)
3.1.2. Calprotectin
3.1.3. Other Activation Signals
3.2. Inhibitory Signals
3.2.1. Silkworm Apolipophorin Protein
3.2.2. Acidic pH and 1M NaCl
3.2.3. Other Potential Sae Inhibitory Molecules
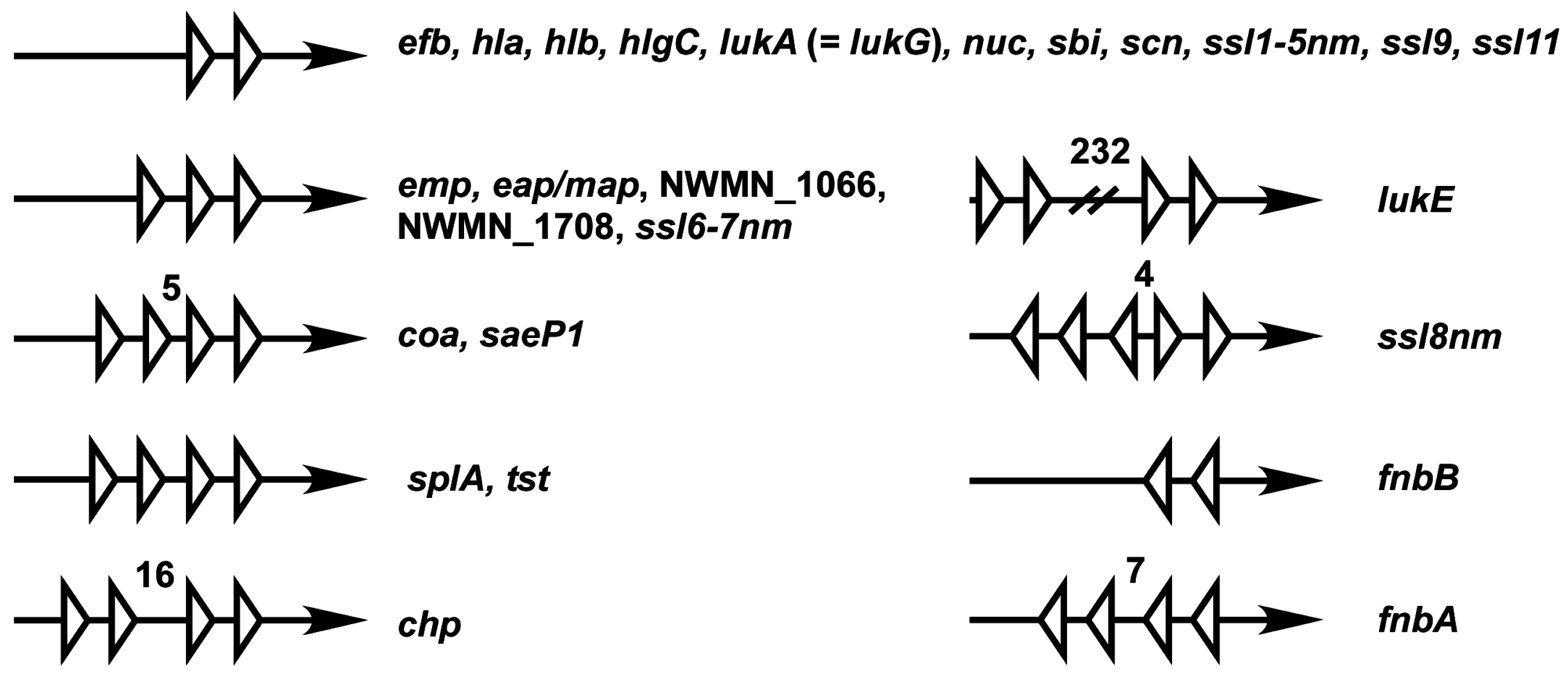
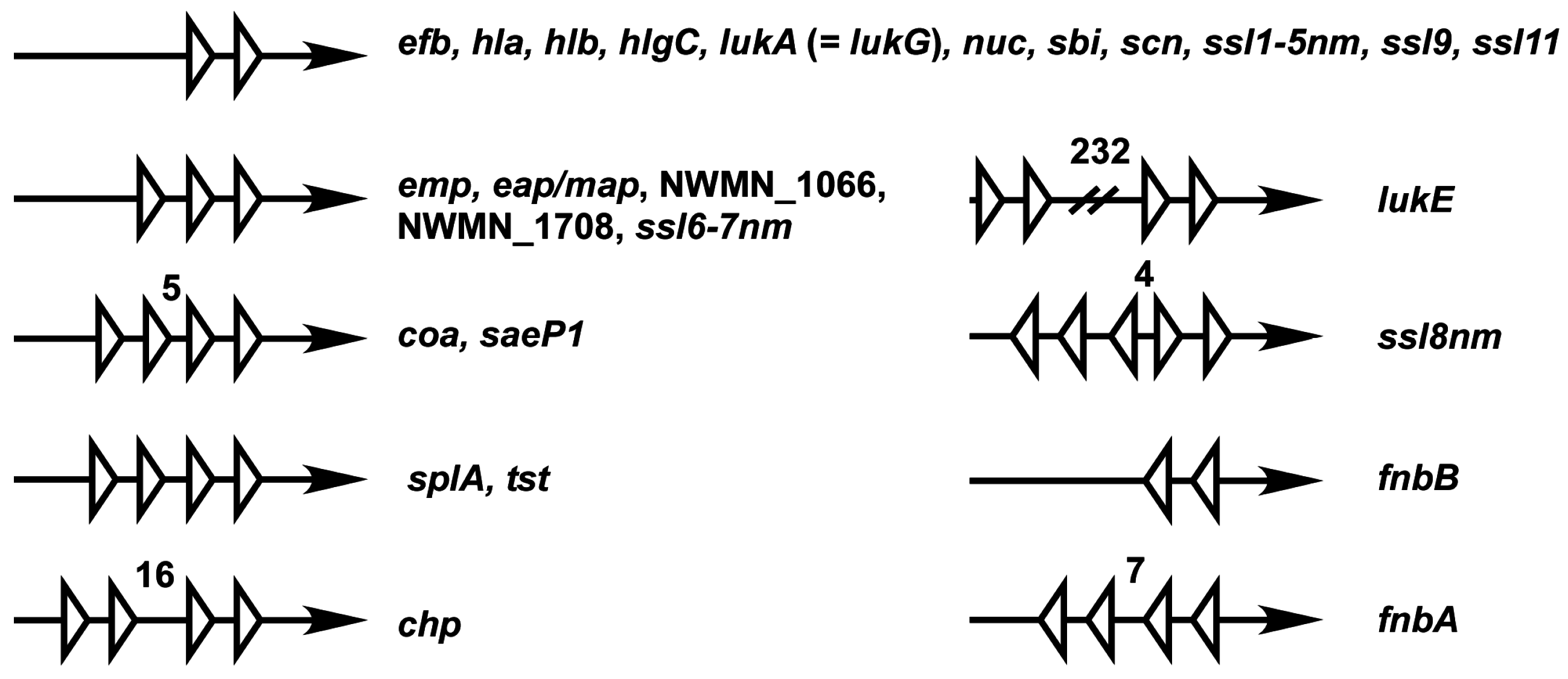
4. The Sae Target Genes
5. Interaction with Other Regulators
5.1. Agr
5.2. WalRK
5.3. SigB
5.4. Fur
5.5. Other Regulators
6. The Role of Sae in the Virulence of S. aureus
6.1. Biofilm Formation
6.2. Invasion of Host
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TCS | Two-component system |
| SBS | SaeR binding sequence |
| HK | Histidine kinase |
| SaeRDB | The DNA binding domain of SaeR |
| LTA | Lipoteichoic acid |
Appendix
References
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.J.; Lowy, F.D. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 2008, 46, S350–S359. [Google Scholar] [CrossRef] [PubMed]
- Recsei, P.; Kreiswirth, B.; O′Reilly, M.; Schlievert, P.; Gruss, A.; Novick, R.P. Regulation of exoprotein gene expression in Staphylococcus aureus by agr. Mol. Gen. Genet. 1986, 202, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; Willard, J.; Yeaman, M.R.; Cheung, A.L.; Bayer, A.S. Regulation of Staphylococcus aureus α-toxin gene (hla) expression by agr, sarA, and sae in vitro and in experimental infective endocarditis. J. Infect. Dis. 2006, 194, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Joo, H.S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins—Critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.L.; Nishina, K.A.; Trotonda, M.P.; Tamber, S. The SarA protein family of Staphylococcus aureus. Int. J. Biochem. Cell Biol. 2008, 40, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Geisinger, E. Quorum sensing in staphylococci. Annu. Rev. Genet. 2008, 42, 541–564. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zheng, L.; Landwehr, C.; Lunsford, D.; Holmes, D.; Ji, Y. Global regulation of gene expression by ArlRS, a two-component signal transduction regulatory system of Staphylococcus aureus. J. Bacteriol. 2005, 187, 5486–5492. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, J.M.; McCormick, J.K.; Schlievert, P.M. Identification of a novel two-component regulatory system that acts in global regulation of virulence factors of Staphylococcus aureus. J. Bacteriol. 2001, 183, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Neoh, H.M.; Nathan, S. Targeting Staphylococcus aureus toxins: A potential form of anti-virulence therapy. Toxins 2016. [Google Scholar] [CrossRef] [PubMed]
- Bronner, S.; Monteil, H.; Prevost, G. Regulation of virulence determinants in Staphylococcus aureus: Complexity and applications. FEMS Microbiol. Rev. 2004, 28, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, A.T.; Raspanti, C.G.; Calzolari, A.; Nagel, R. Characterization of a Tn551-mutant of Staphylococcus aureus defective in the production of several exoproteins. Can. J. Microbiol. 1994, 40, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, A.T.; Calzolari, A.; Cataldi, A.A.; Bogni, C.; Nagel, R. The sae locus of Staphylococcus aureus encodes a two-component regulatory system. FEMS Microbiol. Lett. 1999, 177, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Goerke, C.; Mainiero, M.; Kraus, D.; Wolz, C. The virulence regulator sae of Staphylococcus aureus: Promoter activities and response to phagocytosis-related signals. J. Bacteriol. 2008, 190, 3419–3428. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Jiang, D. The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing. Microbiology 2003, 149, 2709–2717. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Princy, S.A. Exploration of modulated genetic circuits governing virulence determinants in Staphylococcus aureus. Indian. J. Microbiol. 2016, 56, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Haag, A.F.; Bagnoli, F. The role of two-component signal transduction systems in Staphylococcus aureus virulence regulation. Curr. Top. Microbiol. Immunol. 2016. [Google Scholar] [CrossRef]
- Jeong, D.W.; Cho, H.; Lee, H.; Li, C.; Garza, J.; Fried, M.; Bae, T. Identification of P3 promoter and distinct roles of the two promoters of the SaeRS two-component system in Staphylococcus aureus. J. Bacteriol. 2011, 193, 4672–4684. [Google Scholar] [CrossRef] [PubMed]
- Steinhuber, A.; Goerke, C.; Bayer, M.G.; Doring, G.; Wolz, C. Molecular architecture of the regulatory locus sae of Staphylococcus aureus and its impact on expression of virulence factors. J. Bacteriol. 2003, 185, 6278–6286. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cheung, A. The repression of hla by Rot is dependent on sae in Staphylococcus aureus. Infect. Immun. 2008, 76, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Mainiero, M.; Goerke, C.; Geiger, T.; Gonser, C.; Herbert, S.; Wolz, C. Differential target gene activation by the Staphylococcus aureus two-component system saeRS. J. Bacteriol. 2010, 192, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.W.; Cho, H.; Jones, M.B.; Shatzkes, K.; Sun, F.; Ji, Q.; Liu, Q.; Peterson, S.N.; He, C.; Bae, T. The auxiliary protein complex SaePQ activates the phosphatase activity of sensor kinase SaeS in the SaeRS two-component system of Staphylococcus aureus. Mol. Microbiol. 2012, 86, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cho, H.; Yeo, W.S.; Bae, T. The extracytoplasmic linker peptide of the sensor protein SaeS tunes the kinase activity required for staphylococcal virulence in response to host signals. PLoS Pathog. 2015, 11, e1004799. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.P.; Novick, R.P. Regulatory organization of the staphylococcal sae locus. Microbiology 2008, 154, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Mascher, T. Intramembrane-sensing histidine kinases: A new family of cell envelope stress sensors in firmicutes bacteria. FEMS Microbiol. Lett. 2006, 264, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Mascher, T. Bacterial (intramembrane-sensing) histidine kinases: Signal transfer rather than stimulus perception. Trends. Microbiol. 2014, 22, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.; Lam, T.T.; Geiger, T.; Mainiero, M.; Engelmann, S.; Hussain, M.; Bosserhoff, A.; Frosch, M.; Bischoff, M.; Wolz, C.; et al. A point mutation in the sensor histidine kinase SaeS of Staphylococcus aureus strain Newman alters response to biocide exposure. J. Bacteriol. 2009, 191, 7306–7314. [Google Scholar] [CrossRef] [PubMed]
- Flack, C.E.; Zurek, O.W.; Meishery, D.D.; Pallister, K.B.; Malone, C.L.; Horswill, A.R.; Voyich, J.M. Differential regulation of staphylococcal virulence by the sensor kinase SaeS in response to neutrophil-derived stimuli. Proc. Natl. Acad. Sci. USA 2014, 111, E2037–E2045. [Google Scholar] [CrossRef] [PubMed]
- Vandenesch, F.; Kornblum, J.; Novick, R.P. A temporal signal, independent of agr, is required for hla but not spa transcription in Staphylococcus aureus. J. Bacteriol. 1991, 173, 6313–6320. [Google Scholar] [PubMed]
- Wolz, C.; Pohlmann-Dietze, P.; Steinhuber, A.; Chien, Y.T.; Manna, A.; van Wamel, W.; Cheung, A. Agr-independent regulation of fibronectin-binding protein(s) by the regulatory locus sar in Staphylococcus aureus. Mol. Microbiol. 2000, 36, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Li, C.; Jeong, D.; Sohn, C.; He, C.; Bae, T. In the Staphylococcus aureus two-component system sae, the response regulator SaeR binds to a direct repeat sequence and DNA binding requires phosphorylation by the sensor kinase SaeS. J. Bacteriol. 2010, 192, 2111–2127. [Google Scholar] [CrossRef] [PubMed]
- Makgotlho, P.E.; Marincola, G.; Schafer, D.; Liu, Q.; Bae, T.; Geiger, T.; Wasserman, E.; Wolz, C.; Ziebuhr, W.; Sinha, B. SDS interferes with SaeS signaling of Staphylococcus aureus independently of SaePQ. PLoS ONE 2013, 8, e71644. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.E.; Nygaard, T.K.; Ackermann, L.; Watkins, R.L.; Zurek, O.W.; Pallister, K.B.; Griffith, S.; Kiedrowski, M.R.; Flack, C.E.; Kavanaugh, J.S.; et al. Staphylococcus aureus nuclease is an SaeRS-dependent virulence factor. Infect. Immun. 2013, 81, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Ramundo, M.S.; Beltrame, C.O.; Botelho, A.M.; Coelho, L.R.; Silva-Carvalho, M.C.; Ferreira-Carvalho, B.T.; Nicolas, M.F.; Guedes, I.A.; Dardenne, L.E.; O′Gara, J.; et al. A unique SaeS allele overrides cell-density dependent expression of saeR and lukSF-PV in the ST30-SCCmecIV lineage of CA-MRSA. Int. J. Med. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, X.; Zhu, Y.; Niu, L.; Teng, M.; Sun, B.; Li, X. Structure of the DNA-binding domain of the response regulator SaeR from Staphylococcus aureus. Acta. Crystallogr. D Biol. Crystallogr. 2015, 71, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.P.; Huang, C.Y.; Hsieh, T.J.; Chen, S.C.; Chen, Y.R.; Yang, C.S.; Kuo, H.C.; Wang, W.L.; Hsiao, T.H.; Lin, C.H.; et al. Crystal structures of staphylococcal SaeR reveal possible DNA-binding modes. Biochem. Biophys. Res. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, T.K.; Pallister, K.B.; Ruzevich, P.; Griffith, S.; Vuong, C.; Voyich, J.M. SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J. Infect. Dis. 2010, 201, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Joiner, K.A.; Ganz, T.; Albert, J.; Rotrosen, D. The opsonizing ligand on Salmonella Typhimurium influences incorporation of specific, but not azurophil, granule constituents into neutrophil phagosomes. J. Cell Biol. 1989, 109, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.A.; Lilo, S.; Nygaard, T.; Voyich, J.M.; Torres, V.J. Rot and SaeRS cooperate to activate expression of the staphylococcal superantigen-like exoproteins. J. Bacteriol. 2012, 194, 4355–4365. [Google Scholar] [CrossRef] [PubMed]
- Zurek, O.W.; Nygaard, T.K.; Watkins, R.L.; Pallister, K.B.; Torres, V.J.; Horswill, A.R.; Voyich, J.M. The role of innate immunity in promoting SaeR/S-mediated virulence in Staphylococcus aureus. J. Innate. Immun. 2014, 6, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, P.B.; Lehrer, R.I. Mouse neutrophils lack defensins. Infect. Immun. 1992, 60, 3446–3447. [Google Scholar] [PubMed]
- Cho, H.; Jeong, D.W.; Liu, Q.; Yeo, W.S.; Vogl, T.; Skaar, E.P.; Chazin, W.J.; Bae, T. Calprotectin increases the activity of the SaeRS two component system and murine mortality during Staphylococcus aureus infections. PLoS Pathog. 2015, 11, e1005026. [Google Scholar] [CrossRef] [PubMed]
- Foell, D.; Wittkowski, H.; Vogl, T.; Roth, J. S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 2007, 81, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Nemeth, J.; Angel, P.; Hess, J. S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol. 2006, 72, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Corbin, B.D.; Seeley, E.H.; Raab, A.; Feldmann, J.; Miller, M.R.; Torres, V.J.; Anderson, K.L.; Dattilo, B.M.; Dunman, P.M.; Gerads, R.; et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 2008, 319, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Kehl-Fie, T.E.; Chitayat, S.; Hood, M.I.; Damo, S.; Restrepo, N.; Garcia, C.; Munro, K.A.; Chazin, W.J.; Skaar, E.P. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 2011, 10, 158–164. [Google Scholar] [CrossRef] [PubMed]
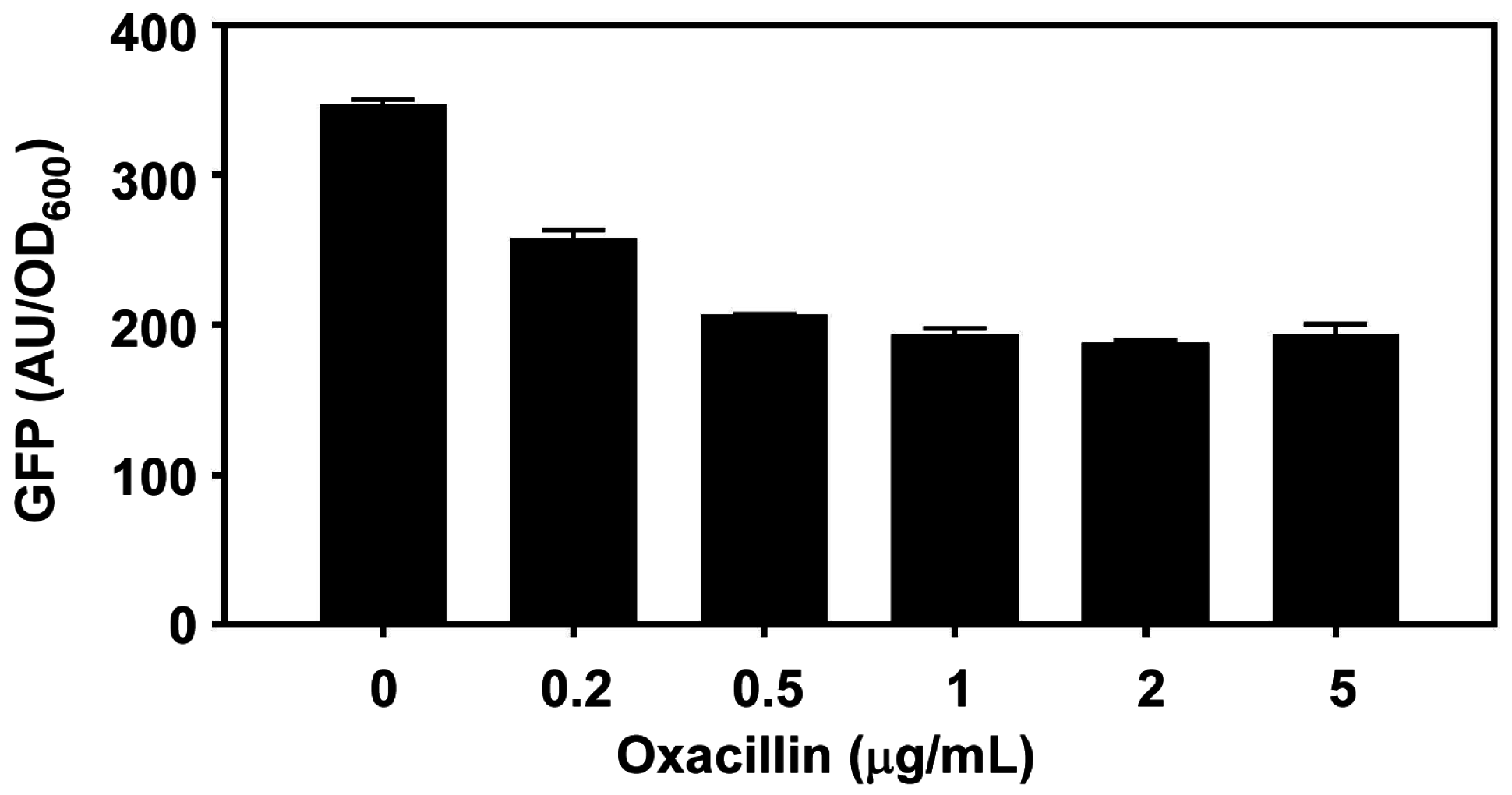
- Ohlsen, K.; Ziebuhr, W.; Koller, K.P.; Hell, W.; Wichelhaus, T.A.; Hacker, J. Effects of subinhibitory concentrations of antibiotics on α-toxin (hla) gene expression of methicillin-sensitive and methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 1998, 42, 2817–2823. [Google Scholar] [PubMed]
- Kuroda, H.; Kuroda, M.; Cui, L.; Hiramatsu, K. Subinhibitory concentrations of β-lactam induce haemolytic activity in Staphylococcus aureus through the SaeRS two-component system. FEMS Microbiol. Lett. 2007, 268, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Hanada, Y.; Sekimizu, K.; Kaito, C. Silkworm apolipophorin protein inhibits Staphylococcus aureus virulence. J. Biol. Chem. 2011, 286, 39360–39369. [Google Scholar] [CrossRef] [PubMed]
- Omae, Y.; Hanada, Y.; Sekimizu, K.; Kaito, C. Silkworm apolipophorin protein inhibits hemolysin gene expression of Staphylococcus aureus via binding to cell surface lipoteichoic acids. J. Biol. Chem. 2013, 288, 25542–25550. [Google Scholar] [CrossRef] [PubMed]
- Weinrick, B.; Dunman, P.M.; McAleese, F.; Murphy, E.; Projan, S.J.; Fang, Y.; Novick, R.P. Effect of mild acid on gene expression in Staphylococcus aureus. J. Bacteriol. 2004, 186, 8407–8423. [Google Scholar] [CrossRef] [PubMed]
- Neumann, Y.; Ohlsen, K.; Donat, S.; Engelmann, S.; Kusch, H.; Albrecht, D.; Cartron, M.; Hurd, A.; Foster, S.J. The effect of skin fatty acids on Staphylococcus aureus. Arch. Microbiol. 2015, 197, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Joost, I.; Skaar, E.P.; Herrmann, M.; Bischoff, M. Haemin represses the haemolytic activity of Staphylococcus aureus in an Sae-dependent manner. Microbiology 2012, 158, 2619–2631. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Sitthisak, S.; Sengupta, M.; Johnson, M.; Jayaswal, R.K.; Morrissey, J.A. Copper stress induces a global stress response in Staphylococcus aureus and represses sae and agr expression and biofilm formation. Appl. Environ. Microbiol. 2010, 76, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Blickwede, M.; Goethe, R.; Wolz, C.; Valentin-Weigand, P.; Schwarz, S. Molecular basis of florfenicol-induced increase in adherence of Staphylococcus aureus strain Newman. J. Antimicrob. Chemother. 2005, 56, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Luo, M.; Fu, Y.J.; Zu, Y.G.; Wang, W.; Zhang, L.; Yao, L.P.; Zhao, C.J.; Sun, Y. Effect of corilagin on membrane permeability of Escherichia coli, Staphylococcus aureus and candida albicans. Phytother. Res. 2012, 27, 1517–1523. [Google Scholar] [PubMed]
- Qiu, J.; Wang, D.; Xiang, H.; Feng, H.; Jiang, Y.; Xia, L.; Dong, J.; Lu, J.; Yu, L.; Deng, X. Subinhibitory concentrations of thymol reduce enterotoxins A and B and α-hemolysin production in Staphylococcus aureus isolates. PLoS ONE 2010, 5, e9736. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.B.; Kukula, M.; Jackson, P.; Pulse, M.; Simecka, J.W.; Valtierra, D.; Weiss, W.J.; Kaplan, N.; Rock, C.O. Perturbation of Staphylococcus aureus gene expression by the enoyl-acyl carrier protein reductase inhibitor AFN-1252. Antimicrob. Agents Chemother. 2013, 57, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Saleh, F.A.; Freer, J.H. Inhibition of secretion of staphylococcal alpha toxin by cerulenin. J. Med. Microbiol. 1984, 18, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, K.; Pakulat, N.; Fleer, S.; Schnaith, A.; Utermohlen, O.; Krut, O.; Muller, S.; Kronke, M. Subinhibitory concentrations of linezolid reduce Staphylococcus aureus virulence factor expression. Antimicrob. Agents Chemother. 2004, 48, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.; Burton, N.; Cooper, R. Proteomic and genomic analysis of methicillin-resistant Staphylococcus aureus (MRSA) exposed to manuka honey in vitro demonstrated down-regulation of virulence markers. J. Antimicrob. Chemother. 2014, 69, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, J.H.; Cho, M.H.; Lee, J. Flavone reduces the production of virulence factors, staphyloxanthin and α-hemolysin, in Staphylococcus aureus. Curr. Microbiol. 2012, 65, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Jeong, D.W.; Li, C.; Bae, T. Organizational requirements of the SaeS binding sites for a functional P1 promoter of the sae operon in Staphylococcus aureus. J. Bacteriol. 2012, 194, 2865–2876. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, A.T.; Cheung, A.L.; Nagel, R. The sae locus of Staphylococcus aureus controls exoprotein synthesis at the transcriptional level. Arch. Microbiol. 1997, 168, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yu, C.; Sun, J.; Liu, H.; Landwehr, C.; Holmes, D.; Ji, Y. Inactivation of a two-component signal transduction system, SaeRS, eliminates adherence and attenuates virulence of Staphylococcus aureus. Infect. Immun. 2006, 74, 4655–4665. [Google Scholar] [CrossRef] [PubMed]
- Rogasch, K.; Ruhmling, V.; Pane-Farre, J.; Hoper, D.; Weinberg, C.; Fuchs, S.; Schmudde, M.; Broker, B.M.; Wolz, C.; Hecker, M.; et al. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J. Bacteriol. 2006, 188, 7742–7758. [Google Scholar] [CrossRef] [PubMed]
- Pantrangi, M.; Singh, V.K.; Wolz, C.; Shukla, S.K. Staphylococcal superantigen-like genes, ssl5 and ssl8, are positively regulated by Sae and negatively by Agr in the Newman strain. FEMS Microbiol. Lett. 2010, 308, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Harraghy, N.; Homerova, D.; Herrmann, M.; Kormanec, J. Mapping the transcription start points of the Staphylococcus aureus eap, emp, and vwb promoters reveals a conserved octanucleotide sequence that is essential for expression of these genes. J. Bacteriol. 2008, 190, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Cockayne, A.; Morrissey, J.A. Iron-regulated biofilm formation in Staphylococcus aureus Newman requires ica and the secreted protein emp. Infect. Immun. 2008, 76, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; Ruyken, M.; van Roon, J.; van Kessel, K.P.; van Strijp, J.A.; van Wamel, W.J. Early expression of SCIN and CHIPS drives instant immune evasion by Staphylococcus aureus. Cell Microbiol. 2006, 8, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Dumont, A.L.; Nygaard, T.K.; Watkins, R.L.; Smith, A.; Kozhaya, L.; Kreiswirth, B.N.; Shopsin, B.; Unutmaz, D.; Voyich, J.M.; Torres, V.J. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol. 2011, 79, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.L.; Malachowa, N.; Hammer, C.H.; Nardone, G.A.; Robinson, M.A.; Kobayashi, S.D.; DeLeo, F.R. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS ONE 2010, 5, e11634. [Google Scholar] [CrossRef] [PubMed]
- Baroja, M.L.; Herfst, C.A.; Kasper, K.J.; Xu, S.X.; Gillett, D.A.; Li, J.; Reid, G.; McCormick, J.K. The SaeRS two-component system is a direct and dominant transcriptional activator of toxic shock syndrome toxin-1 in Staphylococcus aureus. J. Bacteriol. 2016, 198, 2732–2742. [Google Scholar] [CrossRef] [PubMed]
- Cassat, J.E.; Hammer, N.D.; Campbell, J.P.; Benson, M.A.; Perrien, D.S.; Mrak, L.N.; Smeltzer, M.S.; Torres, V.J.; Skaar, E.P. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe 2013, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.; Sau, K.; Roux, C.; Sau, S.; Dunman, P.M.; Lee, C.Y. Staphylococcus aureus ClpC divergently regulates capsule via sae and codY in strain Newman but activates capsule via codY in strain UAMS-1 and in strain Newman with repaired saeS. J. Bacteriol. 2011, 193, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Goerke, C.; Fluckiger, U.; Steinhuber, A.; Zimmerli, W.; Wolz, C. Impact of the regulatory loci agr, sarA and sae of Staphylococcus aureus on the induction of alpha-toxin during device-related infection resolved by direct quantitative transcript analysis. Mol. Microbiol. 2001, 40, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003, 48, 1429–1449. [Google Scholar] [CrossRef] [PubMed]
- Queck, S.Y.; Jameson-Lee, M.; Villaruz, A.E.; Bach, T.H.; Khan, B.A.; Sturdevant, D.E.; Ricklefs, S.M.; Li, M.; Otto, M. RnaШ-independent target gene control by the agr quorum-sensing system: Insight into the evolution of virulence regulation in Staphylococcus aureus. Mol. Cell 2008, 32, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Sengupta, M.; Purves, J.; Tarrant, E.; Williams, P.H.; Cockayne, A.; Muthaiyan, A.; Stephenson, R.; Ledala, N.; Wilkinson, B.J.; et al. Fur is required for the activation of virulence gene expression through the induction of the sae regulatory system in staphylococcus aureus. Int. J. Med. Microbiol. 2011, 301, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, A.T.; Mansilla, C.; Chan, A.; Raspanti, C.; Nagel, R. Studies on the expression of regulatory locus sae in Staphylococcus aureus. Curr. Microbiol. 2003, 46, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Wolz, C.; McDevitt, D.; Foster, T.J.; Cheung, A.L. Influence of agr on fibrinogen binding in Staphylococcus aureus Newman. Infect. Immun. 1996, 64, 3142–3147. [Google Scholar] [PubMed]
- Saravia-Otten, P.; Muller, H.P.; Arvidson, S. Transcription of Staphylococcus aureus fibronectin binding protein genes is negatively regulated by agr and an agr-independent mechanism. J. Bacteriol. 1997, 179, 5259–5263. [Google Scholar] [PubMed]
- Dassy, B.; Hogan, T.; Foster, T.J.; Fournier, J.M. Involvement of the accessory gene regulator (agr) in expression of type 5 capsular polysaccharide by Staphylococcus aureus. J. Gen. Microbiol. 1993, 139, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.; Sau, S.; Gomez, M.; Lee, J.C.; Lee, C.Y. Regulation of Staphylococcus aureus capsular polysaccharide expression by agr and sarA. Infect. Immun. 2002, 70, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Ross, H.F.; Projan, S.J.; Kornblum, J.; Kreiswirth, B.; Moghazeh, S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993, 12, 3967–3975. [Google Scholar] [PubMed]
- Dubrac, S.; Msadek, T. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J. Bacteriol. 2004, 186, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Dubrac, S.; Boneca, I.G.; Poupel, O.; Msadek, T. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J. Bacteriol. 2007, 189, 8257–8269. [Google Scholar] [CrossRef] [PubMed]
- Delaune, A.; Dubrac, S.; Blanchet, C.; Poupel, O.; Mader, U.; Hiron, A.; Leduc, A.; Fitting, C.; Nicolas, P.; Cavaillon, J.M.; et al. The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response. Infect. Immun. 2012, 80, 3438–3453. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Reder, A.; Fuchs, S.; Pagels, M.; Engelmann, S. Physiological proteomics and stress/starvation responses in Bacillus subtilis and Staphylococcus aureus. Res. Microbiol. 2009, 160, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Chen, C.C.; Fang, C.S.; Hsieh, Y.T.; Lin, M.H.; Shu, J.C. Vancomycin activates σ B in vancomycin-resistant Staphylococcus aureus resulting in the enhancement of cytotoxicity. PLoS ONE 2011, 6, e24472. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Fugere, A.; Pepin Gaudreau, K.; Brouillette, E.; Frost, E.H.; Cantin, A.M.; Malouin, F. SigB is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS ONE 2013, 8, e65018. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, M.; Dunman, P.; Kormanec, J.; Macapagal, D.; Murphy, E.; Mounts, W.; Berger-Bachi, B.; Projan, S. Microarray-based analysis of the Staphylococcus aureus σ B regulon. J. Bacteriol. 2004, 186, 4085–4099. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, R.O.; Li, T.; McDevitt, D.; Marra, A.; Sucoloski, S.; Demarsh, P.L.; Gentry, D.R. Isolation and characterization of a sigB deletion mutant of Staphylococcus aureus. Infect Immun 1999, 67, 3667–3669. [Google Scholar] [PubMed]
- Miyazaki, E.; Chen, J.M.; Ko, C.; Bishai, W.R. The Staphylococcus aureus rsbw (orf159) gene encodes an anti-sigma factor of SigB. J. Bacteriol. 1999, 181, 2846–2851. [Google Scholar] [PubMed]
- Romilly, C.; Lays, C.; Tomasini, A.; Caldelari, I.; Benito, Y.; Hammann, P.; Geissmann, T.; Boisset, S.; Romby, P.; Vandenesch, F. A non-coding RNA promotes bacterial persistence and decreases virulence by regulating a regulator in Staphylococcus aureus. PLoS Pathog. 2014, 10, e1003979. [Google Scholar] [CrossRef] [PubMed]
- Goerke, C.; Fluckiger, U.; Steinhuber, A.; Bisanzio, V.; Ulrich, M.; Bischoff, M.; Patti, J.M.; Wolz, C. Role of Staphylococcus aureus global regulators sae and sigmaB in virulence gene expression during device-related infection. Infect. Immun. 2005, 73, 3415–3421. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Helmann, J.D. Functional specialization within the Fur family of metalloregulators. Biometals 2007, 20, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Hantke, K. Regulation of ferric iron transport in Escherichia coli K12: Isolation of a constitutive mutant. Mol. Gen. Genet. 1981, 182, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Litwin, C.M.; Calderwood, S.B. Cloning and genetic analysis of the Vibrio vulnificus fur gene and construction of a fur mutant by in vivo marker exchange. J. Bacteriol. 1993, 175, 706–715. [Google Scholar] [PubMed]
- Johnson, M.; Cockayne, A.; Williams, P.H.; Morrissey, J.A. Iron-responsive regulation of biofilm formation in Staphylococcus aureus involves fur-dependent and fur-independent mechanisms. J. Bacteriol. 2005, 187, 8211–8215. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, M.J.; Ingham, E.; Foster, S.J. In Staphylococcus aureus, fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol. 2001, 183, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.R.; Dunman, P.M.; Fang, F.C. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol. Microbiol. 2006, 61, 927–939. [Google Scholar] [CrossRef] [PubMed]
- McNamara, P.J.; Milligan-Monroe, K.C.; Khalili, S.; Proctor, R.A. Identification, cloning, and initial characterization of rot, a locus encoding a regulator of virulence factor expression in Staphylococcus aureus. J. Bacteriol. 2000, 182, 3197–3203. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.B.; Broussard, T.C.; Bose, J.L.; Rosch, J.W.; Jackson, P.; Subramanian, C.; Rock, C.O. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2014, 111, 10532–10537. [Google Scholar] [CrossRef] [PubMed]
- McCleary, W.R.; Stock, J.B. Acetyl phosphate and the activation of two-component response regulators. J. Biol. Chem. 1994, 269, 31567–31572. [Google Scholar] [PubMed]
- Otto, M. Staphylococcal infections: Mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu. Rev. Med. 2013, 64, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Mack, D.; Fischer, W.; Krokotsch, A.; Leopold, K.; Hartmann, R.; Egge, H.; Laufs, R. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear β-1,6-linked glucosaminoglycan: Purification and structural analysis. J. Bacteriol. 1996, 178, 175–183. [Google Scholar] [PubMed]
- Maira-Litran, T.; Kropec, A.; Abeygunawardana, C.; Joyce, J.; Mark, G., 3rd; Goldmann, D.A.; Pier, G.B. Immunochemical properties of the staphylococcal poly-N-acetylglucosamine surface polysaccharide. Infect. Immun. 2002, 70, 4433–4440. [Google Scholar]
- O’Gara, J.P. Ica and beyond: Biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol. Lett. 2007, 270, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Paharik, A.E.; Horswill, A.R. The staphylococcal biofilm: Adhesins, regulation, and host response. Microbiol. Spectr. 2016. [Google Scholar] [CrossRef]
- Lim, Y.; Jana, M.; Luong, T.T.; Lee, C.Y. Control of glucose- and NaCl-induced biofilm formation by rbf in Staphylococcus aureus. J. Bacteriol. 2004, 186, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Zapotoczna, M.; McCarthy, H.; Rudkin, J.K.; O’Gara, J.P.; O’Neill, E. An essential role for coagulase in Staphylococcus aureus biofilm development reveals new therapeutic possibilities for device-related infections. J. Infect. Dis. 2015, 212, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Cue, D.; Lei, M.G.; Lee, C.Y. Genetic regulation of the intercellular adhesion locus in staphylococci. Front. Cell. Infect. Microbiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Gotz, F. Staphylococcus and biofilms. Mol. Microbiol. 2002, 43, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Boles, B.R.; Horswill, A.R. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 2008, 4, e1000052. [Google Scholar] [CrossRef] [PubMed]
- Cue, D.; Junecko, J.M.; Lei, M.G.; Blevins, J.S.; Smeltzer, M.S.; Lee, C.Y. SaeRS-dependent inhibition of biofilm formation in Staphylococcus aureus Newman. PLoS ONE 2015, 10, e0123027. [Google Scholar] [CrossRef] [PubMed]
- O′Neill, E.; Pozzi, C.; Houston, P.; Humphreys, H.; Robinson, D.A.; Loughman, A.; Foster, T.J.; O′Gara, J.P. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J. Bacteriol. 2008, 190, 3835–3850. [Google Scholar] [CrossRef] [PubMed]
- McCourt, J.; O′Halloran, D.P.; McCarthy, H.; O′Gara, J.P.; Geoghegan, J.A. Fibronectin-binding proteins are required for biofilm formation by community-associated methicillin-resistant Staphylococcus aureus strain lac. FEMS Microbiol. Lett. 2014, 353, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, N.C.; O’Toole, G.A. α-Toxin is required for biofilm formation by Staphylococcus aureus. J. Bacteriol. 2003, 185, 3214–3217. [Google Scholar] [CrossRef] [PubMed]
- Huseby, M.J.; Kruse, A.C.; Digre, J.; Kohler, P.L.; Vocke, J.A.; Mann, E.E.; Bayles, K.W.; Bohach, G.A.; Schlievert, P.M.; Ohlendorf, D.H.; et al. Beta toxin catalyzes formation of nucleoprotein matrix in staphylococcal biofilms. Proc. Natl. Acad. Sci. USA 2010, 107, 14407–14412. [Google Scholar] [CrossRef] [PubMed]
- Beenken, K.E.; Blevins, J.S.; Smeltzer, M.S. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect. Immun. 2003, 71, 4206–4211. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Pozzi, C.; Houston, P.; Smyth, D.; Humphreys, H.; Robinson, D.A.; O′Gara, J.P. Association between methicillin susceptibility and biofilm regulation in Staphylococcus aureus isolates from device-related infections. J. Clin. Microbiol. 2007, 45, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Mrak, L.N.; Zielinska, A.K.; Beenken, K.E.; Mrak, I.N.; Atwood, D.N.; Griffin, L.M.; Lee, C.Y.; Smeltzer, M.S. SaeRS and sarA act synergistically to repress protease production and promote biofilm formation in Staphylococcus aureus. PLoS ONE 2012, 7, e38453. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Kockritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate. Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Munzenmayer, L.; Geiger, T.; Daiber, E.; Schulte, B.; Autenrieth, S.E.; Fraunholz, M.; Wolz, C. Influence of Sae and Agr regulated factors on the escape of Staphylococcus aureus from human macrophages. Cell Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, T.K.; Pallister, K.B.; Zurek, O.W.; Voyich, J.M. The impact of α-toxin on host cell plasma membrane permeability and cytokine expression during human blood infection by CA-MRSA USA300. J. Leukoc. Biol. 2013, 94, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.L.; Pallister, K.B.; Voyich, J.M. The SaeR/S gene regulatory system induces a pro-inflammatory cytokine response during Staphylococcus aureus infection. PLoS ONE 2011, 6, e19939. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S.; Matsumoto, Y.; Sekimizu, K.; Kaito, C. Evaluation of Staphylococcus aureus virulence factors using a silkworm model. FEMS Microbiol. Lett. 2012, 326, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Banger, A.K.; Wallace, A.; Glass, E.M.; Aslund, F.; Schneewind, O.; Missiakas, D.M. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. USA 2004, 101, 12312–12317. [Google Scholar] [CrossRef] [PubMed]
- Voyich, J.M.; Vuong, C.; Dewald, M.; Nygaard, T.K.; Kocianova, S.; Griffith, S.; Jones, J.; Iverson, C.; Sturdevant, D.E.; Braughton, K.R.; et al. The SaeR/S gene regulatory system is essential for innate immune evasion by Staphylococcus aureus. J. Infect. Dis. 2009, 199, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Boyle-Vavra, S.; Daum, R.S. Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS ONE 2010, 5, e15177. [Google Scholar] [CrossRef] [PubMed]
- Beenken, K.E.; Mrak, L.N.; Zielinska, A.K.; Atwood, D.N.; Loughran, A.J.; Griffin, L.M.; Matthews, K.A.; Anthony, A.M.; Spencer, H.J.; Post, G.R.; et al. Impact of the functional status of SaeRS on in vivo phenotypes of Staphylococcus aureus sarA mutants. Mol. Microbiol. 2014, 92, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Kernbauer, E.; Maurer, K.; Torres, V.J.; Shopsin, B.; Cadwell, K. Gastrointestinal dissemination and transmission of Staphylococcus aureus following bacteremia. Infect. Immun. 2015, 83, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Handke, L.D.; Rogers, K.L.; Olson, M.E.; Somerville, G.A.; Jerrells, T.J.; Rupp, M.E.; Dunman, P.M.; Fey, P.D. Staphylococcus epidermidis SaeR is an effector of anaerobic growth and a mediator of acute inflammation. Infect. Immun. 2008, 76, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ravcheev, D.A.; Best, A.A.; Tintle, N.; Dejongh, M.; Osterman, A.L.; Novichkov, P.S.; Rodionov, D.A. Inference of the transcriptional regulatory network in Staphylococcus aureus by integration of experimental and genomics-based evidence. J. Bacteriol. 2011, 193, 3228–3240. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus ID 1 | Name | Product | SaeR Binding Sequence | Evidence 2 | References 3 |
|---|---|---|---|---|---|
| NWMN_0166 | coa | coagulase | GTTAATGCTTTGTTTA GTTAATATATAGTTAA | EMSA, MA, NB, RA | [21,48,63,64,65] |
| NWMN_0388 | ssl1nm | enterotoxin-like toxin | GTTAAATGAGGTTTAA | 2D | [66] |
| NWMN_0389 | ssl2nm | enterotoxin-like toxin | GTTAAAAACAGGTTAA | 2D | [66] |
| NWMN_0390 | ssl3nm | enterotoxin-like toxin | GTTAAAAGGGGTTTAA | ||
| NWMN_0391 | ssl4nm | enterotoxin-like toxin | GTTAAACAAGGTTTAA | ||
| NWMN_0392 | ssl5nm | enterotoxin-like toxin | ATTAAACATGGTTTAA | MA, RT | [65,67] |
| NWMN_0393 | ssl6nm | enterotoxin-like toxin | GTTCAAAAATAGTTAA GTTAAAAAGAGGTTAA | ||
| NWMN_0394 | ssl7nm | enterotoxin-like toxin | GTTCAAAAATAGTTAA GTTAAAAAGAGGTTAA | EMSA, 2D, WB | [39,66] |
| NWMN_0395 | ssl8nm | enterotoxin-like toxin | GTTAATGAAGAGCTAA CTTAAATCATTGTTAA ATTAAACGAGTGTTAA | RT | [67] |
| NWMN_0396 | ssl9nm | enterotoxin-like toxin | ATTAAAAATCAGTTAA | EMSA, WB | [39] |
| NWMN_0400 | ss11nm | enterotoxin-like toxin | ATTAATTTTTAGTTAA | EMSA, 2D, WB | [39,66] |
| NWMN_0677 | saeP | SaeP protein | GTTAAGCGATATTTAA GTTAAGAATTAGTTAA | EMSA, MA, NB, RA | [15,19,48] |
| NWMN_0758 | ssr/emp | extracellular matrix binding protein | GTTAAGACAACGTTTA GTTTACTTCAAGTTAA | 1D, MA, RA | [48,68,69] |
| NWMN_0760 | nuc | nuclease | ATTAAATTTTTATTAA | EMSA, 2D, MA, RA,WB | [33,48,66] |
| NWMN_1066 | fibrinogen-binding related protein | ATTAATGTTTAGTTAA GTTAATAAATAGTTAA | MA | [48,65] | |
| NWMN_1069 | efb | similar to fibrinogen binding protein | ATTAATAATTAGTTAA | MA | [48,65] |
| NWMN_1073 | hla | α-hemolysin | GTTAATATATAGTTAA | EMSA, MA,RA | [21,48,64,65] |
| NWMN_1706 | splA | serine protease | TTTAATAAAACGTTAA GTTAATTAATATTTAA | 2D | [66] |
| NWMN_1708 | homologous to ear | GTTAATAGATAGTTAA GTTAATACATTTTTGA | |||
| NWMN_1719 | lukE | leukocidin LukE | TTTAATGAACAGTTAA GTTAATAATCAGTTAA | [27] | |
| NWMN_1872 | map/eap | MHC class II analog protein | ATTAATATTCAGTTAA | 1D, NB, RA | [27,32,68,69] |
| NWMN_1873 | hlb | β-hemolysin (truncated) | ATTAACTGAATATTAA | MA,NB | [12,21,65] |
| NWMN_1876 | scn | staphylococcal complement inhibitor | GTTAATGAATAATTAA | RA | [70] |
| NWMN_1877 | chp | chemotaxis-inhibiting protein | TTTAATTTTTAGTTAA ATTAATTTCAAGTTAA | 1D, MA,RA | [27,48,70] |
| NWMN_1928 | lukA (lukG)4 | leukocidin LukA (LukG) | TTTAATAAATAGTTAA | 1D, MA | [27,65,71,72] |
| NWMN_2317 | sbi | IgG binding protein | GTTAATAATTAGTTAA | 1D, MA | [21,48] |
| NWMN_2319 | hlgC | γ-hemolysin component C | GTTAATGAACAGTTAA | 1D, MA | [27,48,65] |
| NWMN_2397 | fnbB | fibronectin binding protein B | GTTAATAAAAAGTTAA | MA | [48,65] |
| NWMN_2399 | fnbA | fibronectin binding protein A | GTTAATGAAAAGTTAA ATTAATTTTATGTTAA | NB,WB | [19,21] |
| SA1819 | tst | toxic shock syndrome toxin | ATTAATATATATTTAA ATTTAGAGATGGTTAA | EMSA,1D,RA,RT | [73] |
| Possible targets | |||||
| NMTN_0095 | capA | capsular polysaccharide synthesis enzyme | GTTTAAAAGTAATTAA | ||
| NWMN_0157 | conserved hypothetical protein | ATTAATAAATAGTTAA | - | ||
| NWMN_0362 | hypothetical protein | GTTAATCAAGAGTTAA GTTAAGATGAATTTAA | - | ||
| NWMN_0403 | lpl1nm | lipoprotein | TTTAATAAATAGTTAA | - | |
| NWMN_1533 | his | histidyl-t-RNA synthetase | GTTAAACGTACGTTAA | - | |
| NWMN_1880 | sak5 | staphylokinase | GTTAAATATTTGTTAA GTTAATTATTTTTTAA | [12,70] | |
| NWMN_2536 | aur | zinc metalloproteinase aureolysin | TTTAAAATATAATTAA | [24,66,74]. | |
| NWMN_2592 | 2-oxoglutarate/malate translocator | GTTAACAACACGTTAA | - | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Yeo, W.; Bae, T. The SaeRS Two‐Component System of Staphylococcus aureus. Genes 2016, 7, 81. https://doi.org/10.3390/genes7100081
Liu Q, Yeo W, Bae T. The SaeRS Two‐Component System of Staphylococcus aureus. Genes. 2016; 7(10):81. https://doi.org/10.3390/genes7100081
Chicago/Turabian StyleLiu, Qian, Won‐Sik Yeo, and Taeok Bae. 2016. "The SaeRS Two‐Component System of Staphylococcus aureus" Genes 7, no. 10: 81. https://doi.org/10.3390/genes7100081
APA StyleLiu, Q., Yeo, W., & Bae, T. (2016). The SaeRS Two‐Component System of Staphylococcus aureus. Genes, 7(10), 81. https://doi.org/10.3390/genes7100081