Abstract
Inherited mutations in the DNA mismatch repair genes (MMR) can cause MMR deficiency and increased susceptibility to colorectal and endometrial cancer. Microsatellite instability (MSI) is the defining molecular signature of MMR deficiency. The clinical classification of identified MMR gene sequence variants has a direct impact on the management of patients and their families. For a significant proportion of cases sequence variants of uncertain clinical significance (also known as unclassified variants) are identified, constituting a challenge for genetic counselling and clinical management of families. The effect on protein function of these variants is difficult to interpret. The presence or absence of MSI in tumours can aid in determining the pathogenicity of associated unclassified MMR gene variants. However, there are some considerations that need to be taken into account when using MSI for variant interpretation. The use of MSI and other tumour characteristics in MMR gene sequence variant classification will be explored in this review.
1. Introduction
Identification of cancer-causing germline variants in high-risk cancer genes in cancer cases, directs the clinical management of not only the individual, but also the whole family. These measures include presymptomatic surveillance, prophylactic surgery and chemotherapy regimens [1]. Thus it is very important to identify carriers of pathogenic (disease causing) variants in these cancer genes.
Germline variants in the mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) that abrogate protein function cause the cancer susceptibility disorder, Lynch Syndrome [2]. Affected families often present clinically with multiple cases of early onset colorectal and endometrial cancers across several generations [3,4]. It is the most common hereditary form of these cancers, accounting for about 1%–3% [5,6,7,8]. Consistent with mutations in genes integral to DNA mismatch repair, the tumours develop through a mutator phenotype pathway, which is characterized by clonal global expansion or contraction of genomic short repetitive elements (microsatellites) producing the defining molecular signature of widespread somatic microsatellite instability (MSI) [9,10,11].
Identification of Lynch syndrome cases is very important, because these individuals have an increased risk of developing multiple metachronous and synchronous tumours [12,13]. Once an at-risk individual is identified for diagnostic testing, an algorithmic approach is generally used to detect MMR mutations. MSI testing and/or MMR protein immunohistochemistry (IHC) are used to identify MMR deficiency in tumours [14,15,16,17,18]. Somatic acquired alterations, such as MLH1 promoter CpG island methylation and biallelic somatic mutations in the MMR genes are increasingly being used to identify so-called spontaneous MMR deficient colorectal and endometrial tumours [19,20,21,22,23,24,25]. Presence of the oncogenic BRAF V600E mutation in colorectal tumours can also be used to indicate spontaneous cases, but it is not a useful marker for triaging endometrial tumours [21,26]. The remaining cases are prioritised for genetic testing. When a pathogenic variant is identified in a case there are obvious clinical ramifications, so accurate variant classification is important. Tumour MSI status and other clinicopathological characteristics can aid in variant interpretation. Here, we describe the role MSI plays in MMR gene sequence variant classification.
2. MMR Gene Sequence Variant Classification
MMR gene sequence variants that are predicted to lead to premature termination of translation or a truncated protein are assumed to alter gene/protein function, and are thus classified as pathogenic (disease-causing). The remainder start as unclassified variants, as the effect on protein function of these variants is difficult to interpret. Such sequence variants include nucleotide changes predicted to cause missense substitutions, small in-frame insertions/deletions, or intragenic/intergenic sequence changes that may alter splicing or gene regulation. The issue of variants of uncertain clinical significance poses a major problem for clinicians, researchers and families that are suspected to have Lynch syndrome [1]. Recently, the International Society for Gastrointestinal Tumours (InSiGHT) has adopted a five-tiered variant classification scheme for interpretation of sequence variants identified in the four MMR genes [27]. The five classes are as follows: Class 5 (pathogenic), class 4 (likely pathogenic), class 3 (uncertain), class 2 (likely not pathogenic), and class 1 (not pathogenic). Pathogenicity is defined as being clinically relevant in a genetic counselling setting. The scheme incorporates qualitative or quantitative evidence to achieve sequence variant classification. Both approaches require multiple points of evidence, and are linked to clinical recommendations. The strength of the five-tiered scheme is its ability to utilize multiple sources of data readily available in the clinical setting.
2.1. Qualitative Classification Scheme
The InSiGHT classification rules include sequence-based criterion to annotate mutations that are widely accepted as pathogenic in standard clinical practice, i.e., nonsense/frameshift mutations and partial gene deletions. In contrast, for MMR variants of unknown clinical significance there are several criteria that can be assessed for qualitative classification, none of which should be used alone. These include analysis of tumour characteristics, such as MSI and IHC (loss of expression of the appropriate protein/s) [28,29,30], which could demonstrate an association of the variant with Lynch syndrome. Non-tumour characteristics associated with pathogenic mutations include, segregation with disease, absence in control individuals, and a deleterious effect in functional assays (protein function or splicing) [28,29,30,31,32]. Furthermore, co-occurrence in trans with a pathogenic mutation, without an abnormal phenotype (i.e., constitutional MMR deficiency—CMMR-D [33]) also suggests that a variant is likely not pathogenic.
2.2. Multifactorial Likelihood Model
Multifactorial likelihood analysis was established to tackle the issue of unclassified variants in the familial breast cancer genes BRCA1/2 [34,35], and has since been applied to variants in CDKN2A [36] and the MMR genes [27,37]. It provides a quantitative measure of probability in favour of pathogenicity linked to clinical recommendations [38]. The model integrates different lines of genetic evidence using Bayesian analysis [34,39]. See an outline of the MMR multifactorial model in Figure 1. Each variant starts with a “prior probability” of pathogenicity, which can be based on in silico analyses. The approach used for the MMR genes to measure the prior probabilities for missense substitutions was to calibrate in silico algorithm outputs predicting the effects based on conservation and physicochemical properties of the residues [40,41], against a reference set of variants that were classified with confidence using the types of data mentioned above [42].

Figure 1.
Outline of the mismatch repair genes (MMR) multifactorial likelihood model.
A “posterior probability” of pathogenicity is derived by updating the prior probability with likelihood ratios (LR) or odds ratios (OR) for pathogenicity determined from statistical analyses of observational data by comparing cases with known pathogenic variants to individuals with no pathogenic variants in a large dataset [34]. The posterior probability is used to classify a sequence variant based on the cut-offs for the classes (5, >0.99; 4, 0.95–0.99; 3, 0.05–0.949; 2, 0.001–0.049; 1, <0.001) [27,38]. Currently, statistical measures based on colorectal tumour features (outlined below) and variant segregation are incorporated into the MMR multifactorial model as LRs [37], to ensure at least one additional piece of evidence is combined with the bioinformatically derived probabilities. Importantly, the individual components must be independent [34], thus highly correlated/overlapping information cannot be used together, such as the MSI and IHC status from the same tumour.
3. Tumour Features Suitable for Use in MMR Gene Variant Classification
Tumour MSI status can be used as both a positive predictor (MSI) and negative predictor (microsatellite stable—MSS) for MMR gene pathogenic variant status. Similarly loss of protein expression identified using IHC can be a positive predictor and intact protein expression a negative predictor. MSI/IHC status usually triggers MMR gene mutation screening in an index case, particularly with the implementation of universal testing for CRCs [5,43,44], thus ascertainment of the cases needs to be taken into account when using this type of tumour information in variant classification. If only unstable or tumours with loss of protein expression are tested for germline sequence variation, then only variants associated with MSI/immunoloss will be identified. However, an independent metachronous colorectal tumour or additional clinicopathological features not used in ascertainment (BRAF, MLH1 promoter methylation) could be assayed in the index tumour to aid in the interpretation of uncertain variants. Likewise, tumour features of relatives known to carry the same variant may be considered. Other features of MSI colorectal tumours, such as mucinous histology, lymphocytic infiltration, or anatomic location [45,46] may also be able to contribute to MMR gene sequence variant interpretation. However, large-scale studies with detailed pathology features recorded and in-depth MMR gene testing would be required to determine LRs.
Non-independence of tumour features needs to be considered when using this information in variant classification. For example, due to the high correlation between MSI and IHC [47,48] only one of these measures should be included for an individual tumour. When MSI and IHC results are both available but discordant, the result indicating MMR deficiency should be considered as the summary result for consideration in modelling, since there are appropriate explanations for such discordance. For example, some tumours from deleterious missense variant carriers can be MSI and have immunostable protein expression [49,50]. In contrast, sometimes IHC can be a better predictor of MMR deficiency, e.g., especially for MSH6 pathogenic variant carriers. The latter occurs particularly when the Bethesda/National Cancer Institute (NCI) panel [51], which contains mononucleotide and dinucleotide markers, is used to test MSI. MSH6 as part of the MutSα heterodimer is primarily involved in the repair of mismatches and single nucleotide IDLs [52]. Accordingly, MSI analysis using a quasimonomorphic mononucleotide microsatellite marker panel has been shown to be more effective at identifying MSH6 pathogenic variant carriers [53], and demonstrates superior performance compared to the NCI panel [54,55].
Both the mononucleotide and NCI panels are currently used to detect MSI in tumours. These microsatellite marker panels have subsequently been used to test for MSI in extracolonic tumours [53,56,57,58], but were originally developed to distinguish MSI and MSS colorectal tumours. Presence of MSI (and loss of protein expression) can also be used to characterize MMR deficiency in the extracolonic Lynch spectrum tumours. As the risk of endometrial cancer can be as high as CRC in Lynch syndrome [59,60,61,62,63,64,65], incorporating endometrial tumour features would increase information available for classifying MMR gene sequence variants. It has been demonstrated that there are MSI events shared between endometrial and colorectal tumours (and specific to tumour type) leading to the development of more informative mononucleotide marker panels [55,66]. Widespread use of these and similar tumour-generic MSI panels will produce a more reliable measure of MSI status in both tumour types and more data to derive tumour-generic LRs for the quantitative model.
MSI is also detected in ~15% of spontaneous types of both colorectal and endometrial tumours [8,67,68]. Accordingly, colorectal tumour BRAF V600E status, or colorectal/endometrial tumour MLH1 promoter methylation (that are rarely detected in Lynch Syndrome associated tumours [21,69]), are very informative for identifying probable non-causal variants among mismatch-repair-deficient tumours. The successful application of IHC for identification of BRAF V600E [70,71] makes it more readily utilized in molecular and genetic testing algorithms. However, MLH1 promoter methylation has been shown to be superior to BRAF as a negative predictor in colorectal tumours [72], and the main indicator of non-pathogenicity in endometrial tumours where BRAF V600E status has no predictive value at all [21]. Thus, MLH1 methylation would appear to be the better tumour test to pursue in the clinical setting.
Acquired MLH1 promoter hypermethylation is also frequent in other MMR gene mutation-negative MSI Lynch spectrum tumours: Ovarian [73,74], gastric [75] and urothelial tumours [76]. Thus, there is value in using large studies of other Lynch spectrum cancer to explore the positive or negative predictive value of tumour features of additional LS cancers for interpreting the clinical significance of MMR gene variants.
With the recent technological advances in tumour sequencing, the hypermutated molecular tumour signature [55,77,78] could be used as an alternative to MSI testing to identify MMR deficient tumours. Additionally, acquired events in both alleles of an MMR gene (pathogenic mutation or loss of heterozygosity) have been demonstrated to cause MMR deficiency in more than 50% of colorectal and endometrial tumours without a germline MMR gene mutation or MLH1 promoter methylation [24,25]. Therefore, there is potential value as a predictor for in vivo loss of function, e.g., if a variant is also seen as a somatic second hit with a known pathogenic mutation in a tumour with IHC loss or MSI then that could be seen as equivalent to a functional assay.
4. Qualitative and Quantitative Use of MSI in MMR Gene Variant Classification
The quantitative approach (multifactorial likelihood model) is the preferable one for MMR gene variant classification, because the final output is a quantitative probability of pathogenicity. The model currently includes LRs already calculated for MSI together with BRAF tumour status [37], and pending further analysis of datasets it is quite feasible to implement other tumour features mentioned in the previous section. For use in the interim, qualitative experience-based criteria have been developed to ensure that as much information could be used to aid in variant classification. Figure 2 outlines the process of variant classification using tumour characteristics, such as MSI.
In the InSiGHT qualitative rules, two or more independent MMR deficient tumours are required to contribute to classification of a MMR variant as pathogenic [27]. This can include multiple primary tumours from a single individual (as long as they are proven metachronous). The need for multiple tumour results for carriers of a given variant is implemented to minimize the chance of acquired somatic changes, such as MLH1 promoter methylation and now biallelic somatic mutations [24,25], being the cause of tumour MMR deficiency rather than the presence of a pathogenic mutation. This strategy also in some way minimizes mis-classification due to potential technical issues and the issues of poor sensitivity of MSI reported for MSH6 variants, mentioned above.
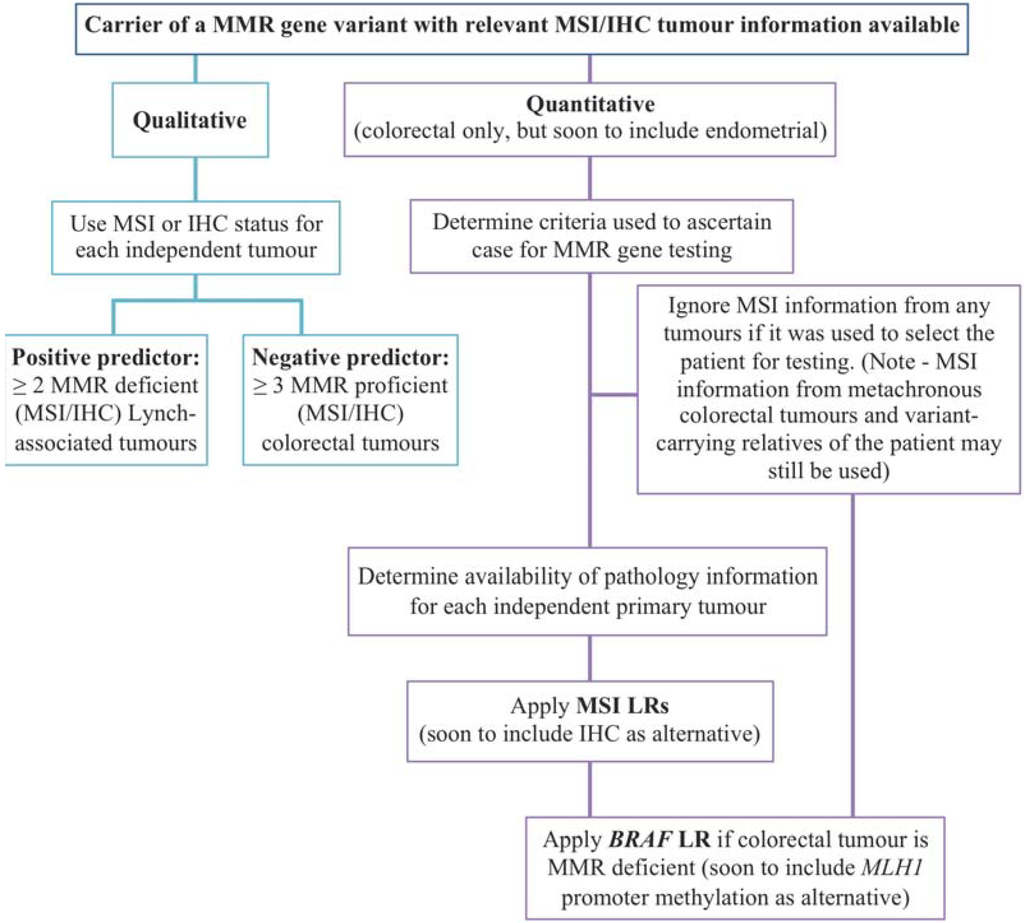
Figure 2.
Flowchart of variant classification process using tumour features. MMR—mismatch repair; MSI—microsatellite instability; IHC—immunohistochemistry; LR—likelihood ratio.
Few studies have addressed using MSI/IHC in a statistical approach for classification of MMR variants of uncertain clinical significance. The first report of a Bayesian integrated approach to incorporate MSI/IHC [79] included an LR for clinicopathologic features (IHC/MSI) derived using a large dataset [80]. However, ascertainment bias was not taken into account, in that only MSI tumours were tested for germline variants in the reference set, leading to overestimation of the likelihood of pathogenicity with an OR of ~27:1. Whereas, in a previous study we derived LRs for colorectal tumour MSI and BRAF V600E status using a large and well-characterised dataset (where testing for germline alterations was performed on a random subset of MSS tumours), and applied them carefully considering issues relating to ascertainment bias [37]. These analyses demonstrated that a MMR gene variant present in an MSI-H colorectal tumour has ~7–9-fold chance of being disease-causing, and if also wild-type BRAF the likelihood of being disease-causing increases to 12 to13-fold. This LR has since been used in additional studies [27,81]. The measures of LS-associated tumour features played an integral part in the categorization of over 60% MMR gene variants analysed using multifactorial likelihood analysis into classes associated with management recommendations (i.e., classes 1, 2, 4 and 5).
In the future it would be beneficial if a population-based study could be used to estimate statistical measures for tumour features (i.e., IHC and MLH1 promoter methylation) of endometrial tumours [8]. If MSI were to be incorporated into the model for endometrial tumours, a mononucleotide marker panel would be more suitable than the NCI panel for testing endometrial cases [55,82].
5. Conclusions
Tumour MSI and IHC status can be very useful in aiding in the interpretation of MMR gene variants of uncertain clinical significance. Patient ascertainment for gene testing on the basis of tumour phenotype can limit their usefulness in multifactorial likelihood analyses and could lead to misinterpretation of variant pathogenicity. As the cost of panel gene testing for cancer predisposition falls and the perceived utility of testing multiple genes in parallel increases, there may well be a move to a universal testing system where essentially all colon (and possibly endometrial) cancer cases get a panel test prior to any testing for tumour features. For the purpose of utility of tumour features for multifactorial modelling, subjects would no longer be ascertained based on personal/family history and/or MSI/IHC. Thus, ascertainment-based confounding would be reduced or even removed. The appropriate tumour test could then become a reflex assay and the result used to help evaluate unclassified variants. In the interim, tumour MSI (and IHC) status can still be a useful tool, in combination with other clinical, molecular, and functional assay information, to aid in the interpretation of MMR gene variants of uncertain clinical significance.
Acknowledgments
Amanda B. Spurdle is a National Health and Medical Research Council Senior Research Fellow. Bryony A. Thompson is a National Health and Medical Research Council Early Career Fellow.
Author Contributions
Both authors contributed equally.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of european experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet. 1999, 36, 801–818. [Google Scholar] [PubMed]
- Lynch, H.T.; Smyrk, T.; Lynch, J.F. Molecular genetics and clinical-pathology features of hereditary nonpolyposis colorectal carcinoma (Lynch syndrome): Historical journey from pedigree anecdote to molecular genetic confirmation. Oncology 1998, 55, 103–108. [Google Scholar] [CrossRef]
- Jass, J.R. Diagnosis of hereditary non-polyposis colorectal cancer. Histopathology 1998, 32, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef]
- Ward, R.L.; Hicks, S.; Hawkins, N.J. Population-based molecular screening for Lynch syndrome: Implications for personalized medicine. J. Clin. Oncol. 2013, 31, 2554–2562. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Panescu, J.; Lockman, J.; Sotamaa, K.; Fix, D.; Comeras, I.; LaJeunesse, J.; Nakagawa, H.; Westman, J.A.; Prior, T.W.; et al. Comment on: Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2007, 67, 9603. [Google Scholar] [CrossRef]
- Buchanan, D.D.; Tan, Y.Y.; Walsh, M.D.; Clendenning, M.; Metcalf, A.M.; Ferguson, K.; Arnold, S.T.; Thompson, B.A.; Lose, F.A.; Parsons, M.T.; et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J. Clin. Oncol. 2014, 32, 90–100. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Risinger, J.I.; Berchuck, A.; Kohler, M.F.; Watson, P.; Lynch, H.T.; Boyd, J. Genetic instability of microsatellites in endometrial carcinoma. Cancer Res. 1993, 53, 5100–5103. [Google Scholar] [PubMed]
- Win, A.K.; Lindor, N.M.; Winship, I.; Tucker, K.M.; Buchanan, D.D.; Young, J.P.; Rosty, C.; Leggett, B.; Giles, G.G.; Goldblatt, J.; et al. Risks of colorectal and other cancers after endometrial cancer for women with Lynch syndrome. J. Natl. Cancer Inst. 2013, 105, 274–279. [Google Scholar] [CrossRef]
- Win, A.K.; Lindor, N.M.; Young, J.P.; Macrae, F.A.; Young, G.P.; Williamson, E.; Parry, S.; Goldblatt, J.; Lipton, L.; Winship, I.; et al. Risks of primary extracolonic cancers following colorectal cancer in Lynch syndrome. J. Natl. Cancer Inst. 2012, 104, 1363–1372. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Salovaara, R.; Kristo, P.; Canzian, F.; Hemminki, A.; Peltomaki, P.; Chadwick, R.B.; Kaariainen, H.; Eskelinen, M.; Jarvinen, H.; et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N. Engl. J. Med. 1998, 338, 1481–1487. [Google Scholar] [CrossRef]
- Lindor, N.M.; Burgart, L.J.; Leontovich, O.; Goldberg, R.M.; Cunningham, J.M.; Sargent, D.J.; Walsh-Vockley, C.; Petersen, G.M.; Walsh, M.D.; Leggett, B.A.; et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J. Clin. Oncol. 2002, 20, 1043–1048. [Google Scholar] [CrossRef]
- Salovaara, R.; Loukola, A.; Kristo, P.; Kaariainen, H.; Ahtola, H.; Eskelinen, M.; Harkonen, N.; Julkunen, R.; Kangas, E.; Ojala, S.; et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J. Clin. Oncol. 2000, 18, 2193–2200. [Google Scholar]
- Terdiman, J.P.; Gum, J.R., Jr.; Conrad, P.G.; Miller, G.A.; Weinberg, V.; Crawley, S.C.; Levin, T.R.; Reeves, C.; Schmitt, A.; Hepburn, M.; et al. Efficient detection of hereditary nonpolyposis colorectal cancer gene carriers by screening for tumor microsatellite instability before germline genetic testing. Gastroenterology 2001, 120, 21–30. [Google Scholar] [CrossRef]
- Schofield, L.; Grieu, F.; Goldblatt, J.; Amanuel, B.; Iacopetta, B. A state-wide population-based program for detection of Lynch syndrome based upon immunohistochemical and molecular testing of colorectal tumours. Fam. Cancer 2011, 11, 1–6. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.; Panescu, J.; Lockman, J.; Sotamaa, K.; Fix, D.; Comeras, I.; La Jeunesse, J.; Nakagawa, H.; Westman, J.A.; et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006, 66, 7810–7817. [Google Scholar] [CrossRef]
- Metcalf, A.M.; Spurdle, A.B. Endometrial tumour BRAF mutations and MLH1 promoter methylation as predictors of germline mismatch repair gene mutation status: A literature review. Fam. Cancer 2014, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.M.; Christensen, E.R.; Tester, D.J.; Kim, C.Y.; Roche, P.C.; Burgart, L.J.; Thibodeau, S.N. Hypermethylation of the HMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998, 58, 3455–3460. [Google Scholar] [PubMed]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.P.; Markowitz, S.; Willson, J.K.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of HMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef]
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst-Stams, W.A.; Goossens, M.; Ouchene, H.; Hendriks-Cornelissen, S.J.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology 2014, 146, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014, 147, 1308–1316. [Google Scholar] [CrossRef]
- Gudgeon, J.M.; Williams, J.L.; Burt, R.W.; Samowitz, W.S.; Snow, G.L.; Williams, M.S. Lynch syndrome screening implementation: Business analysis by a healthcare system. Am. J. Manag. Care 2011, 17, e288–300. [Google Scholar] [PubMed]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capell, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef]
- Barnetson, R.A.; Cartwright, N.; van Vliet, A.; Haq, N.; Drew, K.; Farrington, S.; Williams, N.; Warner, J.; Campbell, H.; Porteous, M.E.; et al. Classification of ambiguous mutations in DNA mismatch repair genes identified in a population-based study of colorectal cancer. Hum. Mutat. 2008, 29, 367–374. [Google Scholar] [CrossRef]
- Hofstra, R.M.; Spurdle, A.B.; Eccles, D.; Foulkes, W.D.; de Wind, N.; Hoogerbrugge, N.; Hogervorst, F.B. Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum. Mutat. 2008, 29, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Lucci-Cordisco, E.; Boccuto, L.; Neri, G.; Genuardi, M. The use of microsatellite instability, immunohistochemistry and other variables in determining the clinical significance of MLH1 and MSH2 unclassified variants in Lynch syndrome. Cancer Biomark. 2006, 2, 11–27. [Google Scholar] [PubMed]
- Kansikas, M.; Kariola, R.; Nystrom, M. Verification of the three-step model in assessing the pathogenicity of mismatch repair gene variants. Hum. Mutat. 2011, 32, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Couch, F.J.; Hogervorst, F.B.; Radice, P.; Sinilnikova, O.M. Prediction and assessment of splicing alterations: Implications for clinical testing. Hum. Mutat. 2008, 29, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the european consortium “care for CMMRD” (C4CMMRD). J. Med. Genet. 2014, 51, 355–365. [Google Scholar] [CrossRef]
- Goldgar, D.E.; Easton, D.F.; Deffenbaugh, A.M.; Monteiro, A.N.; Tavtigian, S.V.; Couch, F.J. Integrated evaluation of DNA sequence variants of unknown clinical significance: Application to BRCA1 and BRCA2. Am. J. Hum. Genet. 2004, 75, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Easton, D.F.; Byrnes, G.B.; Spurdle, A.B.; Iversen, E.S.; Greenblatt, M.S. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008, 29, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.J.; Duraisamy, S.; Newell, J.A.; Chan, P.A.; Tie, M.M.; Rogers, A.E.; Ankuda, C.K.; von Walstrom, G.M.; Bond, J.P.; Greenblatt, M.S. Classifying variants of CDKN2A using computational and laboratory studies. Hum. Mutat. 2011, 32, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.A.; Goldgar, D.E.; Paterson, C.; Clendenning, M.; Walters, R.; Arnold, S.; Parsons, M.T.; Michael, D.W.; Gallinger, S.; Haile, R.W.; et al. A multifactorial likelihood model for MMR gene variant classification incorporating probabilities based on sequence bioinformatics and tumor characteristics: A report from the Colon Cancer Family Registry. Hum. Mutat. 2013, 34, 200–209. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wenstrup, R.J.; Allen-Brady, K.; Tavtigian, S.V.; Monteiro, A.N.; Iversen, E.S.; Couch, F.J.; et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am. J. Hum. Genet. 2007, 81, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Byrnes, G.B.; Goldgar, D.E.; Thomas, A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum. Mutat. 2008, 29, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Greenblatt, M.S.; Lesueur, F.; Byrnes, G.B. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008, 29, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.A.; Greenblatt, M.S.; Vallee, M.P.; Herkert, J.C.; Tessereau, C.; Young, E.L.; Adzhubey, I.A.; Li, B.; Bell, R.; Feng, B.; et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum. Mutat. 2013, 34, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kidambi, T.D.; Blanco, A.; Myers, M.; Conrad, P.; Loranger, K.; Terdiman, J.P. Selective versus universal screening for Lynch syndrome: A six-year clinical experience. Dig. Dis. Sci. 2014. [Google Scholar] [CrossRef]
- Perez-Carbonell, L.; Ruiz-Ponte, C.; Guarinos, C.; Alenda, C.; Paya, A.; Brea, A.; Egoavil, C.M.; Castillejo, A.; Barbera, V.M.; Bessa, X.; et al. Comparison between universal molecular screening for Lynch syndrome and revised bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut 2012, 61, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Jass, J.R. HNPCC and sporadic MSI-H colorectal cancer: A review of the morphological similarities and differences. Fam. Cancer 2004, 3, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.A.; Hayashi, S.; O'Shea, A.M.; Burgart, L.J.; Smyrk, T.C.; Shimizu, D.; Waring, P.M.; Ruszkiewicz, A.R.; Pollett, A.F.; Redston, M.; et al. Pathology features in bethesda guidelines predict colorectal cancer microsatellite instability: A population-based study. Gastroenterology 2007, 133, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Cicek, M.S.; Lindor, N.M.; Gallinger, S.; Bapat, B.; Hopper, J.L.; Jenkins, M.A.; Young, J.; Buchanan, D.; Walsh, M.D.; le Marchand, L.; et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J. Mol. Diagn. 2011, 13, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Loukola, A.; Vilkki, S.; Singh, J.; Launonen, V.; Aaltonen, L.A. Germline and somatic mutation analysis of MLH3 in MSI-positive colorectal cancer. Am. J. Pathol. 2000, 157, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Mangold, E.; Pagenstecher, C.; Friedl, W.; Fischer, H.P.; Merkelbach-Bruse, S.; Ohlendorf, M.; Friedrichs, N.; Aretz, S.; Buettner, R.; Propping, P.; et al. Tumours from MSH2 mutation carriers show loss of MSH2 expression but many tumours from MLH1 mutation carriers exhibit weak positive MLH1 staining. J. Pathol. 2005, 207, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Van Riel, E.; Ausems, M.G.; Hogervorst, F.B.; Kluijt, I.; van Gijn, M.E.; van Echtelt, J.; Scheidel-Jacobse, K.; Hennekam, E.F.; Stulp, R.P.; Vos, Y.J.; et al. A novel pathogenic MLH1 missense mutation, c.112A > C, p.Asn38His, in six families with Lynch syndrome. Hered. Cancer Clin. Pract. 2010, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Acharya, S.; Wilson, T.; Gradia, S.; Kane, M.F.; Guerrette, S.; Marsischky, G.T.; Kolodner, R.; Fishel, R. HMSH2 forms specific mispair-binding complexes with HMSH3 and HMSH6. Proc. Natl. Acad. Sci. USA 1996, 93, 13629–13634. [Google Scholar] [CrossRef] [PubMed]
- You, J.F.; Buhard, O.; Ligtenberg, M.J.; Kets, C.M.; Niessen, R.C.; Hofstra, R.M.; Wagner, A.; Dinjens, W.N.; Colas, C.; Lascols, O.; et al. Tumours with loss of MSH6 expression are MSI-H when screened with a pentaplex of five mononucleotide repeats. Br. J. Cancer 2010, 103, 1840–1845. [Google Scholar] [CrossRef] [PubMed]
- Xicola, R.M.; Llor, X.; Pons, E.; Castells, A.; Alenda, C.; Pinol, V.; Andreu, M.; Castellvi-Bel, S.; Paya, A.; Jover, R.; et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J. Natl. Cancer Inst. 2007, 99, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Thienpont, B.; Yesilyurt, B.T.; Moisse, M.; Reumers, J.; Coenegrachts, L.; Sagaert, X.; Schrauwen, S.; Smeets, D.; Matthijs, G.; et al. Mismatch repair deficiency endows tumors with a unique mutation signature and sensitivity to DNA double-strand breaks. Elife 2014, 3, e02725. [Google Scholar] [PubMed]
- Williams, A.S.; Huang, W.Y. The analysis of microsatellite instability in extracolonic gastrointestinal malignancy. Pathology 2013, 45, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Segev, Y.; Pal, T.; Rosen, B.; McLaughlin, J.R.; Sellers, T.A.; Risch, H.A.; Zhang, S.; Sun, P.; Narod, S.A.; Schildkraut, J. Risk factors for ovarian cancers with and without microsatellite instability. Int. J. Gynecol. Cancer 2014, 24, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Gylling, A.H.; Nieminen, T.T.; Abdel-Rahman, W.M.; Nuorva, K.; Juhola, M.; Joensuu, E.I.; Jarvinen, H.J.; Mecklin, J.P.; Aarnio, M.; Peltomaki, P.T. Differential cancer predisposition in Lynch syndrome: Insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis 2008, 29, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Barrow, E.; Hill, J.; Evans, D.G. Cancer risk in Lynch syndrome. Fam. Cancer 2013, 12, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, F.; Lasset, C.; Carayol, J.; Bonadona, V.; Perdry, H.; Desseigne, F.; Wang, Q.; Bonaiti-Pellie, C. Estimating cancer risk in HNPCC by the GRL method. Eur. J. Hum. Genet. 2007, 15, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, M.G.; Farrington, S.M.; Carothers, A.D.; Wyllie, A.H.; Sharp, L.; Burn, J.; Liu, B.; Kinzler, K.W.; Vogelstein, B. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum. Mol. Genet. 1997, 6, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.A.; Baglietto, L.; Dowty, J.G.; van Vliet, C.M.; Smith, L.; Mead, L.J.; Macrae, F.A.; St John, D.J.; Jass, J.R.; Giles, G.G.; et al. Cancer risks for mismatch repair gene mutation carriers: A population-based early onset case-family study. Clin. Gastroenterol. Hepatol. 2006, 4, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, F.; Vasen, H.F.; van Houwelingen, H.C. Risk of colorectal and endometrial cancer for carriers of mutations of the HMLH1 and HMSH2 gene: Correction for ascertainment. J. Med. Genet. 2005, 42, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.; Mukherjee, B.; Raymond, V.M.; Tayob, N.; Kastrinos, F.; Sparr, J.; Wang, F.; Bandipalliam, P.; Syngal, S.; Gruber, S.B. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology 2009, 137, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Laird, P.W.; Park, P.J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Curtin, K.; Wolff, R.K.; Tripp, S.R.; Caan, B.J.; Slattery, M.L. Microsatellite instability and survival in rectal cancer. Cancer Causes Control 2009, 20, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.T.; Whiley, P.J.; Beesley, J.; Drost, M.; de Wind, N.; Thompson, B.A.; Marquart, L.; Hopper, J.L.; Jenkins, M.A.; Brown, M.A.; et al. Consequences of germline variation disrupting the constitutional translational initiation codon start sites of MLH1 and BRCA2: Use of potential alternative start sites and implications for predicting variant pathogenicity. Mol. Carcinog 2013. [Google Scholar] [CrossRef]
- Vakiani, E.; Yaeger, R.; Brooke, S.; Zhou, Y.; Klimstra, D.S.; Shia, J. Immunohistochemical detection of the BRAF V600E mutant protein in colorectal neoplasms. Appl. Immunohistochem. Mol. Morphol. 2014. [Google Scholar] [PubMed]
- Roth, R.M.; Hampel, H.; Arnold, C.A.; Yearsley, M.M.; Marsh, W.L.; Frankel, W.L. A modified Lynch syndrome screening algorithm in colon cancer: BRAF immunohistochemistry is efficacious and cost beneficial. Am. J. Clin. Pathol. 2015, 143, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Jorgensen, N.M.; Wallace, A.J.; Buchanan, D.D.; Lalloo, F.; McMahon, R.F.; Hill, J.; Evans, D.G. Tumour MLH1 promoter region methylation testing is an effective prescreen for Lynch syndrome (HNPCC). J. Med. Genet. 2014, 51, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.P.; Goodheart, M.J.; Sood, A.K.; Holmes, R.J.; Hatterman-Zogg, M.A.; Buller, R.E. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer 2003, 98, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Furlan, D.; Carnevali, I.; Marcomini, B.; Cerutti, R.; Dainese, E.; Capella, C.; Riva, C. The high frequency of de novo promoter methylation in synchronous primary endometrial and ovarian carcinomas. Clin. Cancer Res. 2006, 12, 3329–3336. [Google Scholar] [CrossRef] [PubMed]
- Gylling, A.; Abdel-Rahman, W.M.; Juhola, M.; Nuorva, K.; Hautala, E.; Jarvinen, H.J.; Mecklin, J.P.; Aarnio, M.; Peltomaki, P. Is gastric cancer part of the tumour spectrum of hereditary non-polyposis colorectal cancer? A molecular genetic study. Gut 2007, 56, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Furihata, M.; Takeuchi, T.; Ohtsuki, Y.; Terao, N.; Kuwahara, M.; Shuin, T. Genetic analysis of hmlh1 in transitional cell carcinoma of the urinary tract: Promoter methylation or mutation. J. Urol. 2001, 165, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Pastrello, C.; Pin, E.; Marroni, F.; Bedin, C.; Fornasarig, M.; Tibiletti, M.G.; Oliani, C.; Ponz de Leon, M.; Urso, E.D.; Della Puppa, L.; et al. Integrated analysis of unclassified variants in mismatch repair genes. Genet. Med. 2011, 13, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Forberg, J.; Holinski-Feder, E.; Pagenstecher, C.; Plaschke, J.; Kloor, M.; Poremba, C.; Pox, C.P.; Ruschoff, J.; Keller, G.; et al. Novel strategy for optimal sequential application of clinical criteria, immunohistochemistry and microsatellite analysis in the diagnosis of hereditary nonpolyposis colorectal cancer. Int. J. Cancer 2006, 118, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Gonzalez-Acosta, M.; Thompson, B.A.; Sanchez, R.; Gomez, C.; Martinez-Lopez, J.; Perea, J.; Caldes, T.; Rodriguez, Y.; Landolfi, S.; et al. Detailed characterization of MLH1 p.D41H and p.N710D variants coexisting in a Lynch syndrome family with conserved MLH1 expression tumors. Clin. Genet. 2014. [Google Scholar] [CrossRef]
- Wong, Y.F.; Cheung, T.H.; Lo, K.W.; Yim, S.F.; Chan, L.K.; Buhard, O.; Duval, A.; Chung, T.K.; Hamelin, R. Detection of microsatellite instability in endometrial cancer: Advantages of a panel of five mononucleotide repeats over the national cancer institute panel of markers. Carcinogenesis 2006, 27, 951–955. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).