
Characterization of DNA Methylation in Circulating Tumor Cells


Abstract
1. Introduction
2. The Utility of CTCs as Tumor Biomarkers
3. Biological and Clinical Relevance of DNA Methylation in Cancer
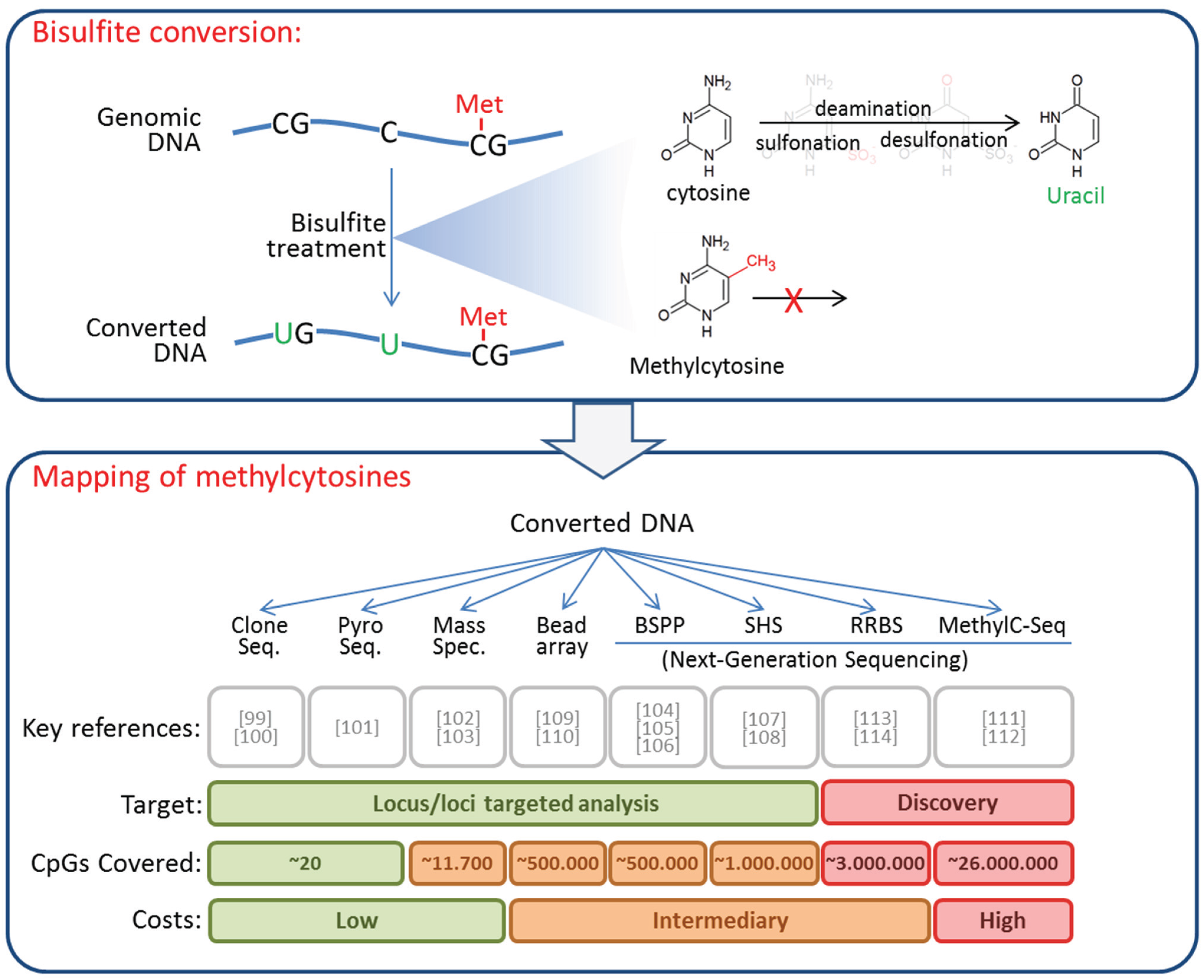
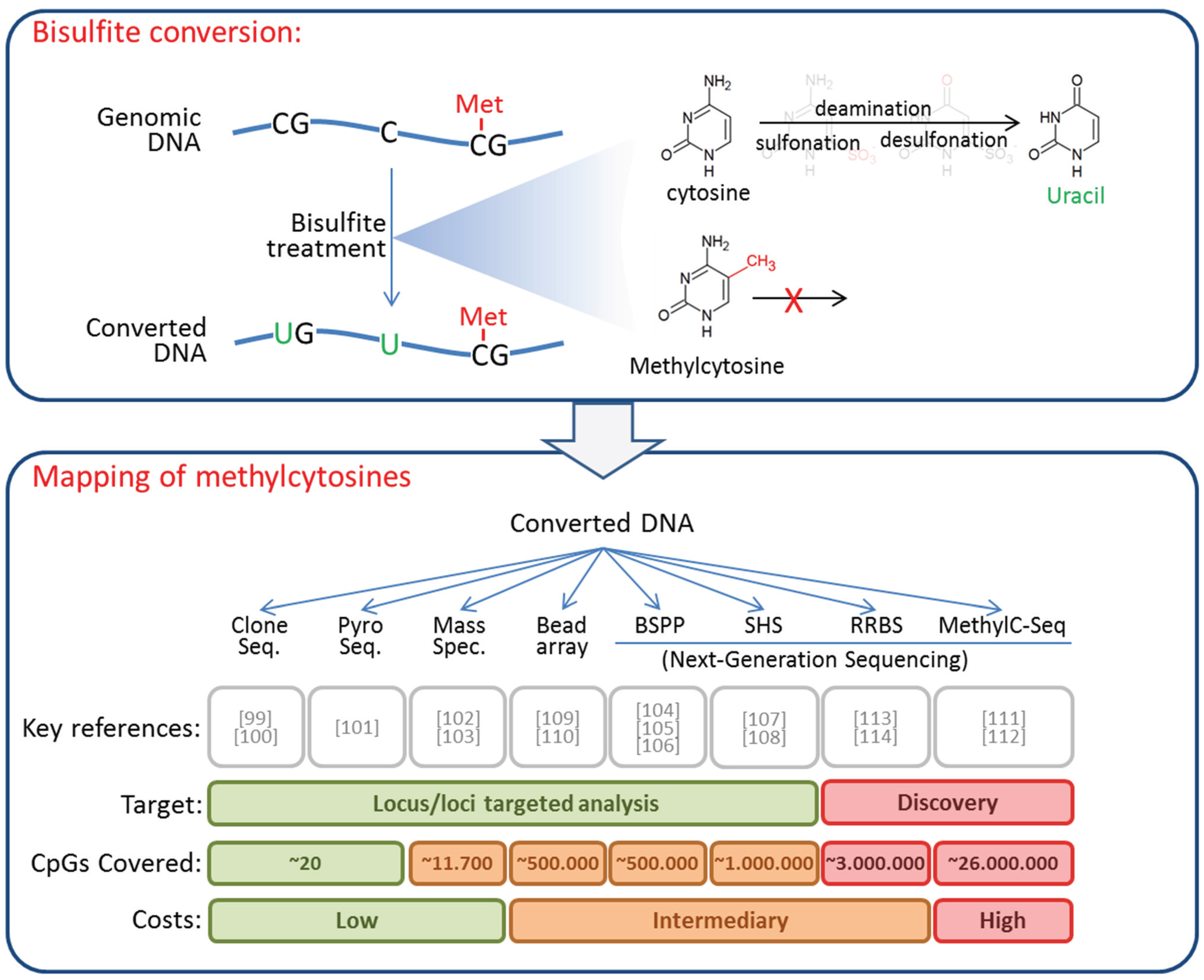
4. DNA Methylation in CTCs

{kind=link}
{kind=link}
| Reference | Entity | CTC Preparation | Methylation Analysis | Key Findings | ||
|---|---|---|---|---|---|---|
| Enrichment | Detection and Isolation | Locus | Method | |||
| Chimonidou, Strati et al., 2011 [117] | BC (Human) | Density centrifugation + EpCAM-coated beads | - | CST6, BRMS1, SOX17 | MSP | Positive correlation with presence and stage of disease. |
| Chimonidou, Strati et al., 2013 [118] | BC (Human) | Density centrifugation + EpCAM-coated beads | - | SOX17 | MSP (real-time) | Strong correlation between methylation detected in CTCs and cfDNA. |
| Chimonidou, Kallergi et al., 2013 [119] | BC (Human) | Density centrifugation | Detection of CK+ cells on cytospins by ICC and scratching | BRMS1 | MSP | Weak correlation between methylation and protein expression. Discordant methylation in CTCs and patient-matched PTs. |
| Friedlander, Ngo et al., 2014 [134] | mCRPC (Human) | CAM | - | Genome wide (27000 CpG) | Methylation array | Correlation between methylation in CTCs and non-matched PTs. Hypermethylation in apoptosis, angiogenesis, and VEGF pathway genes. |
| Ogunwobi, Pussyk et al., 2013 [135] | HCC (Murine cell line) | Implantation of tumor cells in mice, and establishment of CTC lines | HGF, c-Met | HRM and Pyrosequencing | HGF and c-MET overexpression in the CTC-lines correlated with hypomethylation of their promoters. | |
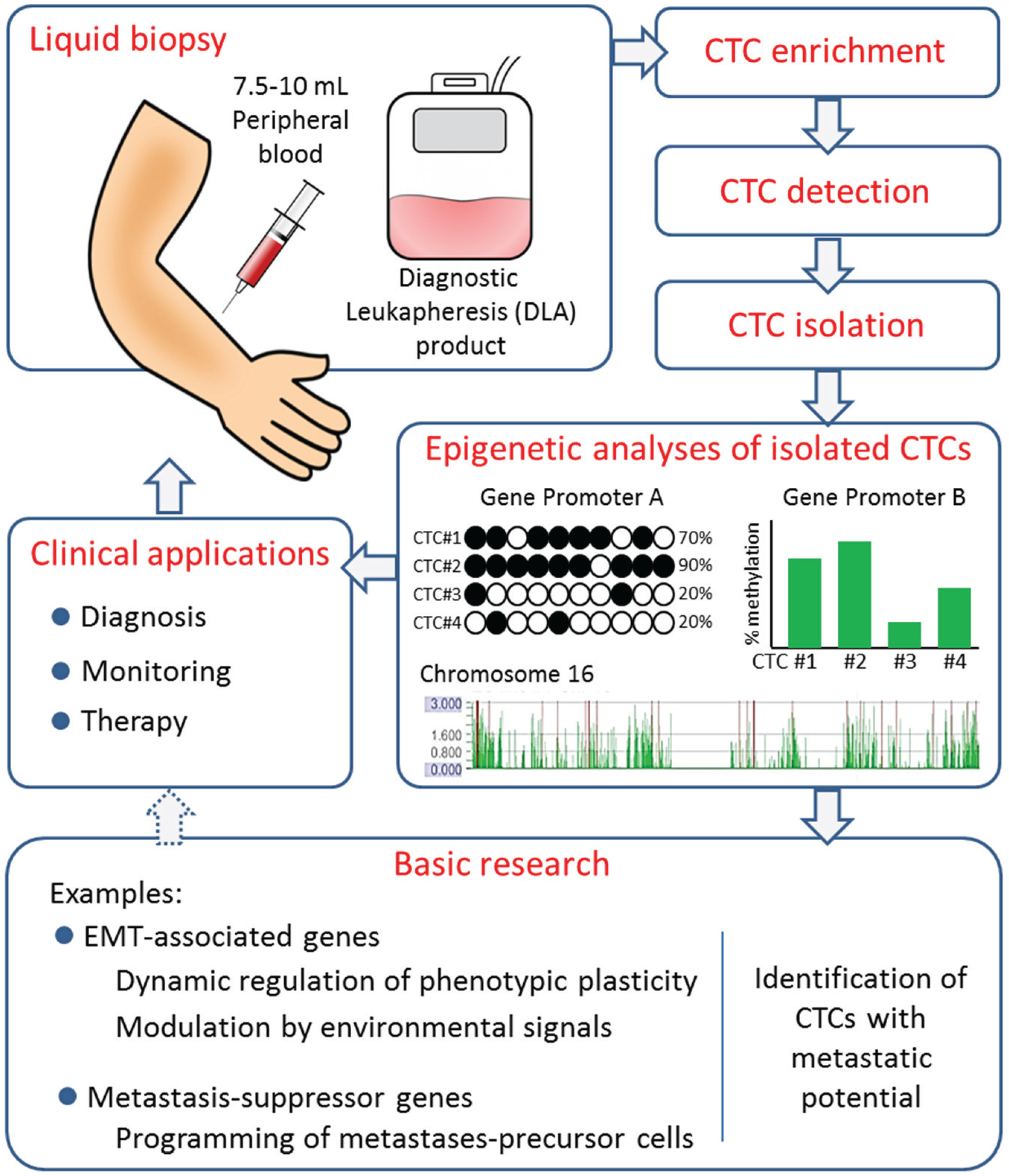
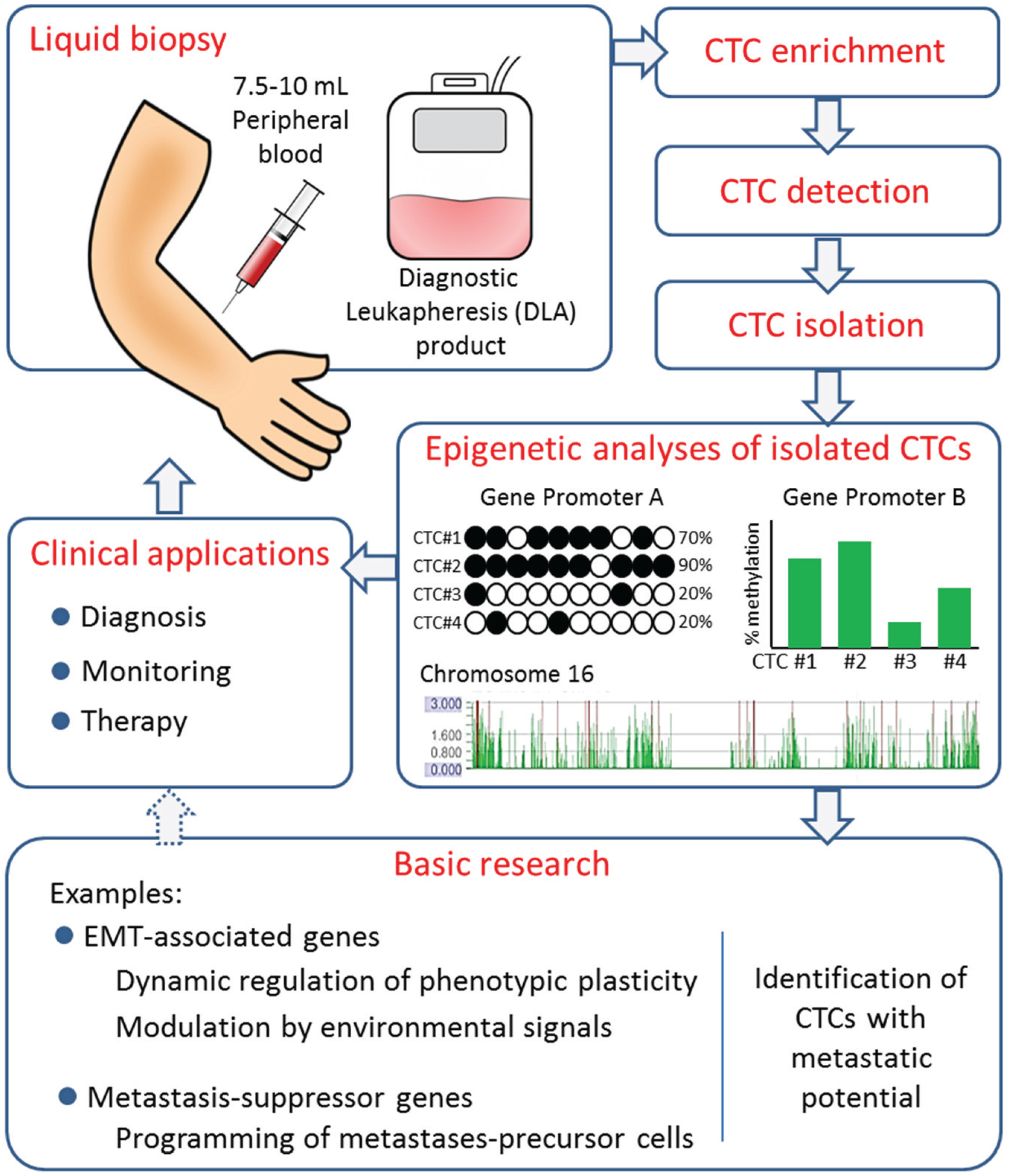
5. Outlook and Perspectives
Acknowledgments
Conflicts of Interest
References
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Speicher, M.R. The biology of circulating tumor cells. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Mani, S.A.; Cristofanilli, M. Molecular mechanisms of metastasis in breast cancer—Clinical applications. Nat. Rev. Clin. Oncol. 2010, 7, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, M.; Schmidt, B. Circulating nucleic acids (CNAS) and cancer—A survey. Biochim. Biophys. Acta 2007, 1775, 181–232. [Google Scholar] [CrossRef] [PubMed]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res. 2006, 12, 4218–4224. [Google Scholar] [CrossRef] [PubMed]
- Nole, F.; Munzone, E.; Zorzino, L.; Minchella, I.; Salvatici, M.; Botteri, E.; Medici, M.; Verri, E.; Adamoli, L.; Rotmensz, N.; et al. Variation of circulating tumor cell levels during treatment of metastatic breast cancer: Prognostic and therapeutic implications. Ann. Oncol. 2008, 19, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Gruber, I.; Landenberger, N.; Staebler, A.; Hahn, M.; Wallwiener, D.; Fehm, T. Relationship between circulating tumor cells and peripheral T-cells in patients with primary breast cancer. Anticancer Res. 2013, 33, 2233–2238. [Google Scholar] [PubMed]
- Moreno, J.G.; Miller, M.C.; Gross, S.; Allard, W.J.; Gomella, L.G.; Terstappen, L.W. Circulating tumor cells predict survival in patients with metastatic prostate cancer. Urology 2005, 65, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.J.; Rovira, P.S.; Martinez-Zubiaurre, I.; Rodriguez, M.D.; Fernandez, M.; Lorente, J.A. Dynamics of circulating tumor cells in early breast cancer under neoadjuvant therapy. Exp. Ther. Med. 2012, 4, 43–48. [Google Scholar] [PubMed]
- Pierga, J.Y.; Bidard, F.C.; Mathiot, C.; Brain, E.; Delaloge, S.; Giachetti, S.; de Cremoux, P.; Salmon, R.; Vincent-Salomon, A.; Marty, M. Circulating tumor cell detection predicts early metastatic relapse after neoadjuvant chemotherapy in large operable and locally advanced breast cancer in a phase II randomized trial. Clin. Cancer Res. 2008, 14, 7004–7010. [Google Scholar] [CrossRef] [PubMed]
- Rack, B.; Schindlbeck, C.; Juckstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.; Lorenz, R.; Tesch, H.; Fasching, P.A.; et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.M.; Slade, M.J.; English, J.; Graham, H.; Luchtenborg, M.; Sinnett, H.D.; Cross, N.C.; Coombes, R.C. Response of circulating tumor cells to systemic therapy in patients with metastatic breast cancer: Comparison of quantitative polymerase chain reaction and immunocytochemical techniques. J. Clin. Oncol. 2000, 18, 1432–1439. [Google Scholar] [PubMed]
- Liu, M.C.; Shields, P.G.; Warren, R.D.; Cohen, P.; Wilkinson, M.; Ottaviano, Y.L.; Rao, S.B.; Eng-Wong, J.; Seillier-Moiseiwitsch, F.; Noone, A.M.; et al. Circulating tumor cells: A useful predictor of treatment efficacy in metastatic breast cancer. J. Clin. Oncol. 2009, 27, 5153–5159. [Google Scholar] [CrossRef] [PubMed]
- Smerage, J.B.; Barlow, W.E.; Hortobagyi, G.N.; Winer, E.P.; Leyland-Jones, B.; Srkalovic, G.; Tejwani, S.; Schott, A.F.; O’Rourke, M.A.; Lew, D.L.; et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J. Clin. Oncol. 2014, 32, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Heller, G.; Molina, A.; Attard, G.; Danila, D.C.; Jia, X.; Peng, W.; Sandhu, S.K.; Olmos, D.; Riisnaes, R.; et al. Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2015, 33, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Prentice, R.L. Surrogate endpoints in clinical trials: Definition and operational criteria. Stat. Med. 1989, 8, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Tripathy, D.; Shete, S.; Ashfaq, R.; Haley, B.; Perkins, S.; Beitsch, P.; Khan, A.; Euhus, D.; Osborne, C.; et al. HER-2 gene amplification can be acquired as breast cancer progresses. Proc. Natl. Acad. Sci. USA 2004, 101, 9393–9398. [Google Scholar] [CrossRef] [PubMed]
- Pestrin, M.; Bessi, S.; Galardi, F.; Truglia, M.; Biggeri, A.; Biagioni, C.; Cappadona, S.; Biganzoli, L.; Giannini, A.; Di, L.A. Correlation of HER2 status between primary tumors and corresponding circulating tumor cells in advanced breast cancer patients. Breast Cancer Res. Treat. 2009, 118, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Fehm, T.; Muller, V.; Aktas, B.; Janni, W.; Schneeweiss, A.; Stickeler, E.; Lattrich, C.; Lohberg, C.R.; Solomayer, E.; Rack, B.; et al. HER2 status of circulating tumor cells in patients with metastatic breast cancer: A prospective, multicenter trial. Breast Cancer Res. Treat. 2010, 124, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.A.; Talasaz, A.H.; Zhang, H.; Coram, M.A.; Reddy, A.; Deng, G.; Telli, M.L.; Advani, R.H.; Carlson, R.W.; Mollick, J.A.; et al. Single cell profiling of circulating tumor cells: Transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE 2012, 7, e33788. [Google Scholar] [CrossRef] [PubMed]
- Strati, A.; Markou, A.; Parisi, C.; Politaki, E.; Mavroudis, D.; Georgoulias, V.; Lianidou, E. Gene expression profile of circulating tumor cells in breast cancer by RT-qPCR. BMC Cancer 2011. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.C.; Doyle, G.V.; Terstappen, L.W. Significance of circulating tumor cells detected by the cellsearch system in patients with metastatic breast colorectal and prostate cancer. J. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.C.; Niederacher, D.; Topp, S.A.; Honisch, E.; Schumacher, S.; Schmitz, N.; Zacarias Fohrding, L.; Vay, C.; Hoffmann, I.; Kasprowicz, N.S.; et al. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16580–16585. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.Y.; Yang, C.Y.; Liang, S.C.; Liu, R.S.; Jiang, J.K.; Lin, C.H. Dynamic changes in numbers and properties of circulating tumor cells and their potential applications. Cancers 2014, 6, 2369–2386. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Lorente, D.; Omlin, A.; Olmos, D.; de Bono, J.S. Circulating tumor cells: A multifunctional biomarker. Clin. Cancer Res. 2014, 20, 2553–2568. [Google Scholar] [CrossRef] [PubMed]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.P.L.; Raba, K.; Schmidt, O.; Honisch, E.; Meier-Stiegen, F.; Behrens, B.; Möhlendick, B.; Fehm, T.; Neubauer, H.; Klein, C.; et al. Genomic high resolution profiling of single CKpos/CD45neg flow-sorting purified circulating tumour cells from patients with metastatic breast cancer. Clin. Chem. 2014, 60, 1290–1297. [Google Scholar] [PubMed]
- Harb, W.; Fan, A.; Tran, T.; Danila, D.C.; Keys, D.; Schwartz, M.; Ionescu-Zanetti, C. Mutational analysis of circulating tumor cells using a novel microfluidic collection device and qPCR assay. Transl. Oncol. 2013, 6, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Talasaz, A.H.; Powell, A.A.; Huber, D.E.; Berbee, J.G.; Roh, K.H.; Yu, W.; Xiao, W.; Davis, M.M.; Pease, R.F.; Mindrinos, M.N.; et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc. Natl. Acad. Sci. USA 2009, 106, 3970–3975. [Google Scholar] [CrossRef] [PubMed]
- Karabacak, N.M.; Spuhler, P.S.; Fachin, F.; Lim, E.J.; Pai, V.; Ozkumur, E.; Martel, J.M.; Kojic, N.; Smith, K.; Chen, P.I.; et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 2014, 9, 694–710. [Google Scholar] [CrossRef] [PubMed]
- Saucedo-Zeni, N.; Mewes, S.; Niestroj, R.; Gasiorowski, L.; Murawa, D.; Nowaczyk, P.; Tomasi, T.; Weber, E.; Dworacki, G.; Morgenthaler, N.G.; et al. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Int. J. Oncol. 2012, 41, 1241–1250. [Google Scholar] [PubMed]
- Rao, C.G.; Chianese, D.; Doyle, G.V.; Miller, M.C.; Russell, T.; Sanders, R.A., Jr.; Terstappen, L.W. Expression of epithelial cell adhesion molecule in carcinoma cells present in blood and primary and metastatic tumors. Int. J. Oncol. 2005, 27, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Gorges, T.M.; Tinhofer, I.; Drosch, M.; Rose, L.; Zollner, T.M.; Krahn, T.; von Ahsen, O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer 2012. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Stoecklein, N.H. Dynamic EpCAM expression on circulating and disseminating tumor cells: Causes and consequences. Cell. Mol. Life Sci. 2014, 71, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Gradilone, A.; Naso, G.; Vincenzi, B.; Petracca, A.; Nicolazzo, C.; Palazzo, A.; Saltarelli, R.; Spremberg, F.; Cortesi, E.; et al. Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res. Treat. 2011, 130, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, S.; Liu, Z.; Huang, J.; Pu, X.; Li, J.; Yang, D.; Deng, H.; Yang, N.; Xu, J. Classification of circulating tumor cells by epithelial-mesenchymal transition markers. PLoS ONE 2015, 10, e0123976. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Zoli, W.; Rigaud, M. Circulating tumor cells and epithelial, mesenchymal and stemness markers: Characterization of cell subpopulations. Ann. Transl. Med. 2014, 2. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Kraan, J.; Bolt, J.; van der Spoel, P.; Elstrodt, F.; Schutte, M.; Martens, J.W.; Gratama, J.W.; Sleijfer, S.; Foekens, J.A. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J. Natl. Cancer Inst. 2009, 101, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Low, W.S.; Wan Abas, W.A. Benchtop technologies for circulating tumor cells separation based on biophysical properties. BioMed Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.; Schutze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M.; et al. Isolation by size of epithelial tumor cells: A new method for the immunomorphological and molecular characterization of circulatingtumor cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Desitter, I.; Guerrouahen, B.S.; Benali-Furet, N.; Wechsler, J.; Janne, P.A.; Kuang, Y.; Yanagita, M.; Wang, L.; Berkowitz, J.A.; Distel, R.J.; et al. A new device for rapid isolation by size and characterization of rare circulating tumor cells. Anticancer Res. 2011, 31, 427–441. [Google Scholar] [PubMed]
- Adams, D.L.; Zhu, P.; Makarova, O.V.; Martin, S.S.; Charpentier, M.; Chumsri, S.; Li, S.; Amstutz, P.; Tang, C.M. The systematic study of circulating tumor cell isolation using lithographic microfilters. RSC Adv. 2014, 9, 4334–4342. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhao, Q.; Chen, J.J.; Chen, W.T.; Pearl, M.L. Clinical significance of circulating tumor cells detected by an invasion assay in peripheral blood of patients with ovarian cancer. Gynecol. Oncol. 2009, 112, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.A.; Wu, X.; Zhong, X.; Riggs, A.D.; Pfeifer, G.P. A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A.D.; Dokun, O.Y. DNA methylation and human diseases: An overview. In DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution; Grosjean, H., Ed.; Landes Bioscience: Austin, TX, USA, 2009; pp. 103–116. [Google Scholar]
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta 2007, 1775, 138–162. [Google Scholar] [CrossRef] [PubMed]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Assenov, Y.; Martin-Subero, J.I.; Balint, B.; Siebert, R.; Taniguchi, H.; Yamamoto, H.; Hidalgo, M.; Tan, A.C.; Galm, O.; et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012, 22, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Carmona, F.J.; Davalos, V.; Vidal, E.; Gomez, A.; Heyn, H.; Hashimoto, Y.; Vizoso, M.; Martinez-Cardus, A.; Sayols, S.; Ferreira, H.J.; et al. A comprehensive DNA methylation profile of epithelial-to-mesenchymal transition. Cancer Res. 2014, 74, 5608–5619. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Pantel, K. Tumor cell dissemination: Emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Vrba, L.; Jensen, T.J.; Garbe, J.C.; Heimark, R.L.; Cress, A.E.; Dickinson, S.; Stampfer, M.R.; Futscher, B.W. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS ONE 2010, 5, e8697. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.S.; Qu, Y.; Cheng, Y.; Li, W.C.; Rotter, V.; Oyan, A.M.; Kalland, K.H. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics 2010. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.; Fan, C.; Knafl, G.; Wu, M.J.; Coleman, B.; Perou, C.M.; Swift-Scanlan, T. Vimentin DNA methylation predicts survival in breast cancer. Breast Cancer Res. Treat. 2013, 137, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Fackler, M.J.; McVeigh, M.; Evron, E.; Garrett, E.; Mehrotra, J.; Polyak, K.; Sukumar, S.; Argani, P. DNA methylation of RASSF1A, HIN-1, RAR-β, Cyclin D2 and Twist in in situ and invasive lobular breast carcinoma. Int. J. Cancer 2003, 107, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.; Scheel, C.; Weinhold, S.; Honisch, E.; Iwaniuk, K.M.; Trompeter, H.I.; Niederacher, D.; Wernet, P.; Santourlidis, S.; Uhrberg, M. Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Res. Notes 2010. [Google Scholar] [CrossRef] [PubMed]
- Davalos, V.; Moutinho, C.; Villanueva, A.; Boque, R.; Silva, P.; Carneiro, F.; Esteller, M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 2011, 31, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S.; Ouatas, T.; Halverson, D.; Palmieri, D.; Salerno, M. Metastasis suppressor genes: Basic biology and potential clinical use. Clin. Breast Cancer 2003, 4, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.; Haack, H.; Kissil, J.L.; Barry, M.; Bronson, R.T.; Shen, S.S.; Whittaker, C.A.; Crowley, D.; Hynes, R.O. Protein 4.1 B suppresses prostate cancer progression and metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 12784–12789. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A.; Alexa, A.; Jung, V.; Hader, C.; Hoffmann, M.J.; Yamanaka, M.; Fritzsche, S.; Wlazlinski, A.; Muller, M.; Lengauer, T.; et al. Factor interaction analysis for chromosome 8 and DNA methylation alterations highlights innate immune response suppression and cytoskeletal changes in prostate cancer. Mol. Cancer 2007. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Fenaux, P.; Gattermann, N.; Seymour, J.F.; Hellstrom-Lindberg, E.; Mufti, G.J.; Duehrsen, U.; Gore, S.D.; Ramos, F.; Beyne-Rauzy, O.; List, A.; et al. Prolonged survival with improved tolerability in higher-risk myelodysplastic syndromes: Azacitidine compared with low dose ARA-C. Br. J. Haematol. 2010, 149, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Garzon, R.; Klisovic, R.B.; Schwind, S.; Walker, A.; Geyer, S.; Liu, S.; Havelange, V.; Becker, H.; Schaaf, L.; et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc. Natl. Acad. Sci. USA 2010, 107, 7473–7478. [Google Scholar] [CrossRef] [PubMed]
- Cashen, A.F.; Schiller, G.J.; O’Donnell, M.R.; DiPersio, J.F. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Balch, C.; Schilder, J.; Breen, T.; Zhang, S.; Shen, C.; Li, L.; Kulesavage, C.; Snyder, A.J.; Nephew, K.P.; et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer 2010, 116, 4043–4053. [Google Scholar] [CrossRef] [PubMed]
- Glasspool, R.M.; Brown, R.; Gore, M.E.; Rustin, G.J.; McNeish, I.A.; Wilson, R.H.; Pledge, S.; Paul, J.; Mackean, M.; Hall, G.D.; et al. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br. J. Cancer 2014, 110, 1923–1929. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Lu, X.; Wang, X.; Liu, Y.; Guo, B.; Zhang, Y.; Zhang, W.; Nie, J.; Feng, K.; Chen, M.; et al. Low-dose decitabine-based chemoimmunotherapy for patients with refractory advanced solid tumors: A phase I/II report. J. Immunol. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, R.; Jewell, R.; van den Oord, J.J.; Wolter, P.; Stierner, U.; Lindholm, C.; Hertzman Johansson, C.; Linden, D.; Johansson, H.; Frostvik Stolt, M.; et al. Mgmt promoter methylation is associated with temozolomide response and prolonged progression-free survival in disseminated cutaneous melanoma. Int. J. Cancer 2015, 136, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Tabatabai, G.; Kastner, B.; Felsberg, J.; Steinbach, J.P.; Wick, A.; Schnell, O.; Hau, P.; Herrlinger, U.; Sabel, M.C.; et al. MGMT promoter methylation is a strong prognostic biomarker for benefit from dose-intensified temozolomide rechallenge in progressive glioblastoma: The DIRECTOR trial. Clin. Cancer Res. 2015, 21, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Moutinho, C.; Belotti, A.; Bencardino, K.; Chirico, G.; Cassingena, A.; Rusconi, F.; Esposito, A.; Nichelatti, M.; et al. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.F.; Xu, W.; Wang, X.; Sun, J.S.; Xiang, J.J.; Li, Z.S.; Zhang, X.F. Negative methylation status of vimentin predicts improved prognosis in pancreatic carcinoma. World J. Gastroenterol. 2014, 20, 13172–13177. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Jeong, E.M.; Kim, J.H.; Rho, S.B.; Lee, E.J. Aberrant methylation of the VIM promoter in uterine cervical squamous cell carcinoma. Oncology 2014, 86, 359–368. [Google Scholar] [PubMed]
- Itzkowitz, S.; Brand, R.; Jandorf, L.; Durkee, K.; Millholland, J.; Rabeneck, L.; Schroy, P.C., 3rd; Sontag, S.; Johnson, D.; Markowitz, S.; et al. A simplified, noninvasive stool DNA test for colorectal cancer detection. Am. J. Gastroenterol. 2008, 103, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.L.; Henrique, R.; Danielsen, S.A.; Duarte-Pereira, S.; Eknaes, M.; Skotheim, R.I.; Rodrigues, A.; Magalhaes, J.S.; Oliveira, J.; Lothe, R.A.; et al. Three epigenetic biomarkers, GDF15, TMEFF2, and VIM, accurately predict bladder cancer from DNA-based analyses of urine samples. Clin. Cancer Res. 2010, 16, 5842–5851. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Borre, M.; Christiansen, A.; Hermann, G.G.; Orntoft, T.F.; Dyrskjot, L. Diagnosis of bladder cancer recurrence based on urinary levels of EOMES, HOXA9, POU4F2, TWIST1, VIM, and ZNF154 hypermethylation. PLoS ONE 2012, 7, e46297. [Google Scholar] [CrossRef] [PubMed]
- Schuebel, K.E.; Chen, W.; Cope, L.; Glockner, S.C.; Suzuki, H.; Yi, J.M.; Chan, T.A.; van Neste, L.; van Criekinge, W.; van den Bosch, S.; et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007, 3, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Waterland, R.A. Methods of DNA methylation analysis. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Harrison, J.; Paul, C.L.; Frommer, M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994, 22, 2990–2997. [Google Scholar] [PubMed]
- Uhlmann, K.; Brinckmann, A.; Toliat, M.R.; Ritter, H.; Nurnberg, P. Evaluation of a potential epigenetic biomarker by quantitative methyl-single nucleotide polymorphism analysis. Electrophoresis 2002, 23, 4072–4079. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Nelson, M.R.; Stanssens, P.; Zabeau, M.; Liloglou, T.; Xinarianos, G.; Cantor, C.R.; Field, J.K.; van den Boom, D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc. Natl. Acad. Sci. USA 2005, 102, 15785–15790. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Turner, J.; Gibbs, P.; Lipton, L.; Giovanneti, M.; Cantor, C.; van den Boom, D. Cytosine methylation profiling of cancer cell lines. Proc. Natl. Acad. Sci. USA 2008, 105, 4844–4849. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.H.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Diep, D.; Plongthongkum, N.; Gore, A.; Fung, H.L.; Shoemaker, R.; Zhang, K. Library-free methylation sequencing with bisulfite padlock probes. Nat. Methods 2012, 9, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Pei, L.; Srivastava, G.; Joshi, T.; Kushwaha, G.; Choi, J.H.; Robertson, K.D.; Wang, X.; Colbourne, J.K.; Zhang, L.; et al. Targeted bisulfite sequencing by solution hybrid selection and massively parallel sequencing. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, H.; Ji, G.; Gao, F.; Wu, M.; Sun, J.; Luo, H.; Wu, J.; Wu, R.; Zhang, X. High resolution profiling of human exon methylation by liquid hybridization capture-based bisulfite sequencing. BMC Genomics 2011. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Wu, X.; Li, X.; Wen, L.; Tang, F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013, 23, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Gravina, S.; Ganapathi, S.; Vijg, J. Single-cell, locus-specific bisulfite sequencing (SLBS) for direct detection of epimutations in DNA methylation patterns. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chimonidou, M.; Strati, A.; Tzitzira, A.; Sotiropoulou, G.; Malamos, N.; Georgoulias, V.; Lianidou, E.S. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin. Chem. 2011, 57, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Chimonidou, M.; Strati, A.; Malamos, N.; Georgoulias, V.; Lianidou, E.S. SOX17 promoter methylation in circulating tumor cells and matched cell-free DNA isolated from plasma of patients with breast cancer. Clin. Chem. 2013, 59, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Chimonidou, M.; Kallergi, G.; Georgoulias, V.; Welch, D.R.; Lianidou, E.S. Breast cancer metastasis suppressor-1 promoter methylation in primary breast tumors and corresponding circulating tumor cells. Mol. Cancer Res. 2013, 11, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Abrahamson, M.; Zhang, M.; Fernandez, M.A.; Grubb, A.; Su, J.; Yu, G.L.; Li, Y.; Parmelee, D.; Xing, L.; et al. Cystatin E is a novel human cysteine proteinase inhibitor with structural resemblance to family 2 cystatins. J. Biol. Chem. 1997, 272, 10853–10858. [Google Scholar] [PubMed]
- Sotiropoulou, G.; Anisowicz, A.; Sager, R. Identification, cloning, and characterization of cystatin M, a novel cysteine proteinase inhibitor, down-regulated in breast cancer. J. Biol. Chem. 1997, 272, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shridhar, R.; Dai, Q.; Song, J.; Barlow, S.C.; Yin, L.; Sloane, B.F.; Miller, F.R.; Meschonat, C.; Li, B.D.; et al. Cystatin M: A novel candidate tumor suppressor gene for breast cancer. Cancer Res. 2004, 64, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Kim, W.J.; Kim, T.Y.; Fields, C.R.; Massoll, N.A.; Robertson, K.D.; Brown, K.D. Epigenetic silencing of the tumor suppressor cystatin M occurs during breast cancer progression. Cancer Res. 2006, 66, 7899–7909. [Google Scholar] [CrossRef] [PubMed]
- Kioulafa, M.; Balkouranidou, I.; Sotiropoulou, G.; Kaklamanis, L.; Mavroudis, D.; Georgoulias, V.; Lianidou, E.S. Methylation of cystatin M promoter is associated with unfavorable prognosis in operable breast cancer. Int. J. Cancer 2009, 125, 2887–2892. [Google Scholar] [CrossRef] [PubMed]
- Seraj, M.J.; Samant, R.S.; Verderame, M.F.; Welch, D.R. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000, 60, 2764–2769. [Google Scholar] [PubMed]
- Metge, B.J.; Frost, A.R.; King, J.A.; Dyess, D.L.; Welch, D.R.; Samant, R.S.; Shevde, L.A. Epigenetic silencing contributes to the loss of BRMS1 expression in breast cancer. Clin. Exp. Metastasis 2008, 25, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Yamamoto, Y.; Kawasoe, T.; Iwase, H. Reduced expression of the breast cancer metastasis suppressor 1 mRNA is correlated with poor progress in breast cancer. Clin. Cancer Res. 2006, 12, 6410–6414. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stark, A.M.; Tongers, K.; Maass, N.; Mehdorn, H.M.; Held-Feindt, J. Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. J. Cancer Res. Clin. Oncol. 2005, 131, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.G.; Yoder, B.J.; Short, S.; Tarr, S.; Prescott, N.; Crowe, J.P.; Dawson, A.E.; Budd, G.T.; Sizemore, S.; Cicek, M.; et al. Loss of breast cancer metastasis suppressor 1 protein expression predicts reduced disease-free survival in subsets of breast cancer patients. Clin. Cancer Res. 2006, 12, 6702–6708. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, W.; Glockner, S.C.; Guo, M.; Machida, E.O.; Wang, D.H.; Easwaran, H.; Van Neste, L.; Herman, J.G.; Schuebel, K.E.; Watkins, D.N.; et al. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res. 2008, 68, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Sinner, D.; Kordich, J.J.; Spence, J.R.; Opoka, R.; Rankin, S.; Lin, S.C.; Jonatan, D.; Zorn, A.M.; Wells, J.M. SOX17 AND SOX4 differentially regulate beta-catenin/T-cell factor activity and proliferation of colon carcinoma cells. Mol. Cell. Biol. 2007, 27, 7802–7815. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Tan, H.S.; Wei, J.L.; Zhu, C.R.; Jiang, J.X.; Zhu, Y.X.; Cai, F.L.; Chong, M.H.; Ren, C.L. Decreased expression of SOX17 is associated with tumor progression and poor prognosis in breast cancer. Tumour Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, G.; Cheng, Y.; Tang, Y.; Dong, Z.; McElwee, K.J.; Li, G. Reduced expression of SRY-box containing gene 17 correlates with an unfavorable melanoma patient survival. Oncol. Rep. 2014, 32, 2571–2579. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Friedlander, T.W.; Ngo, V.T.; Dong, H.; Premasekharan, G.; Weinberg, V.; Doty, S.; Zhao, Q.; Gilbert, E.G.; Ryan, C.J.; Chen, W.T.; et al. Detection and characterization of invasive circulating tumor cells derived from men with metastatic castration-resistant prostate cancer. Int. J. Cancer 2014, 134, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Puszyk, W.; Dong, H.J.; Liu, C. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS ONE 2013, 8, e63765. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Choi, J.Y.; Lee, K.M.; Sung, H.; Park, S.K.; Oze, I.; Pan, K.F.; You, W.C.; Chen, Y.X.; Fang, J.Y.; et al. DNA methylation in peripheral blood: A potential biomarker for cancer molecular epidemiology. J. Epidemiol. 2012, 22, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Fan, T.; Zhao, Q.; Zeng, W.; Zaslavsky, E.; Chen, J.J.; Frohman, M.A.; Golightly, M.G.; Madajewicz, S.; Chen, W.T. Isolation of circulating epithelial and tumor progenitor cells with an invasive phenotype from breast cancer patients. Int. J. Cancer 2010, 126, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Paris, P.L.; Kobayashi, Y.; Zhao, Q.; Zeng, W.; Sridharan, S.; Fan, T.; Adler, H.L.; Yera, E.R.; Zarrabi, M.H.; Zucker, S.; et al. Functional phenotyping and genotyping of circulating tumor cells from patients with castration resistant prostate cancer. Cancer Lett. 2009, 277, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, T.W.; Roy, R.; Tomlins, S.A.; Ngo, V.T.; Kobayashi, Y.; Azameera, A.; Rubin, M.A.; Pienta, K.J.; Chinnaiyan, A.; Ittmann, M.M.; et al. Common structural and epigenetic changes in the genome of castration-resistant prostate cancer. Cancer Res. 2012, 72, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- DesRochers, T.M.; Shamis, Y.; Alt-Holland, A.; Kudo, Y.; Takata, T.; Wang, G.; Jackson-Grusby, L.; Garlick, J.A. The 3D tissue microenvironment modulates DNA methylation and E-cadherin expression in squamous cell carcinoma. Epigenetics 2012, 7, 34–46. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pixberg, C.F.; Schulz, W.A.; Stoecklein, N.H.; Neves, R.P.L. Characterization of DNA Methylation in Circulating Tumor Cells. Genes 2015, 6, 1053-1075. https://doi.org/10.3390/genes6041053
Pixberg CF, Schulz WA, Stoecklein NH, Neves RPL. Characterization of DNA Methylation in Circulating Tumor Cells. Genes. 2015; 6(4):1053-1075. https://doi.org/10.3390/genes6041053
Chicago/Turabian StylePixberg, Constantin F., Wolfgang A. Schulz, Nikolas H. Stoecklein, and Rui P. L. Neves. 2015. "Characterization of DNA Methylation in Circulating Tumor Cells" Genes 6, no. 4: 1053-1075. https://doi.org/10.3390/genes6041053
APA StylePixberg, C. F., Schulz, W. A., Stoecklein, N. H., & Neves, R. P. L. (2015). Characterization of DNA Methylation in Circulating Tumor Cells. Genes, 6(4), 1053-1075. https://doi.org/10.3390/genes6041053