
Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells

Abstract
:1. Introduction
2. Exploring the Epigenetics of FXS
3. CGG Repeat Instability
4. Neurological Pathology in FXS Neurons Derived from hPSC
5. Therapeutic Strategies in FXS
6. Concluding Remarks
Author Contributions
Conflicts of Interest
References
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu. Rev. Pathol. 2011, 7, 219–245. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, C.; Beaulande, M.; Ehresmann, C.; Ehresmann, B.; Moine, H. The RNA binding protein FMRP: New connections and missing links. Biol. Cell 2003, 95, 221–228. [Google Scholar] [CrossRef]
- Oberle, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boué, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Heitz, D.; Biancalana, V.; Blumenfeld, S.; Kretz, C.; Boué, J.; Tommerup, N.; Van Der Hagen, C.; DeLozier-Blanchet, C.; Croquette, M.F.; et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 1991, 325, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Nolin, S.L.; Lewis, F.A., 3rd; Ye, L.L.; Houck, G.E., Jr.; Glicksman, A.E.; Limprasert, P.; Li, S.Y.; Zhong, N.; Ashley, A.E.; Feingold, E.; et al. Familial transmission of the FMR1 CGG repeat. Am. J. Hum. Genet. 1996, 59, 1252–1261. [Google Scholar] [PubMed]
- Coffee, B.; Zhang, F.; Ceman, S.; Warren, S.T.; Reines, D. Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile X syndrome. Am. J. Hum. Genet. 2002, 71, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Zhang, F.; Warren, S.T.; Reines, D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999, 22, 98–101. [Google Scholar] [PubMed]
- Kumari, D.; Usdin, K. The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum. Mol. Genet. 2010, 19, 4634–4642. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, R.; Tabolacci, E.; Zalfa, F.; Zito, I.; Terracciano, A.; Moscato, U.; Bagni, C.; Oostra, B.; Chiurazzi, P.; Neri, G. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 2005, 14, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Moscato, U.; Zalfa, F.; Bagni, C.; Chiurazzi, P.; Neri, G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008, 16, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Pietrobono, R.; Moscato, U.; Oostra, B.A.; Chiurazzi, P.; Neri, G. Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 2005, 13, 641–648. [Google Scholar] [CrossRef] [PubMed]
- De Graaff, E.; Willemsen, R.; Zhong, N.; de Die-Smulders, C.E.; Brown, W.T.; Freling, G.; Oostra, B. Instability of the CGG repeat and expression of the FMR1 protein in a male fragile X patient with a lung tumor. Am. J. Hum. Genet. 1995, 57, 609–618. [Google Scholar] [PubMed]
- Berman, R.F.; Buijsen, R.A.; Usdin, K.; Pintado, E.; Kooy, F.; Pretto, D.; Pessah, I.N.; Nelson, D.L.; Zalewski, Z.; Charlet-Bergeurand, N.; et al. Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome. J. Neurodev. Disord. 2014, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Bontekoe, C.J.; Bakker, C.E.; Nieuwenhuizen, I.M.; van der Linde, H.; Lans, H.; de Lange, D.; Hirst, M.C.; Oostra, B.A. Instability of a (CGG)98 repeat in the FMR1 promoter. Hum. Mol. Genet. 2001, 10, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Bontekoe, C.J.; de Graaff, E.; Nieuwenhuizen, I.M.; Willemsen, R.; Oostra, B.A. FMR1 premutation allele (CGG)81 is stable in mice. Eur. J. Hum. Genet. 1997, 5, 293–298. [Google Scholar] [PubMed]
- den Broeder, M.J.; van der Linde, H.; Brouwer, J.R.; Oostra, B.A.; Willemsen, R.; Ketting, R.F. Generation and characterization of FMR1 knockout zebrafish. PLoS ONE 2009, 4, e7910. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Luo, T.; Sargent, T.D.; Fallon, J.R. Developmental expression of xenopus fragile X mental retardation-1 gene. Int. J. Dev. Biol. 2005, 49, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Mientjes, E.J.; Nieuwenhuizen, I.; Kirkpatrick, L.; Zu, T.; Hoogeveen-Westerveld, M.; Severijnen, L.; Rife, M.; Willemsen, R.; Nelson, D.L.; Oostra, B.A. The generation of a conditional FMR1 knock out mouse model to study FMRP function in vivo. Neurobiol. Dis. 2006, 21, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Dockendorff, T.C.; Jongens, T.A.; Dreyfuss, G. Characterization of DFMR1, a drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol. 2000, 20, 8536–8547. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.B.; Nosyreva, E.D.; Spencer, C.M.; Volk, L.J.; Musunuru, K.; Zhong, R.; Stone, E.F.; Yuva-Paylor, L.A.; Huber, K.M.; Paylor, R.; et al. A mouse model of the human fragile X syndrome I304N mutation. PLoS Genet. 2009, 5, e1000758. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, J.R.; Mientjes, E.J.; Bakker, C.E.; Nieuwenhuizen, I.M.; Severijnen, L.A.; Van der Linde, H.C.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Elevated FMR1 mrna levels and reduced protein expression in a mouse model with an unmethylated fragile X full mutation. Exp. Cell Res. 2007, 313, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Hunsaker, M.R. Neurocognitive endophenotypes in CGG KI and FMR1 KO mouse models of fragile X-associated disorders: An analysis of the state of the field. F1000Research 2013, 2, 287. [Google Scholar] [CrossRef] [PubMed]
- Kazdoba, T.M.; Leach, P.T.; Silverman, J.L.; Crawley, J.N. Modeling fragile X syndrome in the FMR1 knockout mouse. Intractable Rare Dis. Res. 2014, 3, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Thomson, J.A. Pluripotent stem cell lines. Genes Dev. 2008, 22, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Dvash, T.; Ben-Yosef, D.; Eiges, R. Human embryonic stem cells as a powerful tool for studying human embryogenesis. Pediatr. Res. 2006, 60, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human IPSCS and ESCS. Nat. Biotechnol. 2015, 33, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Narsinh, K.H.; Plews, J.; Wu, J.C. Comparison of human induced pluripotent and embryonic stem cells: Fraternal or identical twins? Mol. Ther. 2011, 19, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Alisch, R.S.; Wang, T.; Chopra, P.; Visootsak, J.; Conneely, K.N.; Warren, S.T. Genome-wide analysis validates aberrant methylation in fragile X syndrome is specific to the FMR1 locus. BMC Med. Genet. 2013, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Brick, D.J.; Nethercott, H.E.; Montesano, S.; Banuelos, M.G.; Stover, A.E.; Schutte, S.S.; O’Dowd, D.K.; Hagerman, R.J.; Ono, M.; Hessl, D.R.; et al. The autism spectrum disorders stem cell resource at children’s hospital of orange county: Implications for disease modeling and drug discovery. Stem Cells Transl. Med. 2014, 3, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- De Esch, C.E.; Ghazvini, M.; Loos, F.; Schelling-Kazaryan, N.; Widagdo, W.; Munshi, S.T.; van der Wal, E.; Douben, H.; Gunhanlar, N.; Kushner, S.A.; et al. Epigenetic characterization of the fmr1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep. 2014, 3, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Doers, M.E.; Musser, M.T.; Nichol, R.; Berndt, E.R.; Baker, M.; Gomez, T.M.; Zhang, S.C.; Abbeduto, L.; Bhattacharyya, A. Ipsc-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014, 23, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-throughput screening using IPSC-derived neuronal progenitors to identify compounds counteracting epigenetic gene silencing in fragile X syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Swaroop, M.; Southall, N.; Huang, W.; Zheng, W.; Usdin, K. High-throughput screening to identify compounds that increase fragile X mental retardation protein expression in neural stem cells differentiated from fragile X syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl. Med. 2015, 4, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE 2011, 6, e26203. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Verlinsky, Y.; Strelchenko, N.; Kukharenko, V.; Rechitsky, S.; Verlinsky, O.; Galat, V.; Kuliev, A. Human embryonic stem cell lines with genetic disorders. Reprod. Biomed. Online 2005, 10, 105–110. [Google Scholar] [CrossRef]
- Turetsky, T.; Aizenman, E.; Gil, Y.; Weinberg, N.; Shufaro, Y.; Revel, A.; Laufer, N.; Simon, A.; Abeliovich, D.; Reubinoff, B.E. Laser-assisted derivation of human embryonic stem cell lines from IVF embryos after preimplantation genetic diagnosis. Hum. Reprod. 2008, 23, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Mateizel, I.; Spits, C.; De Rycke, M.; Liebaers, I.; Sermon, K. Derivation, culture, and characterization of VUB HESC lines. Vitro Cell. Dev. Biol. Anim. 2010, 46, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Tropel, P.; Tournois, J.; Come, J.; Varela, C.; Moutou, C.; Fragner, P.; Cailleret, M.; Laabi, Y.; Peschanski, M.; Viville, S. High-efficiency derivation of human embryonic stem cell lines following pre-implantation genetic diagnosis. Vitro Cell. Dev. Biol. Anim. 2010, 46, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Segal, M.; Ben-Yosef, D. Neural differentiation of fragile X human embryonic stem cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 2013, 374, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, J.; Tomishima, M.J.; Zaninovic, N.; Colak, D.; Yan, Z.; Zhan, Q.; Rosenwaks, Z.; Jaffrey, S.R.; Schildkraut, C.L. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 2013, 53, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Devys, D.; Biancalana, V.; Rousseau, F.; Boue, J.; Mandel, J.L.; Oberle, I. Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am. J. Med. Genet. 1992, 43, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Nakahori, Y.; Tsutsumi, O.; Taketani, Y.; Nakagome, Y. The CPG island of the FMR-1 gene is methylated differently among embryonic tissues: Implication for prenatal diagnosis. Hum. Reprod. 1994, 9, 1471–1473. [Google Scholar] [PubMed]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Suzumori, K.; Yamauchi, M.; Seki, N.; Kondo, I.; Hori, T. Prenatal diagnosis of a hypermethylated full fragile X mutation in chorionic villi of a male fetus. J. Med. Genet. 1993, 30, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Bontekoe, C.J.; Severijnen, L.A.; Oostra, B.A. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat MRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Gafni, O.; Weinberger, L.; Mansour, A.A.; Manor, Y.S.; Chomsky, E.; Ben-Yosef, D.; Kalma, Y.; Viukov, S.; Maza, I.; Zviran, A.; et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013, 504, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Markoulaki, S.; Mitalipova, M.; Cheng, A.W.; Cassady, J.P.; Staerk, J.; Carey, B.W.; Lengner, C.J.; Foreman, R.; Love, J.; et al. Metastable pluripotent states in nod-mouse-derived ESCS. Cell Stem Cell 2009, 4, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Mascetti, V.L.; Pedersen, R.A. Human-mouse chimerism validates human stem cell pluripotency. Cell Stem Cell 2016, 18, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Hagerman, P.J. A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J. Med. Genet. 2001, 38, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Koerner, M.V.; Pauler, F.M.; Huang, R.; Barlow, D.P. The function of non-coding RNAS in genomic imprinting. Development 2009, 136, 1771–1783. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of XIST RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef] [PubMed]
- Lanni, S.; Goracci, M.; Borrelli, L.; Mancano, G.; Chiurazzi, P.; Moscato, U.; Ferre, F.; Helmer-Citterich, M.; Tabolacci, E.; Neri, G. Role of CTCF protein in regulating FMR1 locus transcription. PLoS Genet. 2013, 9, e1003601. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 2005, 20, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Filippova, G.N.; Cheng, M.K.; Moore, J.M.; Truong, J.P.; Hu, Y.J.; Nguyen, D.K.; Tsuchiya, K.D.; Disteche, C.M. Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CPG methylation during early development. Dev. Cell 2005, 8, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Filippova, G.N.; Thienes, C.P.; Penn, B.H.; Cho, D.H.; Hu, Y.J.; Moore, J.M.; Klesert, T.R.; Lobanenkov, V.V.; Tapscott, S.J. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 2001, 28, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Sopher, B.L.; Ladd, P.D.; Pineda, V.V.; Libby, R.T.; Sunkin, S.M.; Hurley, J.B.; Thienes, C.P.; Gaasterland, T.; Filippova, G.N.; La Spada, A.R. CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron 2011, 70, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Dasaradhi, P.V.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. Rna interference: Biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 2003, 67, 657–685. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Alisch, R.S.; Warren, S.T. RNA and micrornas in fragile X mental retardation. Nat. Cell Biol. 2004, 6, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Kelley, K.; Chang, S.J.; Lin, S.L. Mechanism of repeat-associated micrornas in fragile X syndrome. Neural Plast. 2012, 2012, 104796. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, T.; Nogami, M.; Yoshikawa, M.; Ikeno, M.; Okazaki, T.; Takami, Y.; Nakayama, T.; Oshimura, M. Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat. Cell Biol. 2004, 6, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Kanellopoulou, C.; Muljo, S.A.; Kung, A.L.; Ganesan, S.; Drapkin, R.; Jenuwein, T.; Livingston, D.M.; Rajewsky, K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005, 19, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CPG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A.; Jaenisch, R. A shift from reversible to irreversible X inactivation is triggered during es cell differentiation. Mol. Cell 2000, 5, 695–705. [Google Scholar] [CrossRef]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Heitz, D.; Devys, D.; Imbert, G.; Kretz, C.; Mandel, J.L. Inheritance of the fragile X syndrome: Size of the fragile X premutation is a major determinant of the transition to full mutation. J. Med. Genet. 1992, 29, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Wohrle, D.; Salat, U.; Glaser, D.; Mucke, J.; Meisel-Stosiek, M.; Schindler, D.; Vogel, W.; Steinbach, P. Unusual mutations in high functioning fragile X males: Apparent instability of expanded unmethylated cgg repeats. J. Med. Genet. 1998, 35, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Glaser, D.; Wohrle, D.; Salat, U.; Vogel, W.; Steinbach, P. Mitotic behavior of expanded cgg repeats studied on cultured cells: Further evidence for methylation-mediated triplet repeat stability in fragile X syndrome. Am. J. Med. Genet. 1999, 84, 226–228. [Google Scholar] [CrossRef]
- Wohrle, D.; Salat, U.; Hameister, H.; Vogel, W.; Steinbach, P. Demethylation, reactivation, and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am. J. Hum. Genet. 2001, 69, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Harley, H.G.; Rundle, S.A.; MacMillan, J.C.; Myring, J.; Brook, J.D.; Crow, S.; Reardon, W.; Fenton, I.; Shaw, D.J.; Harper, P.S. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am. J. Hum. Genet. 1993, 52, 1164–1174. [Google Scholar] [PubMed]
- Trottier, Y.; Biancalana, V.; Mandel, J.L. Instability of CAG repeats in huntington’s disease: Relation to parental transmission and age of onset. J. Med. Genet. 1994, 31, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Burman, R.W.; Popovich, B.W.; Jacky, P.B.; Turker, M.S. Fully expanded FMR1 CGG repeats exhibit a length- and differentiation-dependent instability in cell hybrids that is independent of DNA methylation. Hum. Mol. Genet. 1999, 8, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Balakumaran, B.S.; Freudenreich, C.H.; Zakian, V.A. CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in saccharomyces cerevisiae. Hum. Mol. Genet. 2000, 9, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Nichol Edamura, K.; Leonard, M.R.; Pearson, C.E. Role of replication and CPG methylation in fragile X syndrome CGG deletions in primate cells. Am. J. Hum. Genet. 2005, 76, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Samadashwily, G.M.; Raca, G.; Mirkin, S.M. Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 1997, 17, 298–304. [Google Scholar] [CrossRef] [PubMed]
- White, P.J.; Borts, R.H.; Hirst, M.C. Stability of the human fragile X (CGG)(N) triplet repeat array in saccharomyces cerevisiae deficient in aspects of DNA metabolism. Mol. Cell Biol. 1999, 19, 5675–5684. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. DNA structures, repeat expansions and human hereditary disorders. Curr. Opin. Struct. Biol. 2006, 16, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M.; Smirnova, E.V. Positioned to expand. Nat. Genet. 2002, 31, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Mitas, M. Trinucleotide repeats associated with human disease. Nucleic Acids Res. 1997, 25, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.; Greenwell, P.W.; Liu, C.P.; Arnheim, N.; Petes, T.D. Triplet repeats form secondary structures that escape DNA repair in yeast. Proc. Natl. Acad. Sci. USA 1999, 96, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Barron, M.D.; Romero, R.M.; Christy, M.; Gold, B.; Dai, J.; Gray, D.M.; Haworth, I.S.; Mitas, M. At physiological PH, D(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry 1997, 36, 3687–3699. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Nichol, K.; Wang, Y.H.; Pearson, C.E. Evidence of CIS-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat. Genet. 2002, 31, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at cgg repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Somma, V.; Nakamura, A.J.; Bonner, W.M.; D’Ambrosio, E.; Usdin, K. The role of DNA damage response pathways in chromosome fragility in fragile X syndrome. Nucleic Acids Res. 2009, 37, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Zhao, X.N.; Usdin, K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2014, 35, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Ogushi, S.; Kurimoto, K.; Shimamoto, S.; Ohta, H.; Saitou, M. Offspring from oocytes derived from in vitro primordial germ cell-like cells in mice. Science 2012, 338, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Bassell, G.J.; Warren, S.T. Fragile X syndrome: Loss of local mrna regulation alters synaptic development and function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Antar, L.N.; Afroz, R.; Dictenberg, J.B.; Carroll, R.C.; Bassell, G.J. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and FMR1 MRNA localization differentially in dendrites and at synapses. J. Neurosci. 2004, 24, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Iliff, A.J.; Renoux, A.J.; Krans, A.; Usdin, K.; Sutton, M.A.; Todd, P.K. Impaired activity-dependent FMRP translation and enhanced mglur-dependent LTD in fragile X premutation mice. Hum. Mol. Genet. 2013, 22, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Braat, S.; D’Hulst, C.; Heulens, I.; De Rubeis, S.; Mientjes, E.; Nelson, D.L.; Willemsen, R.; Bagni, C.; Van Dam, D.; De Deyn, P.P.; et al. The gabaa receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle 2015, 14, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, L.; Chen, J.; Wang, Z.; Li, Z.; Wan, Q. Regulation of gabaa receptors by fragile X mental retardation protein. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 169–176. [Google Scholar] [PubMed]
- Wang, T.; Bray, S.M.; Warren, S.T. New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 2012, 22, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P.J.; Stafstrom, C.E. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009, 9, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J. Fragile-X chromosome and learning disability. J. Am. Acad. Child Adolesc. Psychiatry 1987, 26, 938. [Google Scholar] [CrossRef]
- Willemsen, R.; Oostra, B.A.; Bassell, G.J.; Dictenberg, J. The fragile X syndrome: From molecular genetics to neurobiology. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; McMillan, E.; Wallace, K.; Tubon, T.C., Jr.; Capowski, E.E.; Svendsen, C.N. Normal neurogenesis but abnormal gene expression in human fragile X cortical progenitor cells. Stem Cells Dev. 2008, 17, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Castren, M.; Tervonen, T.; Karkkainen, V.; Heinonen, S.; Castren, E.; Larsson, K.; Bakker, C.E.; Oostra, B.A.; Akerman, K. Altered differentiation of neural stem cells in fragile X syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 17834–17839. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Hagerman, R.J.; Willemsen, R.; Pessah, I.N. Murine hippocampal neurons expressing FMR1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 2010, 19, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Kuznitsov-Yanovsky, L.; Segal, M.; Ben-Yosef, D. Functional deficiencies in fragile X neurons derived from human embryonic stem cells. J. Neurosci. 2015, 35, 15295–15306. [Google Scholar] [CrossRef] [PubMed]
- Suhl, J.A.; Chopra, P.; Anderson, B.R.; Bassell, G.J.; Warren, S.T. Analysis of FMRP mrna target datasets reveals highly associated mrnas mediated by G-quadruplex structures formed via clustered WGGA sequences. Hum. Mol. Genet. 2014, 23, 5479–5491. [Google Scholar] [CrossRef] [PubMed]
- Halevy, T.; Czech, C.; Benvenisty, N. Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem Cell Rep. 2015, 4, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.C. Back to basics: Sox genes. Dev. Dyn. 2007, 236, 2356–2366. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Stolt, C.C. From stem cells to neurons and glia: A soxist’s view of neural development. Trends Neurosci. 2005, 28, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Mayshar, Y.; Amit, A.; Ben-Yosef, D. Molecular mechanisms regulating impaired neurogenesis of fragile X syndrome human embryonic stem cells. Stem Cells Dev. 2015, 24, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Segal, M.; Ben-Yosef, D. Immature responses to GABA in fragile X neurons derived from human embryonic stem cells. Front. Cell. Neurosci. 2016, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Adusei, D.C.; Pacey, L.K.; Chen, D.; Hampson, D.R. Early developmental alterations in gabaergic protein expression in fragile X knockout mice. Neuropharmacology 2010, 59, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Braat, S.; Kooy, R.F. Insights into gabaaergic system deficits in fragile X syndrome lead to clinical trials. Neuropharmacology 2014, 88, 48–54. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Nomura, T.; Xu, J.; Contractor, A. The developmental switch in gaba polarity is delayed in fragile X mice. J. Neurosci. 2014, 34, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Kratovac, S.; Corbin, J.G. Developmental changes in expression of inhibitory neuronal proteins in the fragile X syndrome mouse basolateral amygdala. Brain Res. 2013, 1537, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Paluszkiewicz, S.M.; Martin, B.S.; Huntsman, M.M. Fragile X syndrome: The gabaergic system and circuit dysfunction. Dev. Neurosci. 2011, 33, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Bagni, C.; Oostra, B.A. Fragile X syndrome: From protein function to therapy. Am. J. Med. Genet. A 2013, 161, 2809–2821. [Google Scholar] [CrossRef] [PubMed]
- Smeets, H.J.; Smits, A.P.; Verheij, C.E.; Theelen, J.P.; Willemsen, R.; van de Burgt, I.; Hoogeveen, A.T.; Oosterwijk, J.C.; Oostra, B.A. Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 1995, 4, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.H.; Schaefer, T.L.; Friedmann, K.J.; Fitzpatrick, S.E.; Erickson, C.A. Pharmacotherapy for fragile X syndrome: Progress to date. Drugs 2016, 76, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 2012, 60, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Christman, J.K. 5-azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of fmr1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell. Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Rudek, M.A.; Zhao, M.; He, P.; Hartke, C.; Gilbert, J.; Gore, S.D.; Carducci, M.A.; Baker, S.D. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J. Clin. Oncol. 2005, 23, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Narayanaswamy, R.; Fenn, K.; Szpakowski, S.; Sasaki, C.; Costa, J.; Blancafort, P.; Lizardi, P.M. Sequence-specific biosensors report drug-induced changes in epigenetic silencing in living cells. DNA Cell Biol. 2012, 31, S2–S10. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.P.; Wijetunga, N.A.; McLellan, A.S.; Suzuki, M.; Greally, J.M. DNA demethylation by 5-aza-2′-deoxycytidine is imprinted, targeted to euchromatin, and has limited transcriptional consequences. Epigenetics Chromatin 2015, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, K.; Sattler, N.; Wagner, M.; Lubbert, M.; Dorken, B.; Tamm, I. Induction of gene expression by 5-aza-2′-deoxycytidine in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) but not epithelial cells by DNA-methylation-dependent and -independent mechanisms. Leukemia 2005, 19, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Ananiev, G.E.; Musser, M.; Ness, K.H.; Maglaque, D.L.; Saha, K.; Bhattacharyya, A.; Zhao, X. Establishment of reporter lines for detecting fragile X mental retardation (FMR1) gene reactivation in human neural cells. Stem Cells 2016. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of fmr1 methylation and silencing by editing the triplet repeats in fragile X IPSC-derived neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Dolen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of fragile X syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnrnp A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a drosophila model of fxtas. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-DCAS9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
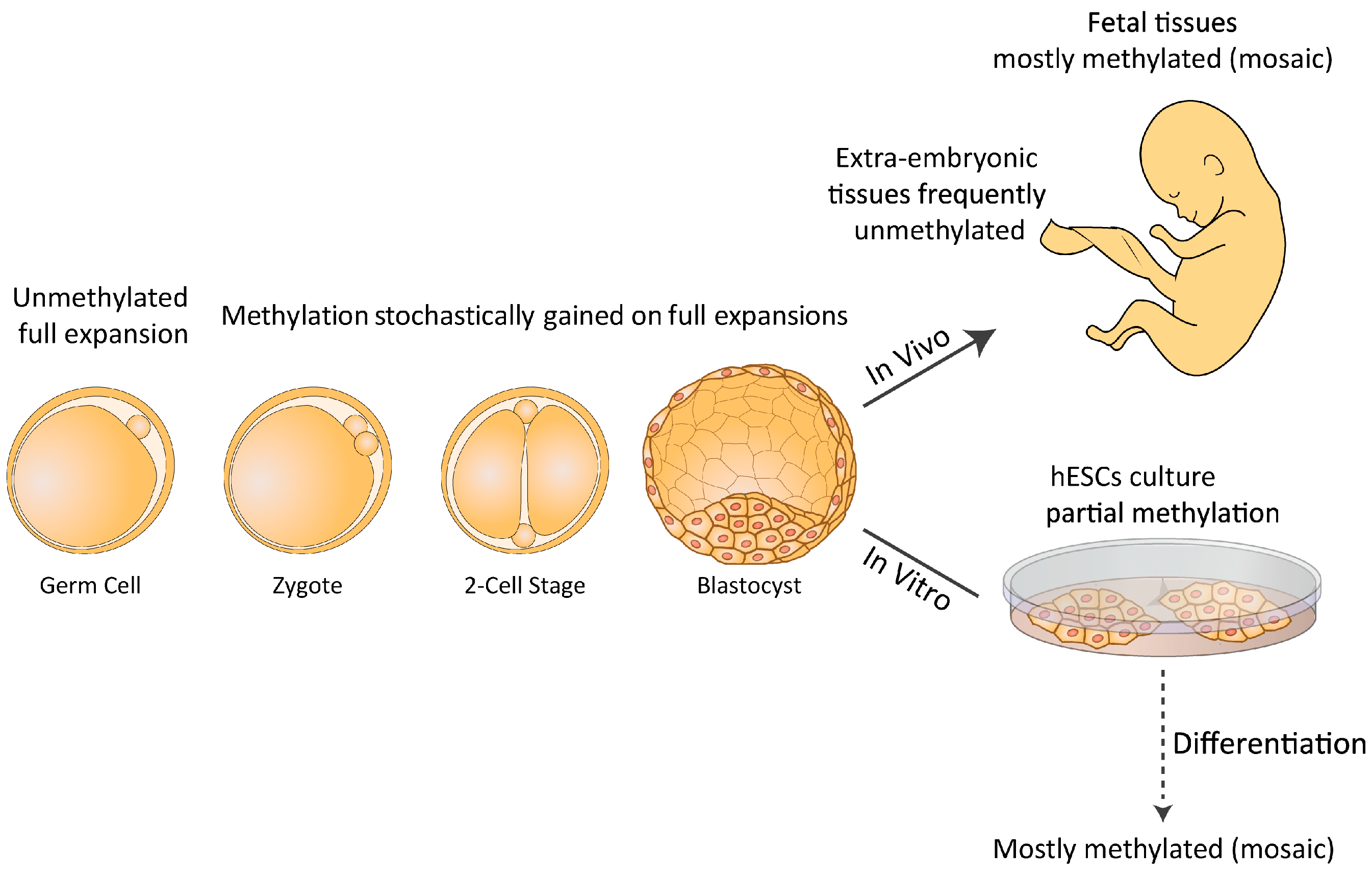

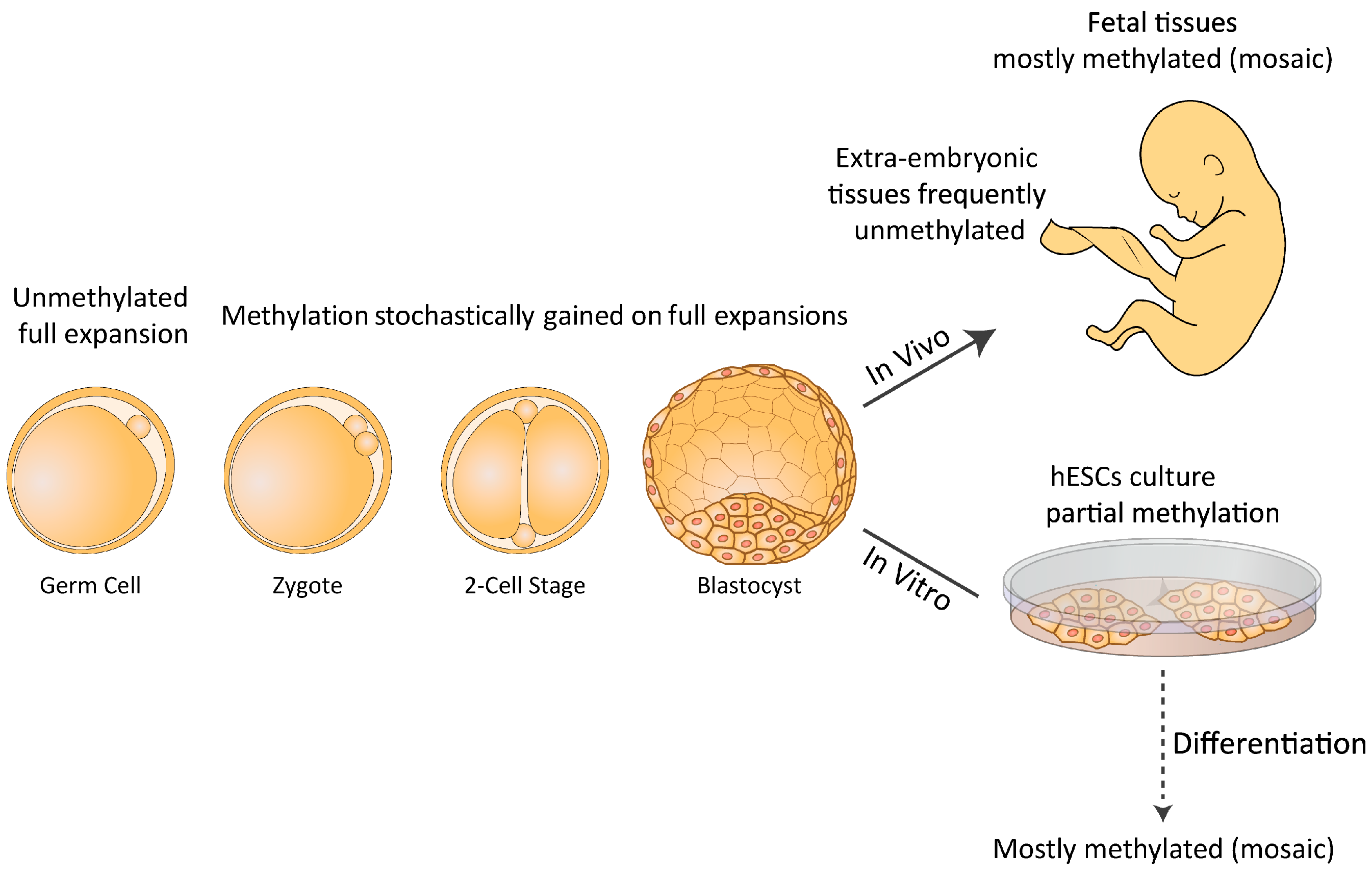{kind=link}
| Cell Line | Karyotype | Number of CGG Repeat | Methylation State | Derivation Center |
|---|---|---|---|---|
| SI-214 | 46, XY | 138, 450 | mostly methylated | RGI [40] |
| Lis01_HEFX1 | 46, XY | 200–650 | unmethylated | TASMC [27] |
| HAD5 | 46, XX | 300 | NA | HHUMC [41] |
| VUB11_FXS | 46, XX | 2000, 2010 | fully methylated | VUB [42] |
| VUB13_FXS | 46, XX | 2000 | fully methylated | VUB [42] |
| STR-189-FRAXA | 46, XX | NA | NA | IGBMC [43] |
| STR-233-FRAXA | 46, XY | NA | NA | IGBMC [43] |
| Lis26_FXS_6 | 46, XY | 50–600 | partially methyalted | TASMC [44] |
| WCMC37 | 46, XY | 142, 167, 179, 450 | mostly methylated | WCMC, [45] |
| SZ-FX1 | 46, XX | 300–600 | mostly methyalted | SZMC [32] |
| SZ-FX3 | 46, XX | 300–600 | partially methyalted | SZMC [32] |
| SZ-FX6 | 46, XY | 200–600 | partially methyalted | SZMC [32] |
| SZ-FX7 | 46, XX | 200–300 | unmethylated | SZMC [32] |
| SZ-FX8 | 46, XY | 200–600 | mostly methyalted | SZMC [32] |
| SZ-FX12 | 46, XX | 150–300 | partially methyalted | SZMC [32] |
| SZ-FX14 | 46, XY | 290–600 | mostly methyalted | SZMC [32] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mor-Shaked, H.; Eiges, R. Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells. Genes 2016, 7, 77. https://doi.org/10.3390/genes7100077
Mor-Shaked H, Eiges R. Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells. Genes. 2016; 7(10):77. https://doi.org/10.3390/genes7100077
Chicago/Turabian StyleMor-Shaked, Hagar, and Rachel Eiges. 2016. "Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells" Genes 7, no. 10: 77. https://doi.org/10.3390/genes7100077
APA StyleMor-Shaked, H., & Eiges, R. (2016). Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells. Genes, 7(10), 77. https://doi.org/10.3390/genes7100077