
Molecular Modeling of Myrosinase from Brassica oleracea: A Structural Investigation of Sinigrin Interaction


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Template Identification and Homology Modeling
2.2. Molecular Dynamics Simulation
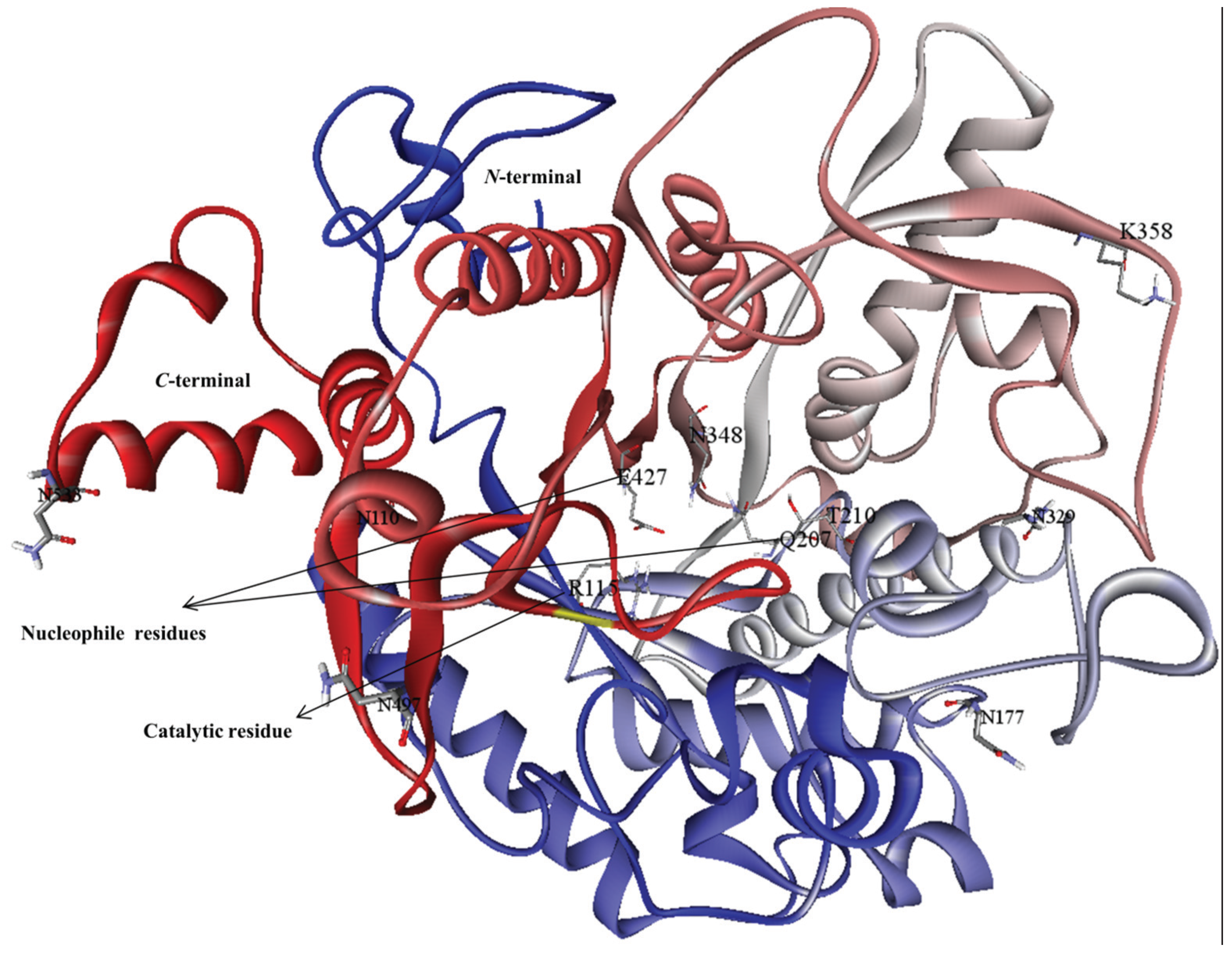
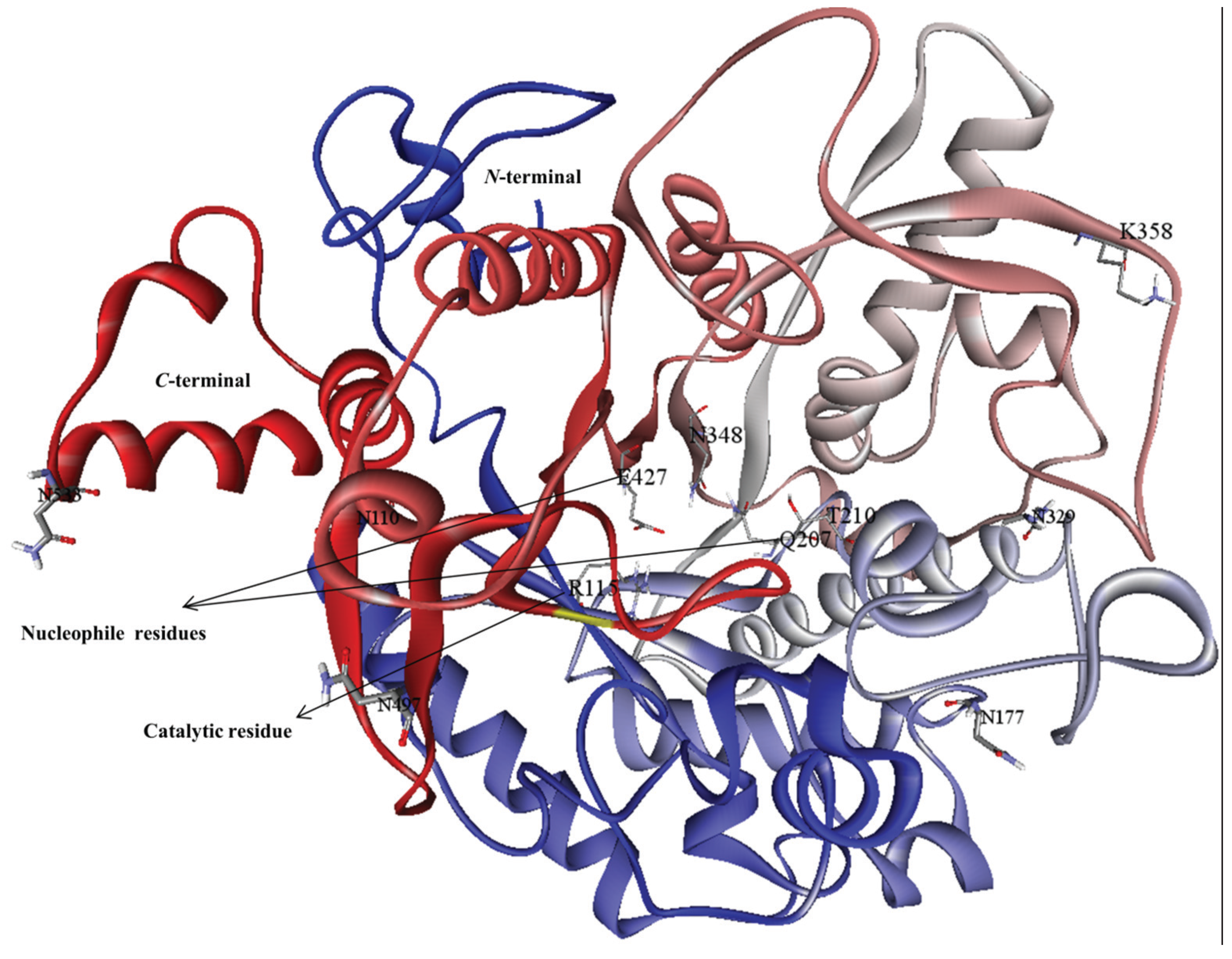
2.3. Analysis of MYR Active Site and N-Glycosylation Sites
2.4. MYR–Sinigrin Interaction
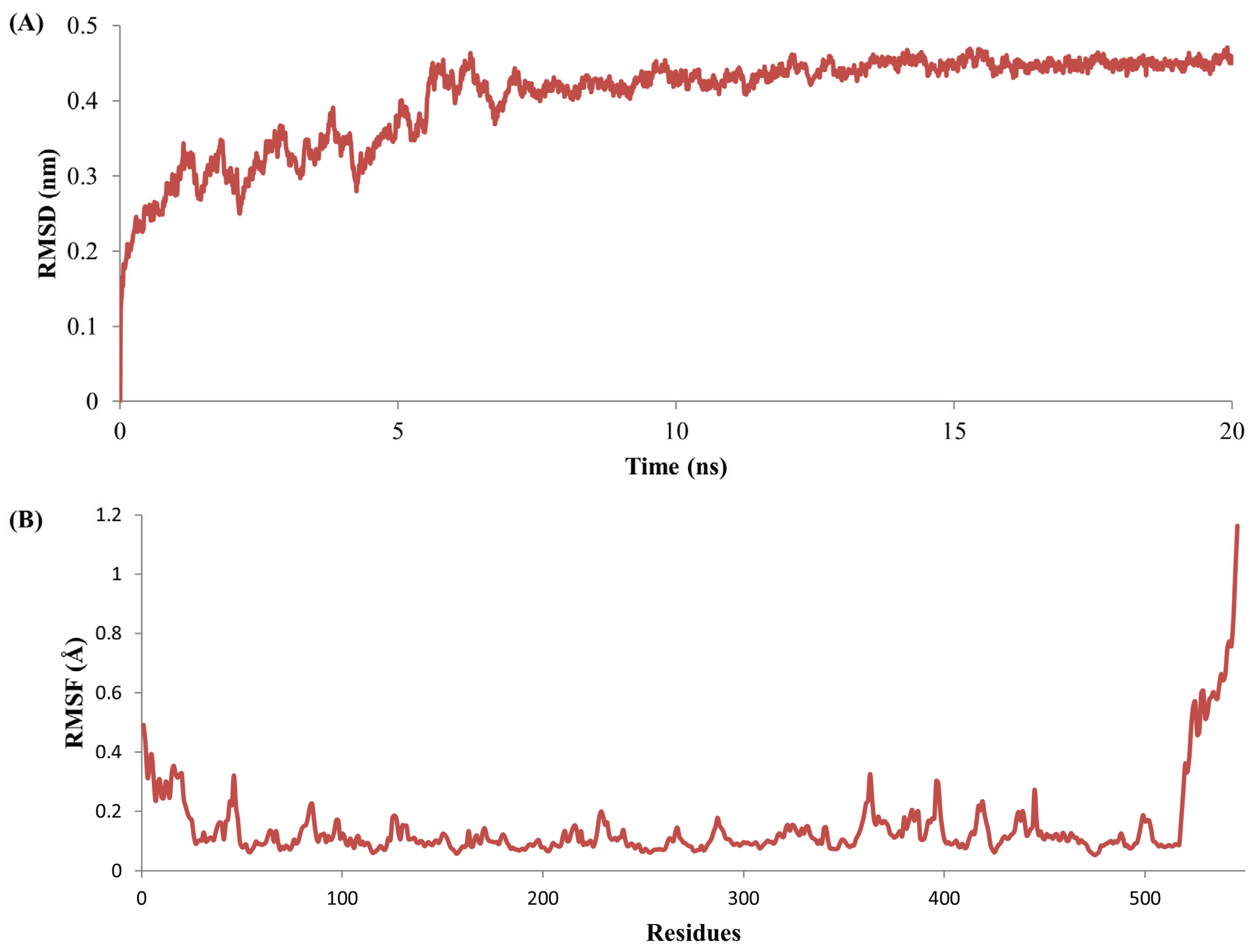
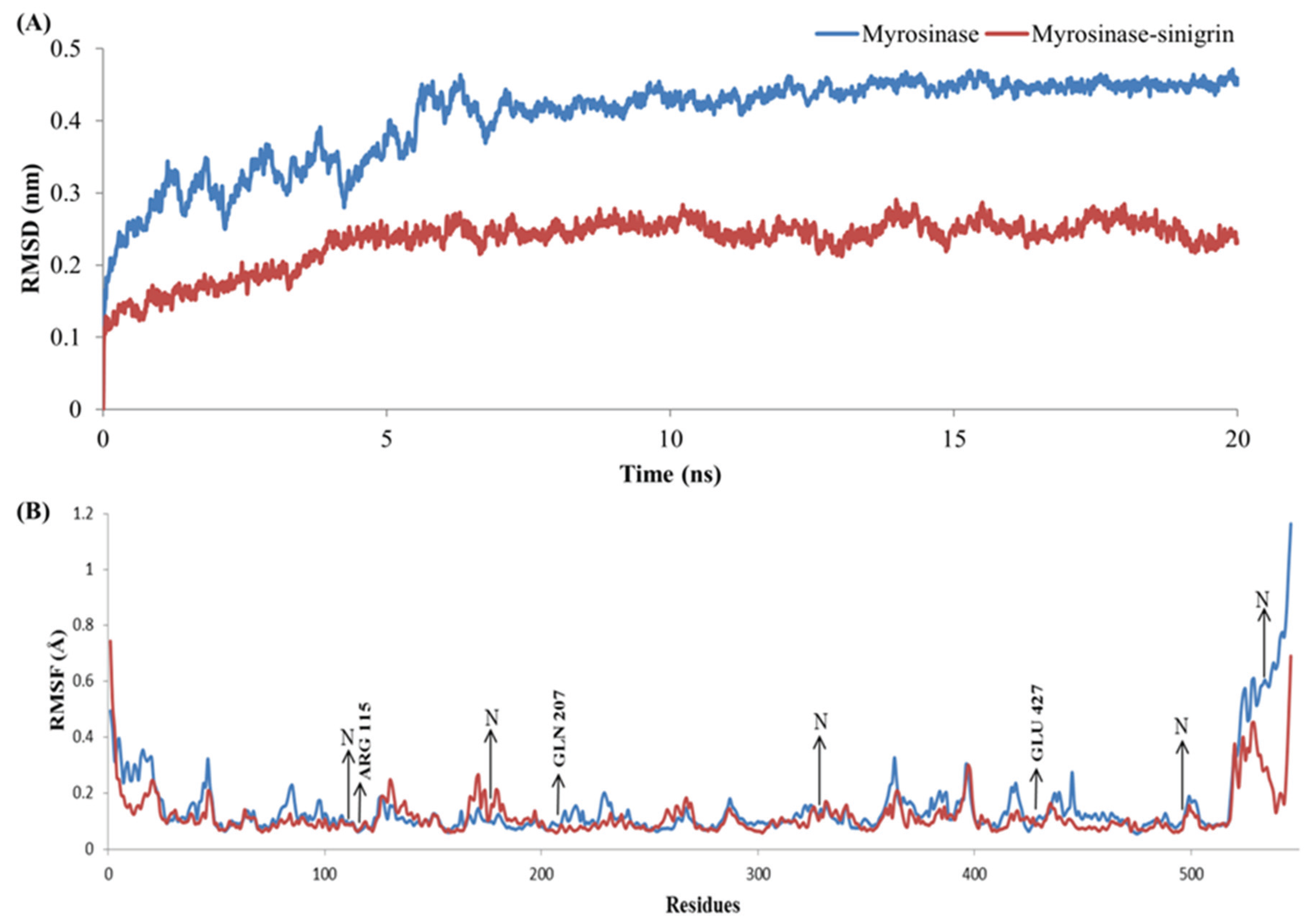
2.5. Complex Stability Refinement by Molecular Dynamics Simulation
3. Results and Discussion
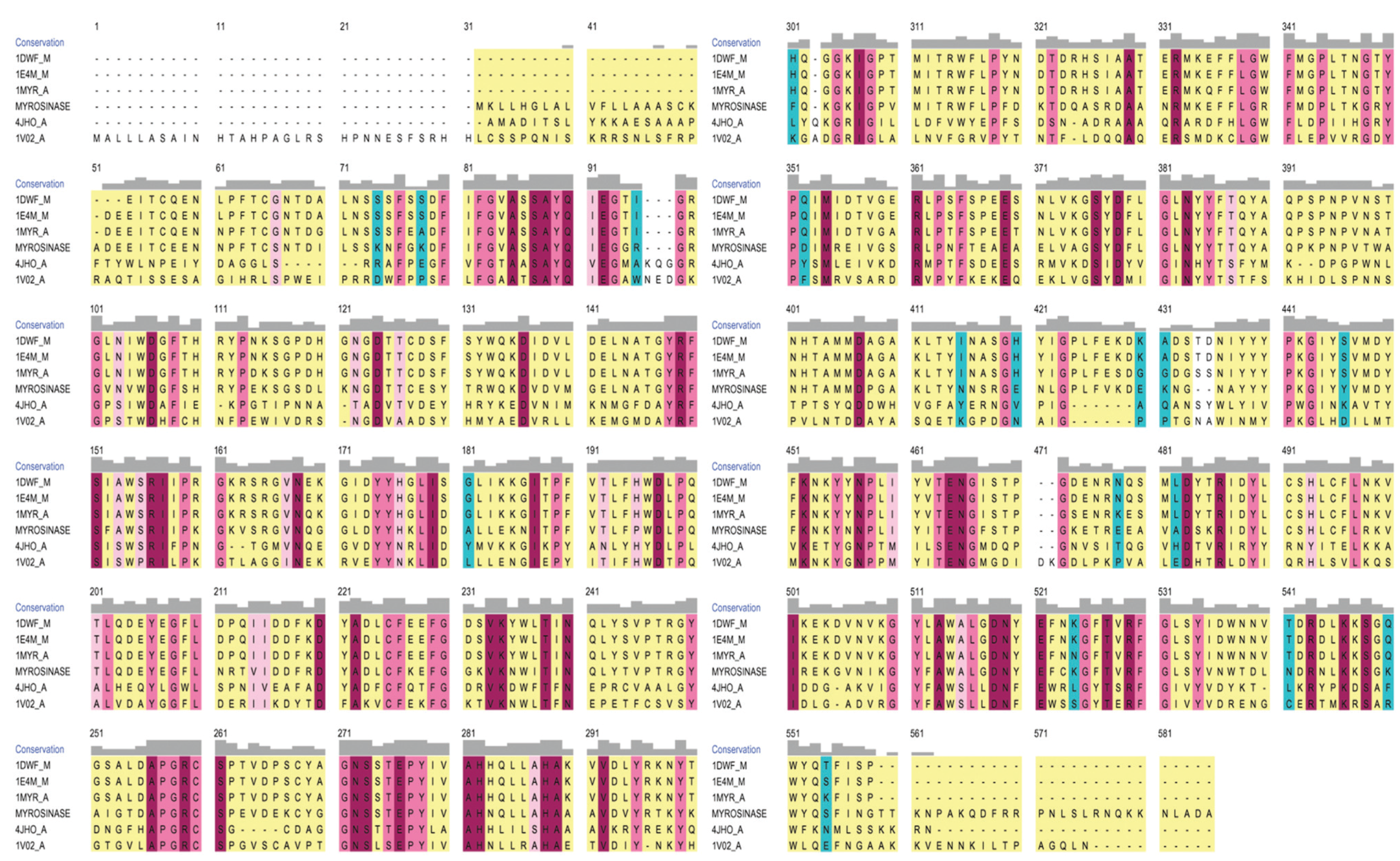
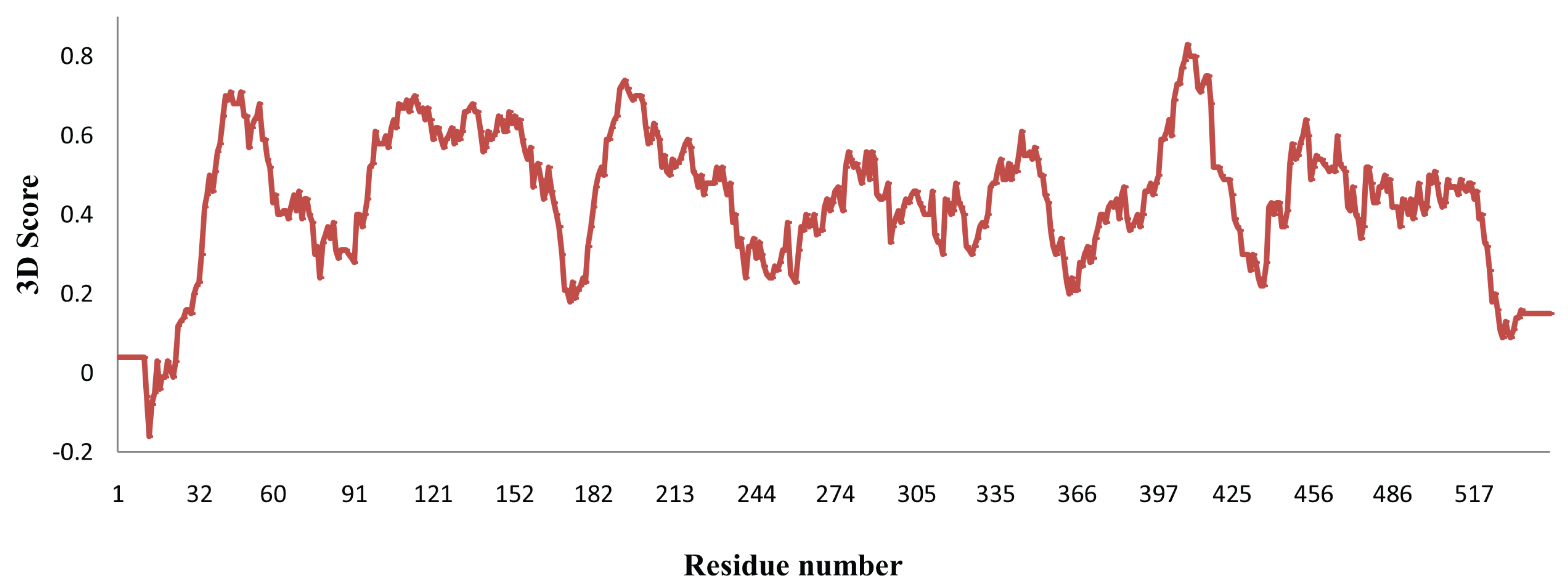
3.1. Homology Modeling
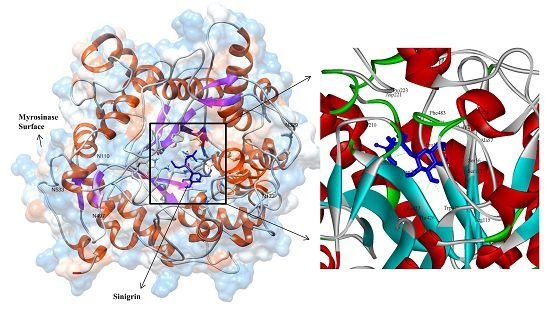
3.2. Model Refinement and Structure of MYR
3.3. Molecular Interaction Studies

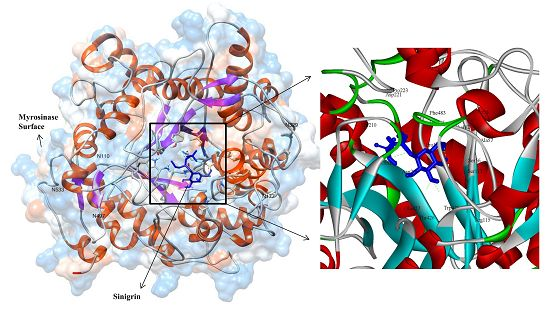{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
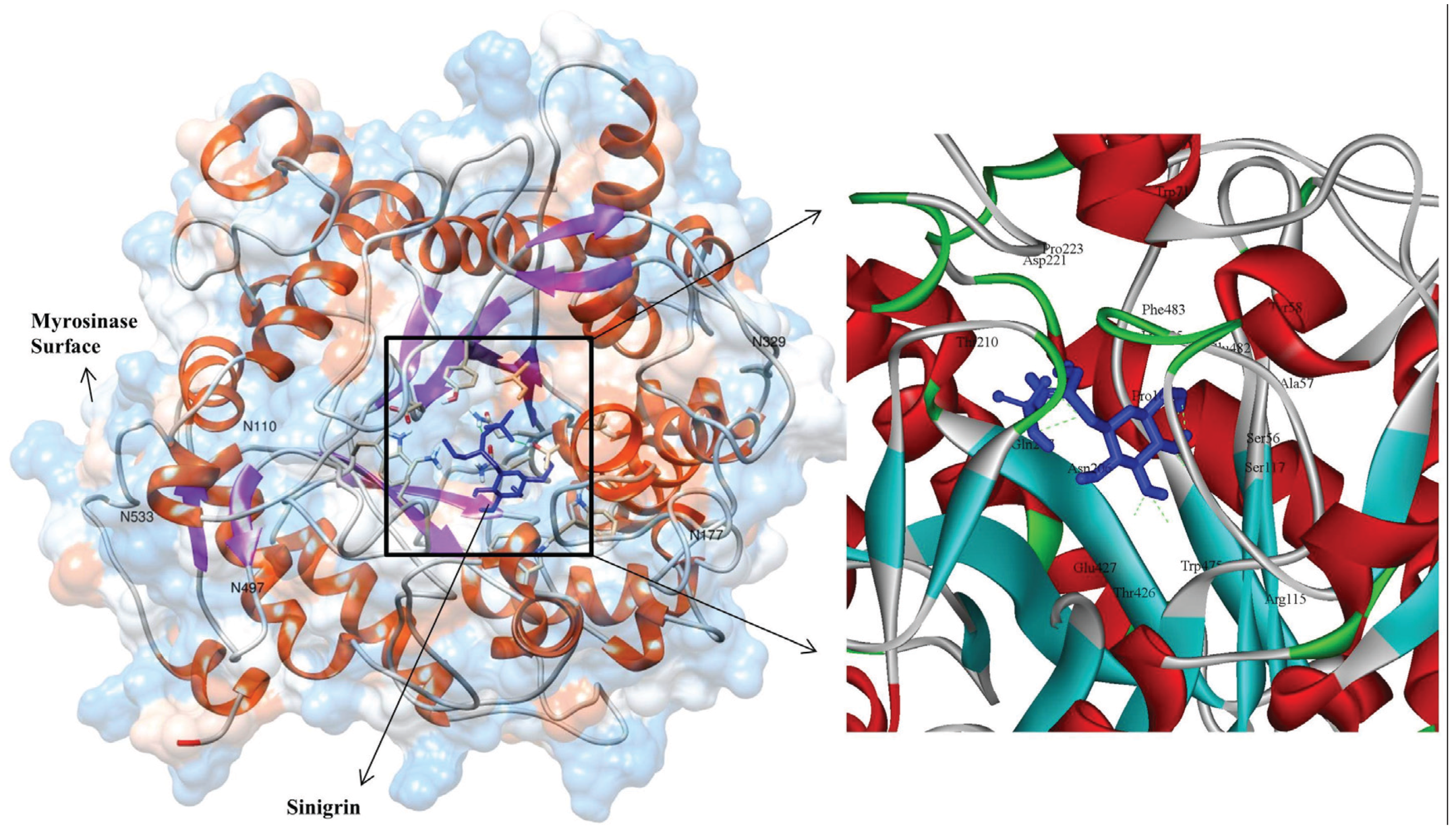{kind=link}
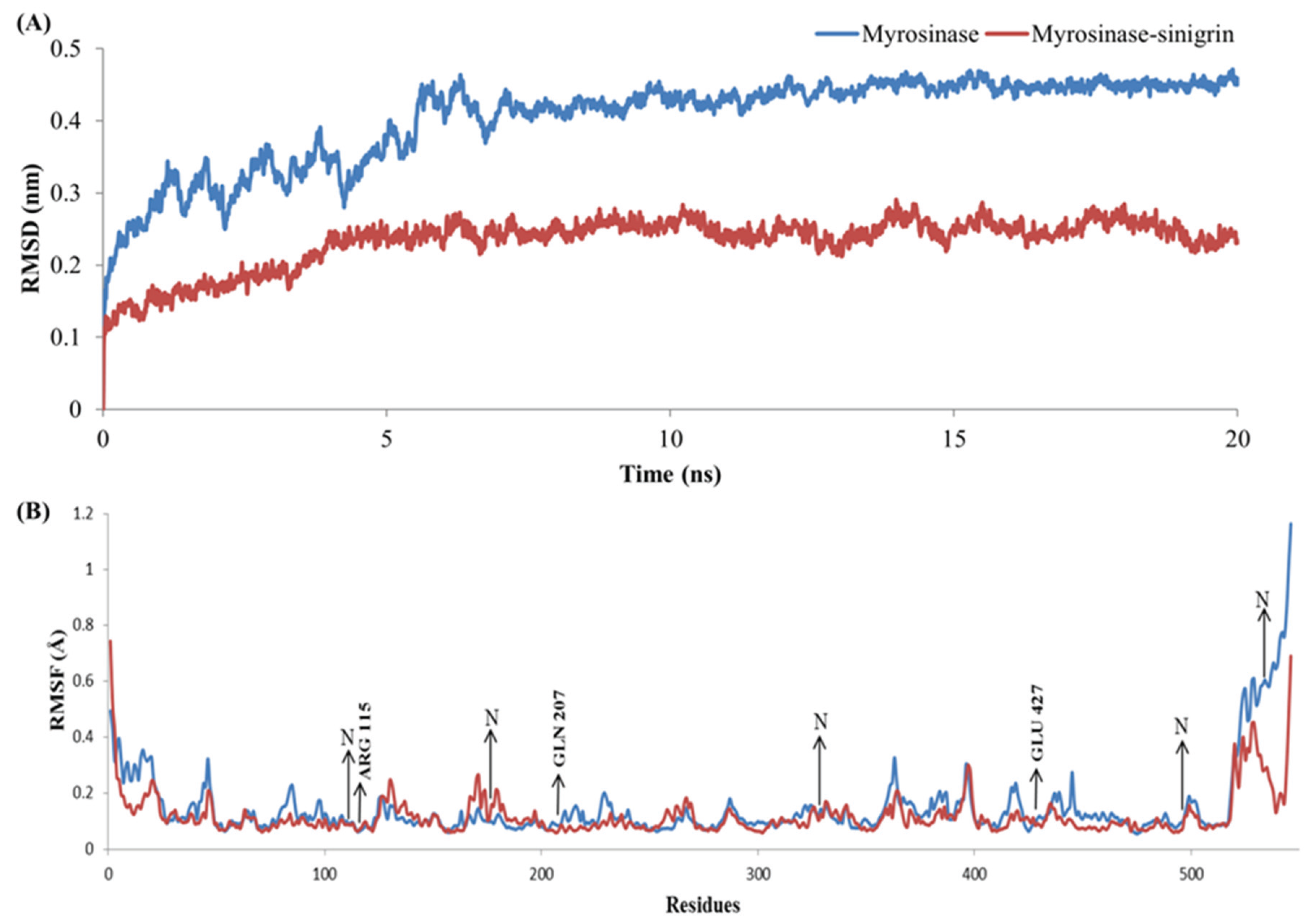{kind=link}
| S. No | Hydrogen Bond Interacting Residue | Hydrogen Bond Donor | Hydrogen Bond Acceptor | Hydrogen Bond Length (Å) | Number of Hydrogen Bonds |
|---|---|---|---|---|---|
| 1 | ARG115 * | ARG115:HH22 | LIG1:O | 2.36 | 1 |
| 2 | SER117 | SER117:HG | LIG1:O | 1.7 | 1 |
| 3 | GLN207 * | GLN207:HE21 | LIG:O | 2.10 | 1 |
| 4 | T221 | LIG1:HN | ASP221:OD1 | 2.27 | 1 |
| 5 | GLU427 * | LIG1:H | GLU427:OE1 | 1.85 | 2 |
| LIG1:H | GLU427:OE2 | 2.34 | - | ||
| 6 | LYS485 | LYS485:HZ2 | LIG1:O | 2.43 | 2 |
| LYS485:HZ2 | LIG1:O | 2.41 | - | ||
| Non-bonded interacted residues: TRP475, THR426, ASN206, PHE483, GLU482, SER56, ALA57, PRO161, TRP71, TYR58 AND PRO223 | |||||
| * Catalytic and nucleophile residues are highlighted in bold. | |||||
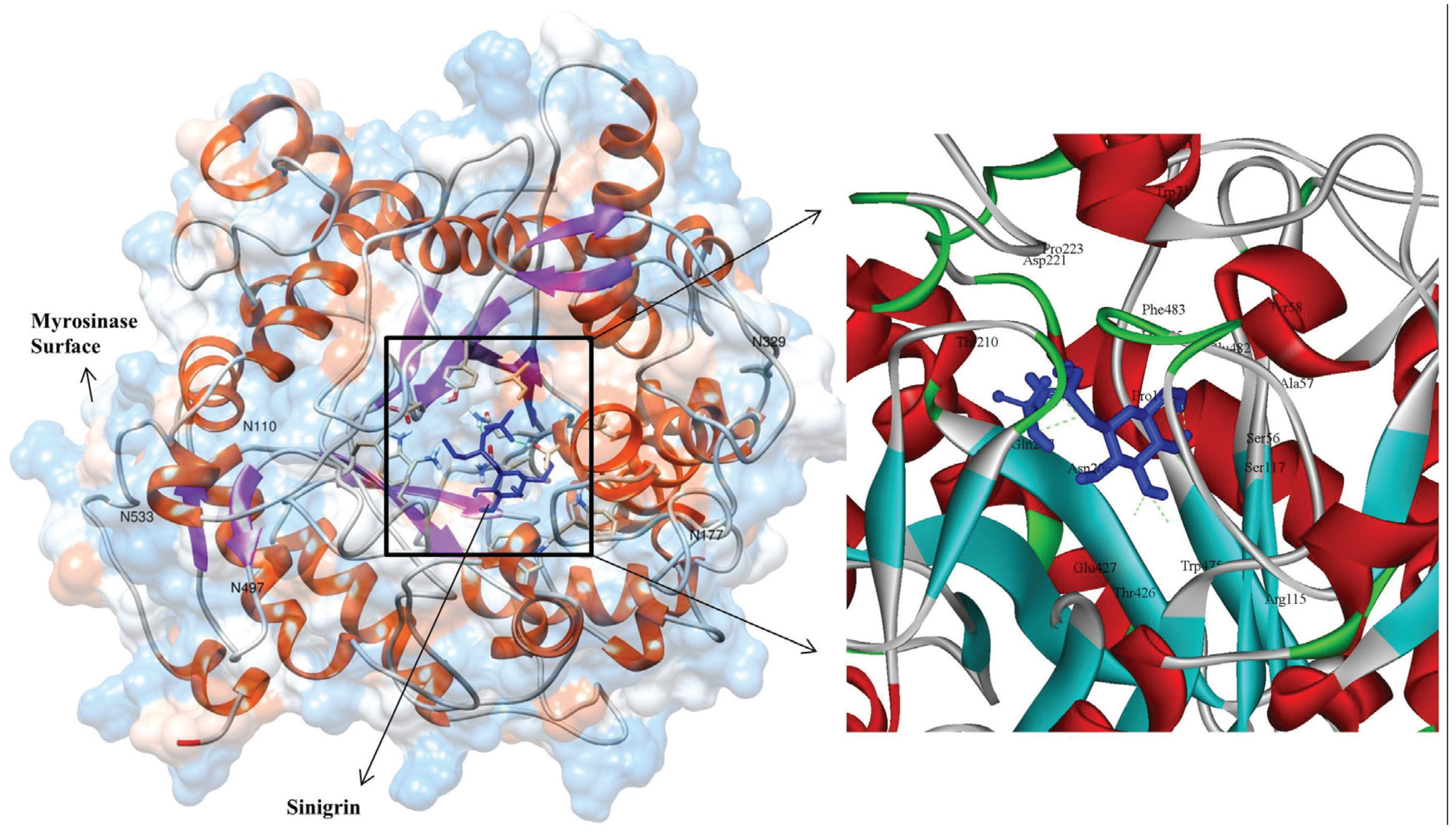
3.4. Stability Evolution of the MYR–Sinigrin Complex
| Molecule | Predicted Binding Affinity (kcal/mol) | Hydrophobic Pair Score (pKd) | Hydrophobic Match Score (pKd) | Hydrophobic Surface Score (pKd) | Predicted Mean Binding Affinity (pKd) |
|---|---|---|---|---|---|
| MYR–sinigrin complex | −6.98 | 5.12 | 5.13 | 5.11 | 5.12 |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflict of Interest
References
- Kelly, P.; Bones, A.; Rossiter, J. Sub-cellular immunolocalization of the glucosinolate sinigrin in seedlings of Brassica juncea. Planta 1998, 206, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Ciska, E.; Martyniak-Przybyszewska, B.; Kozlowska, H. Content of glucosinolates in cruciferous vegetables grown at the same site for two years under different climatic conditions. J. Agric. Food Chem. 2000, 48, 2862–2867. [Google Scholar] [CrossRef] [PubMed]
- Kliebenstein, D.J.; Kroymann, J.; Mitchell-Olds, T. The glucosinolate-myrosinase system in an ecological and evolutionary context. Curr. Opin. Plant Biol. 2005, 8, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ballesta, M.; Carvajal, M. Myrosinase in Brassicaceae: The most important issue for glucosinolate turnover and food quality. Phytochem. Rev. 2015, 14, 1045–1051. [Google Scholar] [CrossRef]
- Ishida, M.; Hara, M.; Fukino, N.; Kakizaki, T.; Morimitsu, Y. Glucosinolate metabolism, functionality and breeding for the improvement of Brassicaceae vegetables. Breed. Sci. 2014, 64, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Tiedink, H.; Davies, J.; van Broekhoven, L.; van der Kamp, H.; Jongen, W. Formation of mutagenic N-nitroso compounds in vegetable extracts upon nitrite treatment: A comparison with the glucosinolate content. Food Chem. Toxicol. 1988, 26, 947–954. [Google Scholar] [CrossRef]
- Avato, P.; Argentieri, M.P. Brassicaceae: A rich source of health improving phytochemicals. Phytochem. Rev. 2015, 14, 1019–1033. [Google Scholar] [CrossRef]
- Bones, A.M.; Rossiter, J.T. The myrosinase-glucosinolate system, its organisation and biochemistry. Physiol. Plant. 1996, 97, 194–208. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Cottaz, S.; Rollin, P.; Vasella, A.; Henrissat, B. High resolution X-ray crystallography shows that ascorbate is a cofactor for myrosinase and substitutes for the function of the catalytic base. J. Biol. Chem. 2000, 275, 39385–39393. [Google Scholar] [CrossRef] [PubMed]
- Gleadow, R.M.; Møller, B.L. Cyanogenic glycosides: Synthesis, physiology, and phenotypic plasticity. Annu. Rev. Plant Biol. 2014, 65, 155–185. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, P.; Osbourn, A. Plant-microbe interactions: Chemical diversity in plant defense. Science 2009, 324, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Meng, Q.; Xu, J.; Jiao, Y.; Zhao, L.; Zhang, X.; Sarkar, F.H.; Brown, M.L.; Dritschilo, A.; Rosen, E.M. DIM (3, 3'-diindolylmethane) confers protection against ionizing radiation by a unique mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 18650–18655. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, R.; Schreiner, M.; Krumbein, A.; Ciska, E.; Holst, B.; Rowland, I.; de Schrijver, R.; Hansen, M.; Gerhäuser, C.; Mithen, R.; et al. Glucosinolates in Brassica vegetables: The influence of the food supply chain on intake, bioavailability and human health. Mol. Nutr. Food Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Kadir, N.H.; David, R.; Rossiter, J.T.; Gooderham, N.J. The selective cytotoxicity of the alkenyl glucosinolate hydrolysis products and their presence in Brassica vegetables. Toxicology 2015, 334, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.I.; Haldar, S.; Porter, S.; Matthews, S.; Sullivan, S.; Coulter, J.; McGlynn, H.; Rowland, I. The effect of cruciferous and leguminous sprouts on genotoxicity, in vitro and in vivo. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 1199–1205. [Google Scholar] [PubMed]
- Porrini, M.; Riso, P.; Oriani, G. Spinach and tomato consumption increases lymphocyte DNA resistance to oxidative stress but this is not related to cell carotenoid concentrations. Eur. J. Nutr. 2002, 41, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Aires, A.; Mota, V.R.; Saavedra, M.J.; Rosa, E.A.; Bennett, R.N. The antimicrobial effects of glucosinolates and their respective enzymatic hydrolysis products on bacteria isolated from the human intestinal tract. J. Appl. Microbiol. 2009, 106, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Riga, E. The effects of Brassica green manures on plant parasitic and free living nematodes used in combination with reduced rates of synthetic nematicides. J. Nematol. 2011, 43, 119–121. [Google Scholar] [PubMed]
- Sotelo, T.; Lema, M.; Soengas, P.; Cartea, M.; Velasco, P. In vitro activity of glucosinolates and their degradation products against brassica-pathogenic bacteria and fungi. Appl. Environ. Microbiol. 2015, 81, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, S.F.; Palmquist, D.E.; Duval, S.M.; Berhow, M.A. Herbicidal activity of glucosinolate-containing seedmeals. 2009, 57, 1821–1826. [Google Scholar] [CrossRef]
- Magrath, R.; Herron, C.; Giamoustaris, A.; Mithen, R. The inheritance of aliphatic glucosinolates in Brassica napus. Plant Breed. 1993, 111, 55–72. [Google Scholar] [CrossRef]
- Devi, J.R.; Thangam, E.B. Mechanisms of anticancer activity of sulforaphane from Brassica oleracea in HEp-2 human epithelial carcinoma cell line. Asian Pac. J. Cancer Prev. 2012, 13, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Lenman, M.; Falk, A.; Rodin, J.; Hoglund, A.; Ek, B.; Rask, L. Differential expression of myrosinase gene families. Plant Physiol. 1993, 103, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Kumar, S.; Sangwan, S.; Yadav, I.S.; Yadav, R. Protein modeling and active site binding mode interactions of myrosinase-sinigrin in Brassica juncea—An in silico approach. J. Mol. Gr. Model. 2011, 29, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Xu, J. Low-homology protein threading. Bioinformatics 2010, 26, i294–i300. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Wunderlich, Z.; Monleon, D.; Tejero, R.; Montelione, G.T. Assessing model accuracy using the homology modeling automatically software. Proteins 2008, 70, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res. 2005, 33, D34–D38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, W.P.; Cottaz, S.; Driguez, H.; Iori, R.; Palmieri, S.; Henrissat, B. The crystal structures of Sinapis alba myrosinase and a covalent glycosyl-enzyme intermediate provide insights into the substrate recognition and active-site machinery of an S-glycosidase. Structure 1997, 5, 663–675. [Google Scholar] [CrossRef]
- Burmeister, W.P. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr. D Biol. Crystallogr. 2000, 56 Pt 3, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Verdoucq, L.; Moriniere, J.; Bevan, D.R.; Esen, A.; Vasella, A.; Henrissat, B.; Czjze, M. Structural determinants of substrate specificity in family 1 beta-glucosidases: Novel insights from the crystal structure of sorghum dhurrinase-1, a plant beta-glucosidase with strict specificity, in complex with its natural substrate. J. Biol. Chem. 2004, 279, 31796–31803. [Google Scholar] [CrossRef] [PubMed]
- Tankrathok, A.; Iglesias-Fernandez, J.; Luang, S.; Robinson, R.C.; Kimura, A.; Rovira, C.; Hrmova, M.; Ketudat Cairns, J.R. Structural analysis and insights into the glycon specificity of the rice GH1 Os7BGlu26 beta-d-mannosidase. Acta Crystallogr. D Biol. Crystallogr. 2013, 69 Pt 10, 2124–2135. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Wallner, B.; Fang, H.; Elofsson, A. Automatic consensus-based fold recognition using Pcons, ProQ, and Pmodeller. Proteins 2003, 53, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 310–322. [Google Scholar]
- Barzilai, J.; Borwein, J.M. Two-Point Step Size Gradient Methods. IMA J. Numer. Anal. 1988, 8, 141–148. [Google Scholar] [CrossRef]
- Wolf, L.K. Digital briefs. Chem. Eng. News Arch. 2009. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.; Goddard, W., III; Skiff, W. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Karpagam, V.; Sathishkumar, N.; Sathiyamoorthy, S.; Rasappan, P.; Shila, S.; Kim, Y.-J.; Yang, D.-C. Identification of BACE1 inhibitors from Panax ginseng saponins—An Insilco approach. Comput. Biol. Med. 2013, 43, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- SchuÈttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Naumoff, D.G. Hierarchical classification of glycoside hydrolases. Biochemistry 2011, 76, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Z.; Liu, H. Deriving chemically essential interactions based on active site alignments and quantum chemical calculations: A case study on glycoside hydrolases. ACS Catal. 2015, 5, 2559–2572. [Google Scholar] [CrossRef]
- Barleben, L.; Panjikar, S.; Ruppert, M.; Koepke, J.; Stockigt, J. Molecular architecture of strictosidine glucosidase: The gateway to the biosynthesis of the monoterpenoid indole alkaloid family. Plant Cell 2007, 19, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Launay, G.; Simonson, T. Homology modelling of protein-protein complexes: A simple method and its possibilities and limitations. BMC Bioinform. 2008. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, S.; Thamilarasan, S.K.; Park, J.-I.; Chung, M.-Y.; Nou, I.-S. Molecular Modeling of Myrosinase from Brassica oleracea: A Structural Investigation of Sinigrin Interaction. Genes 2015, 6, 1315-1329. https://doi.org/10.3390/genes6041315
Natarajan S, Thamilarasan SK, Park J-I, Chung M-Y, Nou I-S. Molecular Modeling of Myrosinase from Brassica oleracea: A Structural Investigation of Sinigrin Interaction. Genes. 2015; 6(4):1315-1329. https://doi.org/10.3390/genes6041315
Chicago/Turabian StyleNatarajan, Sathishkumar, Senthil Kumar Thamilarasan, Jong-In Park, Mi-Young Chung, and Ill-Sup Nou. 2015. "Molecular Modeling of Myrosinase from Brassica oleracea: A Structural Investigation of Sinigrin Interaction" Genes 6, no. 4: 1315-1329. https://doi.org/10.3390/genes6041315
APA StyleNatarajan, S., Thamilarasan, S. K., Park, J.-I., Chung, M.-Y., & Nou, I.-S. (2015). Molecular Modeling of Myrosinase from Brassica oleracea: A Structural Investigation of Sinigrin Interaction. Genes, 6(4), 1315-1329. https://doi.org/10.3390/genes6041315