
Genetics of Type 2 Diabetes—Pitfalls and Possibilities


Abstract
:1. The Diabetes Epidemic
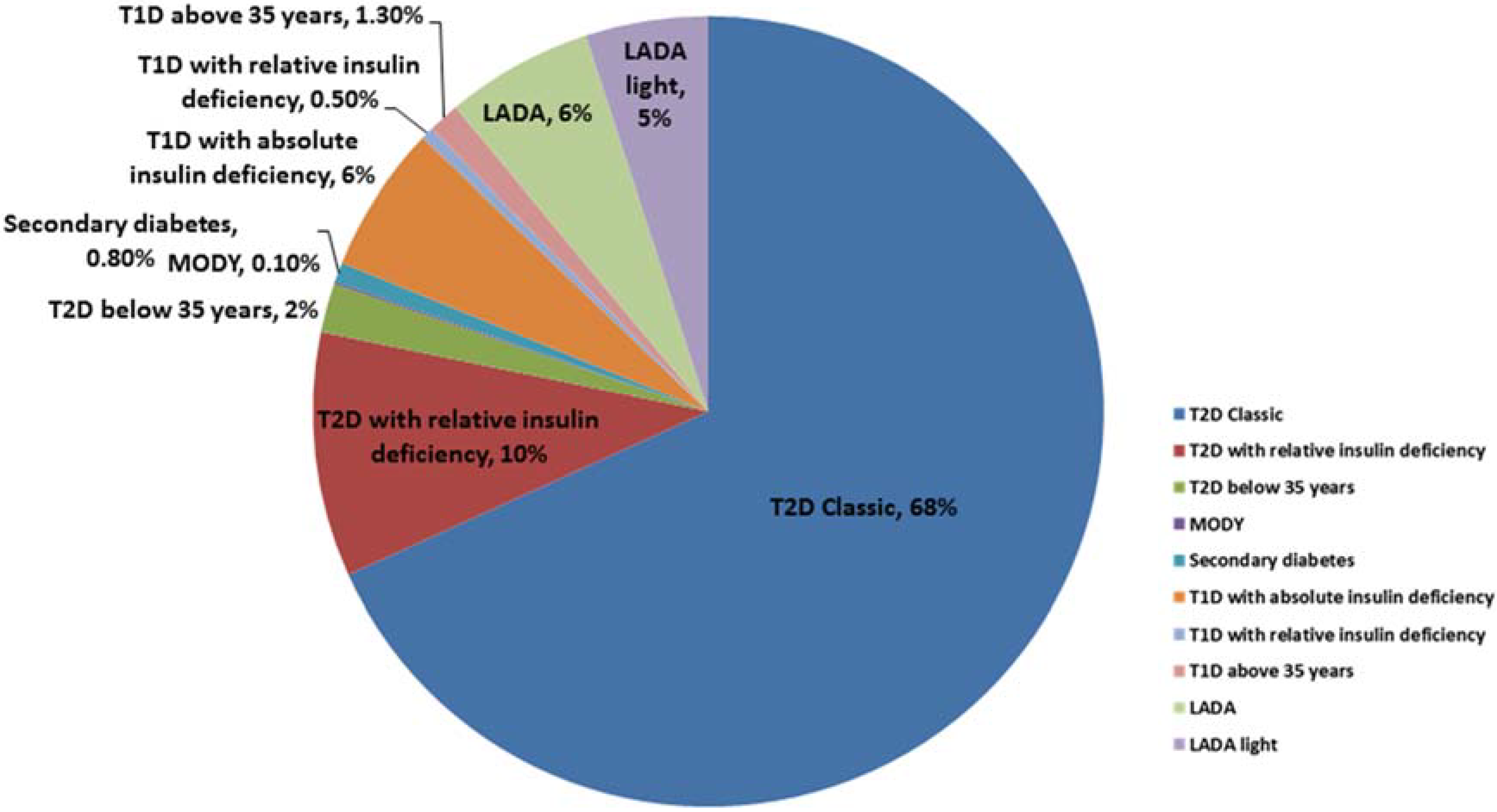
2. The Diabetes Spectrum
3. Heritability of T2D
4. Possibilities
4.1. Search for the Genetic Basis of T2D
4.1.1. Linkage Studies
4.1.2. Candidate Genes for T2D
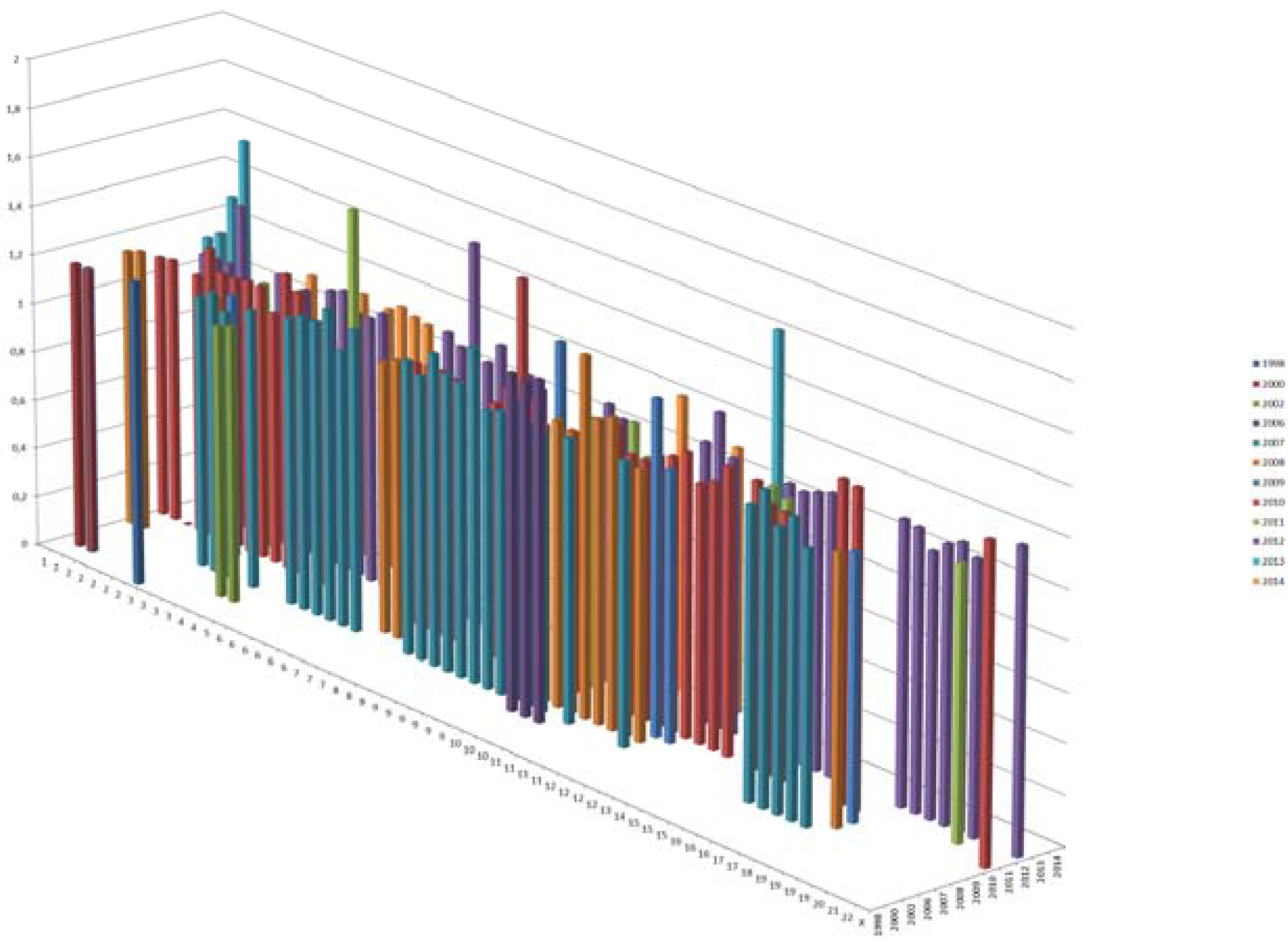
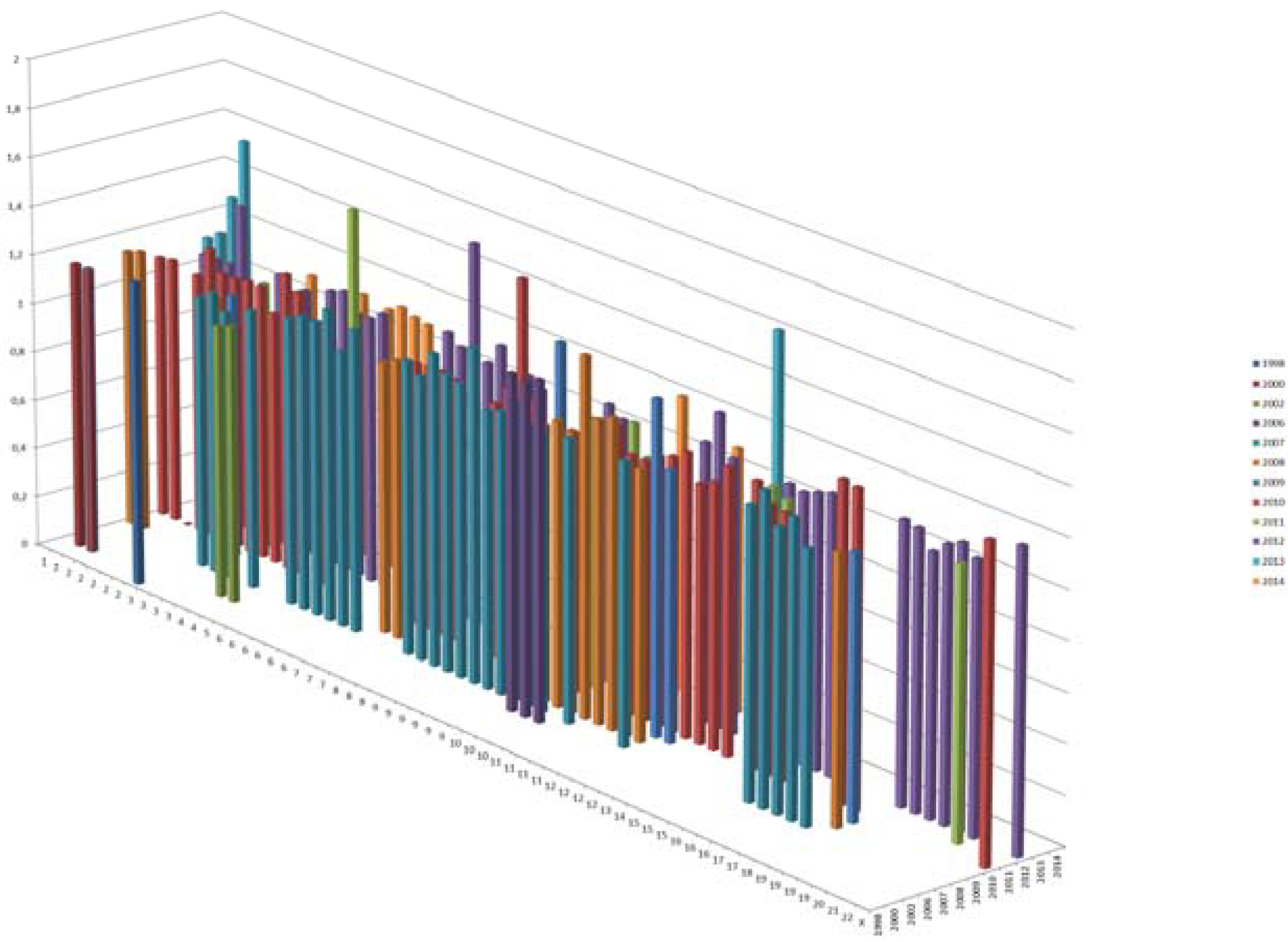
4.1.3. Genome Wide Association Studies (GWAS)

{kind=link}
{kind=link}
{kind=link}
| N | T2D Risk SNP | Gene/Nearest Gene | Gene Location | Chr | RA | OA | OR | TRAIT | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | rs17106184 | FAF1 | intron | 1 | G | A | 1.10 | T2D | [70] |
| 2 | rs2296172 | MACF1 | coding - missense | 1 | G | A | 1.10 | 2D | [79] |
| 3 | rs10923931 | NOTCH2 | intron | 1 | T | G | 1.13 | T2D | [4,80] |
| 4 | rs340874 | PROX1 | intergenic | 1 | C | T | 1.07 | Fasting glucose/HOMA B/T2D | [81] |
| 5 | rs243021 | BCL11A | intergenic | 2 | A | G | 1.08 | T2D | [25] |
| 6 | rs243088 | BCL11A | intergenic | 2 | T | A | 1.07 | T2D | [62] |
| 7 | rs2975760 | CAPN10 | intron | 2 | C | T | 1.17 | T2D | [48,82] |
| 8 | rs3792267 | CAPN10 | intron | 2 | G | A | 1.17 | T2D | [48,82] |
| 9 | rs7607980 | COBLL1 | coding-missense | 2 | T | C | 1.14 | T2D | [79] |
| 10 | rs560887 | G6PC2/ABCB11 | intron | 2 | T | C | 1.03 | Fasting glucose/T2D/HOMA B | [81] |
| 11 | rs780094 | GCKR | intron | 2 | C | T | 1.06 | T2D/Fasting glucose/beta-cell function/triglycerides/fasting insulin | [81] |
| 12 | rs3923113 | GRB14 | intergenic | 2 | A | C | 1.07 | T2D | [62,65] |
| 13 | rs13389219 | GRB14 | intergenic | 2 | C | T | 1.07 | T2D | [62] |
| 14 | rs2943641 | IRS1 | intergenic | 2 | C | T | 1.19 | Fasting glucose/T2D/HOMAB, HOMA IR/AUC ins/AUC ratio/ISI | [83] |
| 15 | rs7578326 | KIAA1486/IRS1 | intron of uncharacterized LOC646736 | 2 | A | G | 1.11 | T2D | [25] |
| 16 | rs7593730 | RBMS1/ITGB6 | intronic | 2 | C | T | 1.11 | T2D | [84] |
| 17 | rs7560163 | RND3 | intergenic | 2 | G | C | 1.33 | T2D | [67] |
| 18 | rs7578597 | THADA | coding-missense | 2 | T | C | 1.15 | T2D | [4,80] |
| 19 | rs10200833 | THADA | intron | 2 | G | C | 1.06 | T2D | [80,85] |
| 20 | rs6723108 | TMEM163 | intergenic | 2 | T | G | 1.31 | Decreased fasting plasma insulin/HOMA-IR/T2D | [74] |
| 21 | rs998451 | TMEM163 | intron | 2 | G | A | 1.56 | Decreased fasting plasma insulin/HOMA-IR/T2D | [74] |
| 22 | rs4607103 | ADAMTS9-AS2 | intron | 3 | C | T | 1.09 | T2D | [4,80] |
| 23 | rs6795735 | ADAMTS9-AS2 | intron | 3 | C | T | 1.09 | T2D | [4,80] |
| 24 | rs11708067 | ADCY5 | intron | 3 | A | G | 1.12 | T2D/2hr glucose/HOMA B | [81,86] |
| 25 | rs2877716 | ADCY5 | intron | 3 | C | T | 1.12 | 2 h insulin adjusted for 2 h glucose/2 h glucose/T2D | [81,86] |
| 26 | rs11071657 | FAM148B | intergenic | 3 | A | G | 1.03 | Fasting glucose/T2D/HOMA B | [81] |
| 27 | rs4402960 | IGF2BP2 | intron | 3 | T | G | 1.11 | T2D | [59] |
| 28 | rs1470579 | IGF2BP2 | intron | 3 | C | A | 1.15 | T2D | [26,59,72,87] |
| 29 | rs6808574 | LPP | intergenic | 3 | C | T | 1.07 | T2D | [70] |
| 30 | rs1801282 | PPARG | coding-missense | 3 | C | G | 1.09 | T2D | [59] |
| 31 | rs13081389 | PPARG | intergenic | 3 | A | G | 1.24 | T2D | [25,53,59,87] |
| 32 | rs17036160 | PPARG | intron | 3 | C | T | 1.11 | T2D | [85] |
| 33 | rs1797912 | PPARG | intron | 3 | A | C | 1.06 | T2D | [85] |
| 34 | rs831571 | PSMD6 | intergenic | 3 | C | T | 1.09 | T2D | [63] |
| 35 | rs7647305 | SFRS10 | intergenic | 3 | C | T | 1.08 | BMI/obesity T2D | [88] |
| 36 | rs16861329 | ST6GAL1 | intron | 3 | G | A | 1.09 | T2D | [65] |
| 37 | rs6780569 | UBE2E2 | intergenic | 3 | G | A | 1.21 | T2D | [73] |
| 38 | rs6815464 | MAEA | intron | 4 | C | G | 1.13 | T2D | [63] |
| 39 | rs7656416 | MAEA | intron | 4 | C | T | 1.15 | T2D | [63,64] |
| 40 | rs6813195 | TMEM154 | intergenic | 4 | C | T | 1.08 | T2D | [70] |
| 41 | rs10010131 | WFS1 | intron | 4 | G | A | 1.14 | T2D | [4,89] |
| 42 | rs4689388 | WFS1 | nearGene-5 | 4 | T | C | 1.16 | T2D | [83] |
| 43 | rs6446482 | WFS1 | intron | 4 | G | C | 1.11 | T2D | [25,89,90] |
| 44 | rs1801214 | WFS1 | coding-missense | 4 | T | C | 1.13 | T2D | [25,89,90] |
| 45 | rs459193 | ANKRD55 | intergenic | 5 | G | A | 1.08 | T2D | [62] |
| 46 | rs702634 | ARL15 | intron | 5 | A | G | 1.06 | T2D | [70] |
| 47 | rs4457053 | ZBED3 | intron of ZBED3-AS1 | 5 | G | A | 1.08 | T2D | [25] |
| 48 | rs1048886 | C6orf57 | coding-missense | 6 | G | A | 1.54 | T2D | [91] |
| 49 | rs7754840 | CDKAL1 | intron | 6 | C | G | 1.17 | T2D | [25,27,59,60,66,73,92,93] |
| 50 | rs7756992 | CDKAL1 | intron | 6 | G | A | 1.20 | T2D | [27] |
| 51 | rs2206734 | CDKAL1 | intron | 6 | T | C | 1.20 | T2D | [25,27,59,60,66,73,92,93] |
| 52 | rs4712523 | CDKAL1 | intron | 6 | G | A | 1.27 | T2D | [25,27,59,60,66,73,83,92,93] |
| 53 | rs10946398 | CDKAL1 | intron | 6 | C | A | 1.12 | T2D | [25,27,59,60,66,73,92,93] |
| 54 | rs7766070 | CDKAL1 | intron | 6 | A | C | 1.23 | T2D | [25,27,59,60,66,73,92,93] |
| 55 | rs2244020 (rs9266650) | HLA-B | intergenic | 6 | G | A | 1.09 | T2D | [94] |
| 56 | rs1535500 | KCNK16 | coding-missense | 6 | T | G | 1.08 | T2D | [63] |
| 57 | rs3130501 | POU5F1-TCF19 | nearGene-5 | 6 | G | A | 1.07 | T2D | [70] |
| 58 | rs9505118 | SSR1-RREB1 | intron | 6 | A | G | 1.06 | T2D | [70] |
| 59 | rs9470794 | ZFAND3 | intron | 6 | C | T | 1.12 | T2D | [63] |
| 60 | rs17168486 | DGKB | intergenic | 7 | T | C | 1.15 | T2D | [62] |
| 61 | rs2191349 | DGKB/TMEM195 | intergenic | 7 | T | G | 1.06 | Fasting glucose, Homa B/T2D | [81] |
| 62 | rs6467136 | GCC1-PAX4 | intergenic | 7 | G | A | 1.11 | T2D | [63] |
| 63 | rs4607517 | GCK | intergenic | 7 | A | G | 1.07 | Fasting glucose/T2D/HOMA B | [81] |
| 64 | rs864745 | JAZF1 | intron | 7 | T | C | 1.10 | T2D | [4,80] |
| 65 | rs849134 | JAZF1 | intron | 7 | A | G | 1.13 | T2D | [25,80] |
| 66 | rs12113122 | JAZF1 | intron | 7 | G | C | 1.55 | T2D | [85] |
| 67 | rs972283 | KLF14 | intergenic | 7 | G | A | 1.07 | Reduced insulin sensitivity T2D | [25] |
| 68 | rs516946 | ANK1 | intron | 8 | C | T | 1.09 | T2D | [62] |
| 69 | rs515071 | ANK1 | intron | 8 | G | A | 1.18 | T2D Reduced beta-cell function | [62,64] |
| 70 | rs13266634 | SLC30A8 | coding-missense | 8 | C | T | 1.19 | T2D | [58] |
| 71 | rs11558471 | SLC30A8 | UTR-3 | 8 | A | G | 1.15 | Fasting glucose, HOMA B T2D | [25,58,59,81,87,92,93] |
| 72 | rs3802177 | SLC30A8 | UTR-3 | 8 | G | A | 1.26 | T2D | [25,58,59,81,87,92,93] |
| 73 | rs896854 | TP53INP1 | intron | 8 | T | C | 1.06 | T2D | [25] |
| 74 | rs10965250 | CDKN2A/2B | intergenic | 9 | G | A | 1.20 | T2D | [25,27,59,60,66,73,92,93] |
| 75 | rs2383208 | CDKN2A/2B | intergenic | 9 | A | G | 1.19 | T2D | [25,27,59,60,66,73,92,93] |
| 76 | rs7018475 | CDKN2A/2B | intergenic | 9 | G | T | 1.35 | T2D | [25,27,59,60,66,73,92,93] |
| 77 | rs564398 | CDKN2A/2B | intergenic | 9 | T | C | 1.12 | T2D | [25,27,59,60,66,73,92,93] |
| 78 | rs10757282 | CDKN2A/2B | intergenic | 9 | C | T | 1.14 | T2D | [25,27,59,60,66,73,92,93] |
| 79 | rs10811661 | CDKN2B | intergenic | 9 | T | C | 1.20 | T2D | [25,59,60,66,68,80,87,92] |
| 80 | rs7034200 | GLIS3 | intron | 9 | A | C | 1.03 | Fasting glucose/T2D/HOMA B | [81] |
| 81 | rs7041847 | GLIS3 | intron | 9 | A | G | 1.10 | T2D | [63,66] |
| 82 | rs10814916 | GLIS3 | intron | 9 | C | A | 1.11 | T2D | [63,66,81] |
| 83 | rs17584499 | PTPRD | intron | 9 | T | C | 1.57 | T2D | [95] |
| 84 | rs2796441 | TLE1 | intergenic | 9 | G | A | 1.07 | T2D | [62] |
| 85 | rs13292136 | TLE4 (CHCHD9) | intergenic | 9 | C | T | 1.11 | T2D | [25] |
| 86 | rs553668 | ADRA2A | UTR-3 | 10 | A | G | 1.42 | T2D | [96] |
| 87 | rs10885122 | ADRA2A | intergenic | 10 | G | T | 1.04 | Fasting glucose/HOMA B/T2D | [81] |
| 88 | rs12779790 | CDC123, CAMK1D | intergenic | 10 | G | A | 1.11 | T2D | [4,80] |
| 89 | rs11257655 | CDC123/CAMK1D | intergenic | 10 | C | T | 1.15 | T2D | [66,69,80] |
| 90 | rs10906115 | CDC123/CAMK1D | intergenic | 10 | A | G | 1.13 | T2D | [66,69,80] |
| 91 | rs10886471 | GRK5 | intron | 10 | C | T | 1.12 | T2D | [66] |
| 92 | rs5015480 | HHEX | intergenic | 10 | C | T | 1.13 | T2D | [25,58,59,92,93] |
| 93 | rs1111875 | HHEX/IDE | intergenic | 10 | C | T | 1.13 | T2D | [59] |
| 94 | rs7903146 | TCF7L2 | intronic/promoter | 10 | T | C | 1.35 | T2D, fasting glucose,.2 h glucose | [51] |
| 95 | rs4506565 | TCF7L2 | intron | 10 | T | A | 1.34 | Fasting glucose, HOMA B T2D | [25,27,51,58,59,60,61,80,87,92,93,97,98,99] |
| 96 | rs7901695 | TCF7L2 | intron | 10 | C | T | 1.37 | T2D | [25,27,51,58,59,60,61,80,87,92,93,97,98,99] |
| 97 | rs1802295 | VPS26A | UTR-3 | 10 | A | G | 1.08 | T2D | [65] |
| 98 | rs12571751 | ZMIZ1 | intron | 10 | A | G | 1.08 | T2D | [62] |
| 99 | rs11603334 | ARAP1 | UTR-5 | 11 | G | A | 1.13 | T2D fasting proinsulin levels/fasting glucose/ | [100] |
| 100 | rs1552224 | CENTD2 | intergenic | 11 | A | C | 1.14 | T2D | [25] |
| 101 | rs11605924 | CRY2 | intron | 11 | A | C | 1.04 | Fasting glucose/HOMA B/T2D | [81] |
| 102 | rs174550 | FADS1 | intron | 11 | T | C | 1.04 | Fasting glucose/T2D/HOMA B | [81] |
| 103 | rs2334499 | HCCA2 | intergenic | 11 | T | C | 1.35 | T2D | [101] |
| 104 | rs3842770 | INS-IGF2 | intron | 11 | A | G | 1.18 | T2D - African American | [94] |
| 105 | rs5219 | KCNJ11 | coding-missense | 11 | T | C | 1.14 | T2D | [54,59,60,87,99] |
| 106 | rs5215 | KCNJ11 | coding-missense | 11 | C | T | 1.14 | T2D | [54,59,60,87,99] |
| 107 | rs2237895 | KCNQ1 | intron | 11 | C | T | 1.45 | T2D | [71] |
| 108 | rs231362 | KCNQ1 | intron | 11 | G | A | 1.08 | T2D | [25] |
| 109 | rs163184 | KCNQ1 | intron | 11 | G | T | 1.22 | T2D | [62,71] |
| 110 | rs2237892 | KCNQ1 | intron | 11 | C | T | 1.25 | Reduced beta-cell function T2D | [25,71,72,92,95] |
| 111 | rs10501320 | MADD | intron | 11 | G | C | 1.01 | T2D fasting proinsulin levels/fasting glucose | [100] |
| 112 | rs10830963 | MTNR1B | intron | 11 | G | C | 1.09 | T2D | [102] |
| 113 | rs1387153 | MTNR1B | intergenic | 11 | T | C | 1.09 | Reduced beta-cell function T2D | [25,95,102] |
| 114 | rs7138803 | BCDIN3D/FAIM2 | intergenic | 12 | A | G | 1.11 | BMI/obesity T2D | [88,103] |
| 115 | rs11063069 | CCND2 | intergenic | 12 | G | A | 1.12 | T2D | [62] |
| 116 | rs1153188 | DCD | intergenic | 12 | A | T | 1.08 | T2D | [80] |
| 117 | rs1531343 | HMGA2 | intron of pseudogene | 12 | C | G | 1.10 | T2D | [25] |
| 118 | rs9668162 | HMGA2 | intron | 12 | G | C | 1.26 | T2D | [85] |
| 119 | rs7305618 | HNF1A | intergenic | 12 | C | T | 1.14 | T2D | [25,68] |
| 120 | rs35767 | IGF1 | nearGene-5 | 12 | G | A | 1.04 | Fasting insulin/T2D/HOMA IR | [81] |
| 121 | rs10842994 | KLHDC5 | intergenic | 12 | C | T | 1.10 | T2D | [62] |
| 122 | rs4275659 | MPHOSPH9 | intron | 12 | C | T | 1.06 | T2D | [70] |
| 123 | rs7957197 | OASL/TCF1/HNF1A | intron of OASL | 12 | T | A | 1.07 | T2D | [25] |
| 124 | rs7961581 | TSPAN8, LGR5 | intergenic | 12 | C | T | 1.09 | T2D | [4,80] |
| 125 | rs9552911 | SGCG | intron | 13 | G | A | 1.63 | T2D | [104] |
| 126 | rs1359790 | SPRY2 | intergenic | 13 | G | A | 1.15 | T2D | [69] |
| 127 | rs2028299 | AP3S2 | UTR-3 | 15 | C | A | 1.10 | T2D | [65] |
| 128 | rs7172432 | C2CD4A/B | intergenic | 15 | A | G | 1.14 | Reduced beta-cell function, T2D | [73] |
| 129 | rs7178572 | HMG20A | intergenic | 15 | A | G | 1.09 | lean T2D | [65,93] |
| 130 | rs7177055 | HMG20A | intergenic | 15 | A | G | 1.08 | T2D | [62] |
| 131 | rs8042680 | PRC1 | intron | 15 | A | C | 1.07 | T2D | [25] |
| 132 | rs7403531 | RASGRP1 | intron | 15 | T | C | 1.10 | T2D | [66] |
| 133 | rs4502156 | VPS13C/C2CD4A/B | intergenic | 15 | T | C | 1.07 | fasting proinsulin levels T2D | [100] |
| 134 | rs11634397 | ZFAND6 | intergenic | 15 | G | A | 1.06 | T2D | [25] |
| 135 | rs7202877 | BCAR1 | intergenic | 16 | T | G | 1.12 | T2D | [62] |
| 136 | rs8050136 | FTO | intron | 16 | A | C | 1.17 | Increased BMI, reduced insulin sensitivity, T2D | [25,60,61,80,87,93,99,105] |
| 137 | rs9939609 | FTO | intron | 16 | A | T | 1.25 | T2D (obese) | [25,60,61,80,87,93,99,105] |
| 138 | rs11642841 | FTO | intron | 16 | A | C | 1.13 | T2D | [25,60,61,80,87,93,99,105] |
| 139 | rs4430796 | HNF1B | intron | 17 | G | A | 1.19 | Reduced beta-cell function T2D | [66,106,66,107,108] |
| 140 | rs7501939 | HNF1B | intron | 17 | T | C | 1.09 | T2D | [106] |
| 141 | rs391300 | SRR | intron | 17 | G | A | 1.28 | T2D | [95] |
| 142 | rs4523957 | SRR | nearGene-5 | 17 | T | 1.27 | T2D | [95] | |
| 143 | rs8090011 | LAMA1 | intron | 18 | G | C | 1.13 | lean T2D | [93] |
| 144 | rs17782313 | MC4R | intergenic | 18 | C | T | 1.06 | BMI/T2D | [88,103] |
| 145 | rs12970134 | MC4R | intergenic | 18 | A | G | 1.08 | T2D/BMI/waist circumference/insulin resistance | [62,109] |
| 146 | rs3794991 | GATAD2A/CILP2 | intron, intergenic | 19 | T | C | 1.12 | T2D | [62,85] |
| 147 | rs8108269 | GIPR | intergenic | 19 | G | T | 1.05 | T2D | [62] |
| 148 | rs3786897 | PEPD | intron | 19 | A | G | 1.10 | T2D | [63] |
| 149 | rs10401969 | SUGP1/CILP2 | intron | 19 | C | T | 1.13 | T2D | [62,85] |
| 150 | rs6017317 | FITM2-R3HDML-HNF4A | intergenic | 20 | G | T | 1.09 | T2D | [63] |
| 151 | rs4812829 | HNF4A | intron | 20 | A | G | 1.09 | T2D | [65] |
| 152 | rs5945326 | DUSP9 | intergenic | X | A | G | 1.27 | T2D | [25] |
| 153 | rs12010175 | FAM58A | intron | X | G | A | 1.21 | T2D | [66] |
| N | SNPs | Gene/Nearest Gene | Gene Location | Chr | Effect Allele | Other Allele | Effect | TRAIT | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | rs9727115 | SNX7 | intron | 1 | G | A | 0.0133 | fasting proinsulin levels adjusted for fasting glucose | [100] |
| 2 | rs2785980 | LYPLAL1 | intergenic | 1 | T | C | 0.017 | fasting insulin | [110] |
| 3 | rs4675095 | IRS1 | intron | 2 | A | T | −0.006/−002 | fasting glucose/HOMA-IR | [81] |
| 4 | rs2943634 | IRS1 | intergenic | 2 | C | A | 0.025 | fasting insulin, CAD | [110] |
| 5 | rs1371614 | DPYSL5 | intron | 2 | T | C | 0.022 | Fasting glucose | [110] |
| 6 | rs11920090 | SLC2A2 | intron | 3 | T | A | 0.02 | Fasting glucose/HOMA B/HBA1C | [81] |
| 7 | rs17046216 | MSMO1 | intron | 4 | A | T | 0.18; 0.19 | Fasting Insulin; insulin resistance | [111] |
| 8 | rs4691380 | PDGFC | intron | 4 | C | T | 0.021 | fasting insulin | [110] |
| 9 | rs6235 | PCSK1 | coding-missense | 5 | G | C | 0.0394/−0.014 | fasting proinsulin levels/Fasting glucose | [100] |
| 10 | rs13179048 | PCSK1 | intergenic | 5 | C | A | 0.018 | Fasting glucose | [110] |
| 11 | rs4646949 | TAF11 | nearGene-3 | 6 | T | G | 0.020 | fasting insulin | [110] |
| 12 | rs6943153 | GRB10 | intron | 7 | C | T | 0.0154 | FG, FI | [26] |
| 13 | rs4841132 | PPP1R3B | intergenic | 8 | A | G | 0.030 | Fasting glucose | [110] |
| 14 | rs7077836 | TCERG1L | intergenic | 10 | T | C | 0.28; 0.34 | Fasting Insulin ; insulin resistance | [111] |
| 15 | rs7944584 | MADD | intron | 11 | A | T | 0.021 | Fasting proinsulin/Fasting glucose/Homa B | [81] |
| 16 | rs10838687 | MADD | intron | 11 | T | G | 0.0253 | fasting proinsulin levels | [100] |
| 17 | rs1483121 | OR4S1 | intergenic | 11 | G | A | 0.015 | Fasting glucose | [110] |
| 18 | rs2074356 | HECTD4/C12orf51 | intron | 12 | 1-h plasma glucose | [112] | |||
| 19 | rs2293941 | PDX1 - AS1 | intron | 13 | A | G | 0.016 | Fasting glucose | [110] |
| 20 | rs17271305 | VPS13C | intron | 15 | G | A | 0.07 | 2hr glucose/2-h insulin, adjusted for 2-h glucose | [86] |
| 21 | rs1549318 | LARP6 | intergenic | 15 | T | C | 0.0192 | fasting proinsulin levels | [100] |
| 22 | rs4790333 | SGSM2 | intron | 17 | T | C | 0.0154 | fasting proinsulin levels | [100] |
| 23 | rs10423928 | GIPR | intron | 19 | A | T | 2 h glucose/Insulinogenic index/AUCins/gluc/2-h insulin, adjusted for 2 h glucose/T2D | [86] | |
| 24 | rs6048205 | FOXA2/LINC00261 | intergenic/nearGene-5 | 20 | A | G | 0.029 | Fasting glucose | [110] |
4.1.4. Rare Variants
| N | SNPs | GENE/Nearest Gene | Gene Location | Chr | Refs. |
|---|---|---|---|---|---|
| 1 | rs35658696 | PAM | coding-missense | 5 | [118] |
| 2 | rs78408340 | PAM | coding-missense | 5 | [118] |
| 3 | rs36046591 | PPIP5K2 | coding-missense | 5 | [118] |
| 4 | p.Lys34Serfs*50 | SLC30A8 | coding-missense | 8 | [119] |
| 5 | p.Arg138* | SLC30A8 | coding-missense | 8 | [119] |
| 6 | rs3824420 | KANK1 | coding-missense | 9 | [118] |
| 7 | rs505922 | ABO | Intronic | 9 | [118] |
| 8 | rs60980157 | GPSM1 | coding-missense | 9 | [118] |
| 9 | p.Leu5Val (20) | ATG13 | coding-missense | 11 | [118] |
| 10 | p.Ile131Val (1) | ATG13 | coding-missense | 11 | [118] |
| 11 | p.Gln249Pro (3) | ATG13 | coding-missense | 11 | [118] |
| 12 | p.Arg392Trp (1) | ATG13 | coding-missense | 11 | [118] |
| 13 | p.Leu427Gln (3) | ATG13 | coding-missense | 11 | [118] |
| 14 | p.Gly434Arg (488) | ATG13 | coding-missense | 11 | [118] |
| 15 | p.X406Gly (200) | ATG13 | coding-missense | 11 | [118] |
| 16 | rs35233100 | MADD | coding-missense | 11 | [118] |
| 17 | p.Arg279Cys (324) | TBC1D30 | coding-missense | 12 | [118] |
| 18 | p.Pro746Leu (427) | TBC1D30 | coding-missense | 12 | [118] |
| 19 | c.1522G > A [p.E508K] | HNF1A | coding-missense | 12 | [117] |
| 20 | rs76895963 | CCND2 | intergenic | 12 | [115] |
| 21 | rs75615236 | CCND2 | intergenic | 12 | [115] |
| 22 | rs150781447 | TBC1D30 | coding-missense | 12 | [118] |
| 23 | rs2650000 | HNF1A | Intergenic | 12 | [118] |
| 24 | Chr. 13: g.27396636delT | PDX1 | coding-missense | 13 | [119] |
| 25 | p.Tyr416Cys (78) | SGSM2 | coding-missense | 17 | [118] |
| 26 | p.Thr789Pro (3) | SGSM2 | coding-missense | 17 | [118] |
| 27 | p.Val996Ile (236) | SGSM2 | coding-missense | 17 | [118] |
| 28 | rs61741902 | SGSM2 | coding-missense | 17 | [118] |
4.1.5. Structural Variants
4.1.6. Protective Variants
4.1.7. The Genetic Architecture of T2D
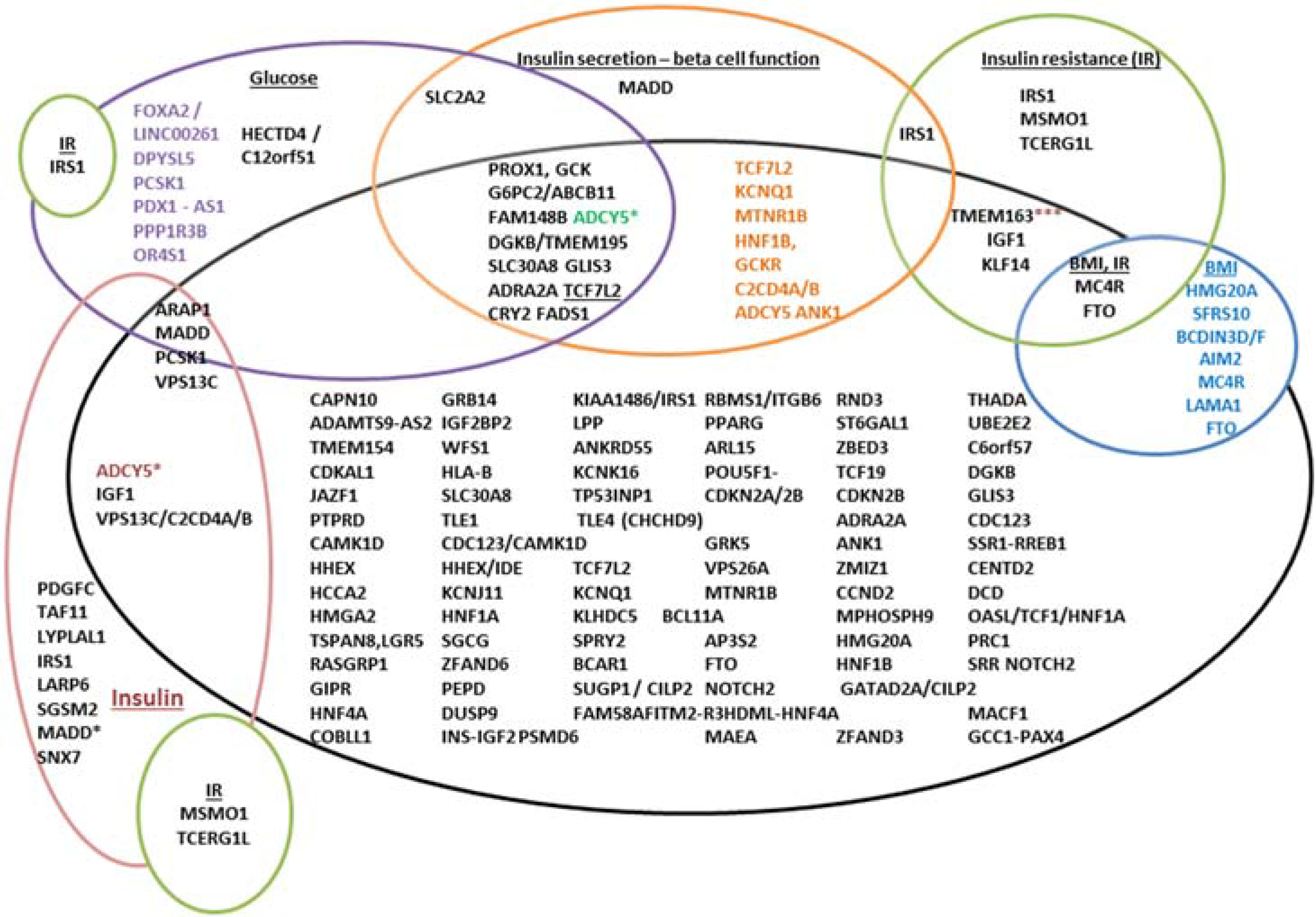
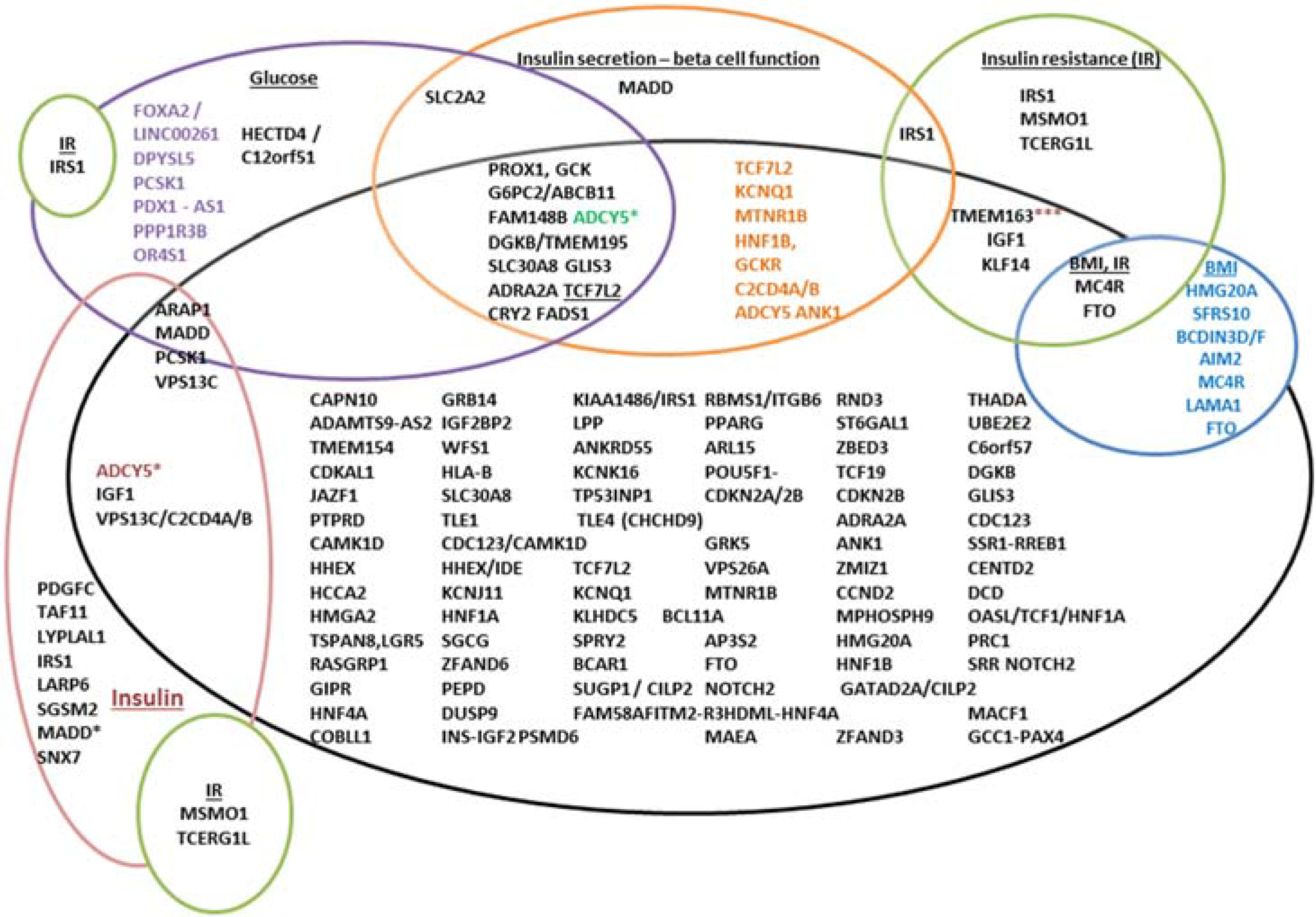
4.2. Functions of Associated Genes
Translational Studies
5. Pitfalls
5.1. Heritability Estimates
5.2. Parent of Origin Effects
5.3. Gene−Gene and Gene−Environment Interactions
5.4. Epigenetics
5.5. Non-Coding RNAs−microRNAs
5.6. The T1D-T2D Paradox
5.7. Systems Biology
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Groop, L.; Lyssenko, V. Genetics of type 2 diabetes. An overview. Endocrinol. Nutr. 2009, 56 (Suppl. 4), 34–37. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Atlas. 2014. Available online: www.Idf.Org/diabetesatlas (accessed on 3 March 2014).
- WHO. WHO report. 2014. Available online: http://www.who.int/gho/publications/world_health_statistics/en/ (accessed on 3 March 2014).
- Lyssenko, V.; Jonsson, A.; Almgren, P.; Pulizzi, N.; Isomaa, B.; Tuomi, T.; Berglund, G.; Altshuler, D.; Nilsson, P.; Groop, L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med. 2008, 359, 2220–2232. [Google Scholar] [CrossRef] [PubMed]
- Groop, L. Pathogenesis of type 2 diabetes: The relative contribution of insulin resistance and impaired insulin secretion. Int. J. Clin. Pract. Suppl. 2000, 113, 3–13. [Google Scholar] [PubMed]
- Steck, A.K.; Rewers, M.J. Genetics of type 1 diabetes. Clin. Chem. 2011, 57, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Menconi, F.; Corathers, S.; Jacobson, E.M.; Tomer, Y. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: From epidemiology to mechanisms. Endocr. Rev. 2008, 29, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Cernea, S.; Dobreanu, M.; Raz, I. Prevention of type 1 diabetes: Today and tomorrow. Diabetes/Metab. Res. Rev. 2010, 26, 602–605. [Google Scholar] [CrossRef]
- Kyvik, K.O.; Green, A.; Beck-Nielsen, H. Concordance rates of insulin dependent diabetes mellitus: A population based study of young Danish twins. BMJ 1995, 311, 913–917. [Google Scholar] [CrossRef]
- Pociot, F.; Norgaard, K.; Hobolth, N.; Andersen, O.; Nerup, J. A nationwide population-based study of the familial aggregation of type 1 (insulin-dependent) diabetes mellitus in Denmark. Danish study group of diabetes in childhood. Diabetologia 1993, 36, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Polychronakos, C.; Li, Q. Understanding type 1 diabetes through genetics: Advances and prospects. Nat. Rev. Genet. 2011, 12, 781–792. [Google Scholar] [CrossRef]
- Hakonarson, H.; Grant, S.F. Genome-wide association studies (GWAS): Impact on elucidating the aetiology of diabetes. Diabetes/Metab. Res. Rev. 2011, 27, 685–696. [Google Scholar] [CrossRef]
- Groop, L.; Pociot, F. Genetics of diabetes—Are we missing the genes or the disease? Mol. Cell. Endocrinol. 2014, 382, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Stankov, K.; Benc, D.; Draskovic, D. Genetic and epigenetic factors in etiology of diabetes mellitus type 1. Pediatrics 2013, 132, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, G.P.; Rewers, M. The epidemic of type 1 diabetes: What is it telling us? Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, S. Genetic susceptibility of childhood type 1 diabetes mellitus in Japan. Pediatr. Endocrinol. Rev. 2012, 10 (Suppl. 1), 62–71. [Google Scholar] [PubMed]
- Thomas, I.H.; Pietropaolo, M. Type 1 diabetes: A genetic pandora’s box? Pediatr. Diabetes 2010, 11, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Tuomi, T.; Rowley, M.; Zimmet, P.; Mackay, I.R. Latent autoimmune diabetes in adults (LADA)—More than a name. Diabetologia 2006, 49, 1996–1998. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, W.V.; Kazlauskas, R.J. Molecular basis for enantioselectivity of lipase from pseudomonas cepacia toward primary alcohols. Modeling, kinetics, and chemical modification of tyr29 to increase or decrease enantioselectivity. J. Organ. Chem. 1999, 64, 2638–2647. [Google Scholar] [CrossRef]
- Tuomi, T.; Zimmet, P.Z.; Rowley, M.J.; Serjeantson, S.W.; Mackay, I.R. Persisting antibodies to glutamic acid decarboxylase in type 1 (insulin-dependent) diabetes mellitus are not associated with neuropathy. Diabetologia 1993, 36, 685. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.; Midthjell, K.; Grill, V. Influence of family history of diabetes on incidence and prevalence of latent autoimmune diabetes of the adult: Results from the nord-trondelag health study. Diabetes Care 2007, 30, 3040–3045. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, R.B. Mild familial diabetes with dominant inheritance. Q. J. Med. 1974, 43, 339–357. [Google Scholar] [PubMed]
- Voight, B.F.; Scott, L.J.; Steinthorsdottir, V.; Morris, A.P.; Dina, C.; Welch, R.P.; Zeggini, E.; Huth, C.; Aulchenko, Y.S.; Thorleifsson, G.; et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010, 42, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Lagou, V.; Welch, R.P.; Wheeler, E.; Montasser, M.E.; Luan, J.; Magi, R.; Strawbridge, R.J.; Rehnberg, E.; Gustafsson, S.; et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012, 44, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Jonsdottir, T.; Walters, G.B.; Styrkarsdottir, U.; Gretarsdottir, S.; Emilsson, V.; Ghosh, S.; et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007, 39, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Turnbull, D.M.; Walker, M.; Hattersley, A.T. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243a>g mitochondrial point mutation. Diabet. Med. 2008, 25, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Ellard, S.; Hattersley, A.T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Van den Ouweland, J.M.; Lemkes, H.H.; Ruitenbeek, W.; Sandkuijl, L.A.; de Vijlder, M.F.; Struyvenberg, P.A.; van de Kamp, J.J.; Maassen, J.A. Mutation in mitochondrial tRNA(leu)(uur) gene in a large pedigree with maternally transmitted type ii diabetes mellitus and deafness. Nat. Genet. 1992, 1, 368–371. [Google Scholar]
- Pearson, E.R.; Flechtner, I.; Njolstad, P.R.; Malecki, M.T.; Flanagan, S.E.; Larkin, B.; Ashcroft, F.M.; Klimes, I.; Codner, E.; Iotova, V.; et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to kir6.2 mutations. N. Engl. J. Med. 2006, 355, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.M.; Black, M.H.; Xiang, A.H.; Allayee, H.; Lawrence, J.M.; Buchanan, T.A. Genetics of gestational diabetes mellitus and type 2 diabetes. Diabetes Care 2007, 30 (Suppl. 2), S134–S140. [Google Scholar] [CrossRef] [PubMed]
- Shaat, N.; Groop, L. Genetics of gestational diabetes mellitus. Curr. Med. Chem. 2007, 14, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, T.H.; Lim, S.; Choi, S.H.; Shin, H.D.; Lee, H.K.; Park, K.S.; Jang, H.C. Type 2 diabetes-associated genetic variants discovered in the recent genome-wide association studies are related to gestational diabetes mellitus in the Korean population. Diabetologia 2009, 52, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Lauenborg, J.; Grarup, N.; Damm, P.; Borch-Johnsen, K.; Jorgensen, T.; Pedersen, O.; Hansen, T. Common type 2 diabetes risk gene variants associate with gestational diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, J.; Grant, A.M. The genetics of gestational diabetes mellitus: Evidence for relationship with type 2 diabetes mellitus. Genet. Med. 2008, 10, 240–250. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.B. Diabetes mellitus after GDM. Diabetes 1991, 40 (Suppl. 2), 131–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Newton, K.M.; Knopp, R.H. Gestational diabetes and the incidence of type 2 diabetes: A systematic review. Diabetes Care 2002, 25, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Young, B.C.; Ecker, J.L. Fetal macrosomia and shoulder dystocia in women with gestational diabetes: Risks amenable to treatment? Curr. Diabetes Rep. 2013, 13, 12–18. [Google Scholar] [CrossRef]
- Group, H.S.C.R.; Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Köbberling, J.; Tillil, H. Empirical risk figures for first-degree relatives of non-insulin dependent diabetics. In The Genetics of Diabetes Mellitus; Academic Press: London, UK, 1982; pp. 201–209. [Google Scholar]
- Kaprio, J.; Tuomilehto, J.; Koskenvuo, M.; Romanov, K.; Reunanen, A.; Eriksson, J.; Stengard, J.; Kesaniemi, Y.A. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 1992, 35, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Newman, B.; Selby, J.V.; King, M.C.; Slemenda, C.; Fabsitz, R.; Friedman, G.D. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia 1987, 30, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.; Kyvik, K.O.; Vaag, A.; Beck-Nielsen, H. Heritability of type ii (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—A population-based twin study. Diabetologia 1999, 42, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Medici, F.; Hawa, M.; Ianari, A.; Pyke, D.A.; Leslie, R.D. Concordance rate for type ii diabetes mellitus in monozygotic twins: Actuarial analysis. Diabetologia 1999, 42, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B.; Cupples, L.A.; Wilson, P.W. Parental transmission of type 2 diabetes: The framingham offspring study. Diabetes 2000, 49, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J. The double puzzle of diabetes. Nature 2003, 423, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.; Oda, N.; Cox, N.J.; Li, X.; Orho-Melander, M.; Hara, M.; Hinokio, Y.; Lindner, T.H.; Mashima, H.; Schwarz, P.E.; et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet. 2000, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, R.; Blangero, J.; Almasy, L.; Dyer, T.D.; Williams, K.L.; Leach, R.J.; O’Connell, P.; Stern, M.P. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am. J. Hum. Genet. 1999, 64, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, I.; Thorleifsson, G.; Benediktsson, R.; Sigurdsson, G.; Emilsson, V.; Einarsdottir, A.S.; Hjorleifsdottir, E.E.; Orlygsdottir, G.T.; Bjornsdottir, G.T.; Saemundsdottir, J.; et al. Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34-q35.2. Am. J. Hum. Genet. 2003, 73, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.F.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Lin, Y.; Zhang, Y.; Yang, J.; Zhang, Y.; Liu, H.; Zhang, B. Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: A large human genome epidemiology (huge) review and meta-analysis. BMC Med. Genet. 2009, 10, e15. [Google Scholar] [CrossRef]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamaki, J.; Mykkanen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A pro12ala substitution in ppargamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hani, E.H.; Boutin, P.; Durand, E.; Inoue, H.; Permutt, M.A.; Velho, G.; Froguel, P. Missense mutations in the pancreatic islet beta cell inwardly rectifying k+ channel gene (kir6.2/bir): A meta-analysis suggests a role in the polygenic basis of type ii diabetes mellitus in Caucasians. Diabetologia 1998, 41, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell katp channel subunits kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 52, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.K.; Nielsen, E.M.; Ek, J.; Andersen, G.; Glumer, C.; Carstensen, B.; Mouritzen, P.; Drivsholm, T.; Borch-Johnsen, K.; Jorgensen, T.; et al. Analysis of separate and combined effects of common variation in KCNJ11 and PPARG on risk of type 2 diabetes. J. Clin. Endocrinol. Metab. 2005, 90, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Cummings, E.A.; Edghill, E.L.; Harries, L.W.; Scott, R.; Costa, T.; Temple, I.K.; Hattersley, A.T.; Ellard, S. Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 gene encoding the kir6.2 subunit of the beta-cell potassium adenosine triphosphate channel. J. Clin. Endocrinol. Metab. 2004, 89, 3932–3935. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT; Lund University; Novartis Institutes of BioMedical Research; Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segre, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Chen, C.H.; Hu, C.; Long, J.; Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in East Asians. Nat. Genet. 2012, 44, 67–72. [Google Scholar] [CrossRef]
- Imamura, M.; Maeda, S.; Yamauchi, T.; Hara, K.; Yasuda, K.; Morizono, T.; Takahashi, A.; Horikoshi, M.; Nakamura, M.; Fujita, H.; et al. A single-nucleotide polymorphism in ANK1 is associated with susceptibility to type 2 diabetes in Japanese populations. Hum. Mol. Genet. 2012, 21, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Kooner, J.S.; Saleheen, D.; Sim, X.; Sehmi, J.; Zhang, W.; Frossard, P.; Been, L.F.; Chia, K.S.; Dimas, A.S.; Hassanali, N.; et al. Genome-wide association study in individuals of south Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet. 2011, 43, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gan, W.; Lu, L.; Dong, X.; Han, X.; Hu, C.; Yang, Z.; Sun, L.; Bao, W.; Li, P.; et al. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese hans. Diabetes 2013, 62, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.D.; McDonough, C.W.; Hicks, P.J.; Roh, B.H.; Wing, M.R.; An, S.S.; Hester, J.M.; Cooke, J.N.; Bostrom, M.A.; Rudock, M.E.; et al. A genome-wide association search for type 2 diabetes genes in African Americans. PLOS ONE 2012, 7, e29202. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.J.; Below, J.E.; Krithika, S.; Valladares, A.; Barta, J.L.; Cox, N.J.; Hanis, C.L.; Wacher, N.; Garcia-Mena, J.; Hu, P.; et al. Genome-wide association study of type 2 diabetes in a sample from Mexico city and a meta-analysis of a Mexican-American sample from starr county, Texas. Diabetologia 2011, 54, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.O.; Long, J.; Cai, Q.; Qi, L.; Xiang, Y.B.; Cho, Y.S.; Tai, E.S.; Li, X.; Lin, X.; Chow, W.H.; et al. Identification of new genetic risk variants for type 2 diabetes. PLOS Genet. 2010, 6, e1001127. [Google Scholar] [CrossRef] [PubMed]
- DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; Mexican American Type 2 Diabetes (MAT2D) Consortium; Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) Consortium; Go, M.J.; Zhang, W.; Below, J.E.; Gaulton, K.J.; et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet. 2014, 46, 234–244. [Google Scholar]
- Yasuda, K.; Miyake, K.; Horikawa, Y.; Hara, K.; Osawa, H.; Furuta, H.; Hirota, Y.; Mori, H.; Jonsson, A.; Sato, Y.; et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008, 40, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jorgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Hara, K.; Maeda, S.; Yasuda, K.; Takahashi, A.; Horikoshi, M.; Nakamura, M.; Fujita, H.; Grarup, N.; Cauchi, S.; et al. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat. Genet. 2010, 42, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Chauhan, G.; Dwivedi, O.P.; Mahajan, A.; Jaiswal, A.; Kaur, I.; Bandesh, K.; Singh, T.; Mathai, B.J.; Pandey, Y.; et al. Genome-wide association study for type 2 diabetes in Indians identifies a new susceptibility locus at 2q21. Diabetes 2013, 62, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Consortium, S.T.D.; Williams, A.L.; Jacobs, S.B.; Moreno-Macias, H.; Huerta-Chagoya, A.; Churchhouse, C.; Marquez-Luna, C.; Garcia-Ortiz, H.; Gomez-Vazquez, M.J.; Burtt, N.P.; et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 2014, 506, 97–101. [Google Scholar] [PubMed]
- Sun, X.; Yu, W.; Hu, C. Genetics of type 2 diabetes: Insights into the pathogenesis and its clinical application. BioMed. Res. Int. 2014, 2014, 926713. [Google Scholar] [PubMed]
- Li, Y.R.; Keating, B.J. Trans-ethnic genome-wide association studies: Advantages and challenges of mapping in diverse populations. Genome Med. 2014, 6, e91. [Google Scholar] [CrossRef]
- Morris, A.P. Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol. 2011, 35, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, A.; Grarup, N.; Li, Y.; Sparso, T.; Tian, G.; Cao, H.; Jiang, T.; Kim, S.Y.; Korneliussen, T.; Li, Q.; et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia 2013, 56, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Scott, L.J.; Saxena, R.; Voight, B.F.; Marchini, J.L.; Hu, T.; de Bakker, P.I.; Abecasis, G.R.; Almgren, P.; Andersen, G.; et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008, 40, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Weedon, M.N.; Schwarz, P.E.; Horikawa, Y.; Iwasaki, N.; Illig, T.; Holle, R.; Rathmann, W.; Selisko, T.; Schulze, J.; Owen, K.R.; et al. Meta-analysis and a large association study confirm a role for calpain-10 variation in type 2 diabetes susceptibility. Am. J. Hum. Genet. 2003, 73, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Rung, J.; Cauchi, S.; Albrechtsen, A.; Shen, L.; Rocheleau, G.; Cavalcanti-Proenca, C.; Bacot, F.; Balkau, B.; Belisle, A.; Borch-Johnsen, K.; et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat. Genet. 2009, 41, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Cornelis, M.C.; Kraft, P.; Stanya, K.J.; Linda Kao, W.H.; Pankow, J.S.; Dupuis, J.; Florez, J.C.; Fox, C.S.; Pare, G.; et al. Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Hum. Mol. Genet. 2010, 19, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Elbers, C.C.; Guo, Y.; Peter, I.; Gaunt, T.R.; Mega, J.L.; Lanktree, M.B.; Tare, A.; Castillo, B.A.; Li, Y.R.; et al. Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am. J. Hum. Genet. 2012, 90, 410–425. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Hivert, M.F.; Langenberg, C.; Tanaka, T.; Pankow, J.S.; Vollenweider, P.; Lyssenko, V.; Bouatia-Naji, N.; Dupuis, J.; Jackson, A.U.; et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010, 42, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.; Rayner, N.W.; Freathy, R.M.; et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Thorleifsson, G.; Walters, G.B.; Gudbjartsson, D.F.; Steinthorsdottir, V.; Sulem, P.; Helgadottir, A.; Styrkarsdottir, U.; Gretarsdottir, S.; Thorlacius, S.; Jonsdottir, I.; et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009, 41, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.S.; Weedon, M.N.; Fawcett, K.A.; Wasson, J.; Debenham, S.L.; Daly, A.; Lango, H.; Frayling, T.M.; Neumann, R.J.; Sherva, R.; et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat. Genet. 2007, 39, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Minton, J.A.; Hattersley, A.T.; Owen, K.; McCarthy, M.I.; Walker, M.; Latif, F.; Barrett, T.; Frayling, T.M. Association studies of genetic variation in the WFS1 gene and type 2 diabetes in U.K. Populations. Diabetes 2002, 51, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Sim, X.; Ong, R.T.; Suo, C.; Tay, W.T.; Liu, J.; Ng, D.P.; Boehnke, M.; Chia, K.S.; Wong, T.Y.; Seielstad, M.; et al. Transferability of type 2 diabetes implicated loci in multi-ethnic cohorts from Southeast Asia. PLOS Genet. 2011, 7, e1001363. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, F.; Serizawa, M.; Yamamoto, K.; Fujisawa, T.; Nakashima, E.; Ohnaka, K.; Ikegami, H.; Sugiyama, T.; Katsuya, T.; Miyagishi, M.; et al. Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes 2009, 58, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Voight, B.F.; Yengo, L.; Amin, N.; Dupuis, J.; Ganser, M.; Grallert, H.; Navarro, P.; Li, M.; Qi, L.; et al. Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLOS Genet. 2012, 8, e1002741. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.C.; Shriner, D.; Chen, B.H.; Li, J.; Chen, W.M.; Guo, X.; Liu, J.; Bielinski, S.J.; Yanek, L.R.; Nalls, M.A.; et al. Meta-analysis of genome-wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLOS Genet. 2014, 10, e1004517. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.J.; Yang, C.F.; Chen, C.C.; Chuang, L.M.; Lu, C.H.; Chang, C.T.; Wang, T.Y.; Chen, R.H.; Shiu, C.F.; Liu, Y.M.; et al. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLOS Genet. 2010, 6, e1000847. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, A.H.; Jokubka, R.; Tojjar, D.; Granhall, C.; Hansson, O.; Li, D.Q.; Nagaraj, V.; Reinbothe, T.M.; Tuncel, J.; Eliasson, L.; et al. Overexpression of alpha2a-adrenergic receptors contributes to type 2 diabetes. Science 2010, 327, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Gianniny, L.; Burtt, N.P.; Lyssenko, V.; Giuducci, C.; Sjogren, M.; Florez, J.C.; Almgren, P.; Isomaa, B.; Orho-Melander, M.; et al. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes 2006, 55, 2890–2895. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.T.; Uimari, P.; Aalto, J.M.; Pirskanen, M.; Kaikkonen, J.; Todorova, B.; Hypponen, J.; Korhonen, V.P.; Asikainen, J.; Devine, C.; et al. Type 2 diabetes whole-genome association study in four populations: The diagen consortium. Am. J. Hum. Genet. 2007, 81, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Timpson, N.J.; Lindgren, C.M.; Weedon, M.N.; Randall, J.; Ouwehand, W.H.; Strachan, D.P.; Rayner, N.W.; Walker, M.; Hitman, G.A.; Doney, A.S.; et al. Adiposity-related heterogeneity in patterns of type 2 diabetes susceptibility observed in genome-wide association data. Diabetes 2009, 58, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Frigge, M.L.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Prokopenko, I.; Langenberg, C.; Florez, J.C.; Saxena, R.; Soranzo, N.; Thorleifsson, G.; Loos, R.J.; Manning, A.K.; Jackson, A.U.; Aulchenko, Y.; et al. Variants in MTNR1B influence fasting glucose levels. Nat. Genet. 2009, 41, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Sanna, S.; Jackson, A.U.; Scuteri, A.; Bonnycastle, L.L.; Clarke, R.; Heath, S.C.; Timpson, N.J.; Najjar, S.S.; Stringham, H.M.; et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008, 40, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Saleheen, D.; Been, L.F.; Garavito, M.L.; Braun, T.; Bjonnes, A.; Young, R.; Ho, W.K.; Rasheed, A.; Frossard, P.; et al. Genome-wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in sikhs of punjabi origin from India. Diabetes 2013, 62, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Sulem, P.; Steinthorsdottir, V.; Bergthorsson, J.T.; Thorleifsson, G.; Manolescu, A.; Rafnar, T.; Gudbjartsson, D.; Agnarsson, B.A.; Baker, A.; et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat. Genet. 2007, 39, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Winckler, W.; Burtt, N.P.; Holmkvist, J.; Cervin, C.; de Bakker, P.I.; Sun, M.; Almgren, P.; Tuomi, T.; Gaudet, D.; Hudson, T.J.; et al. Association of common variation in the hnf1alpha gene region with risk of type 2 diabetes. Diabetes 2005, 54, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Winckler, W.; Graham, R.R.; de Bakker, P.I.; Sun, M.; Almgren, P.; Tuomi, T.; Gaudet, D.; Hudson, T.J.; Ardlie, K.G.; Daly, M.J.; et al. Association testing of variants in the hepatocyte nuclear factor 4alpha gene with risk of type 2 diabetes in 7883 people. Diabetes 2005, 54, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Elliott, P.; Zabaneh, D.; Zhang, W.; Li, Y.; Froguel, P.; Balding, D.; Scott, J.; Kooner, J.S. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat. Genet. 2008, 40, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.K.; Hivert, M.F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.T.; Bielak, L.F.; Prokopenko, I.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Bentley, A.; Adeyemo, A.; Shriner, D.; Zhou, J.; Doumatey, A.; Huang, H.; Ramos, E.; Erdos, M.; Gerry, N.; et al. Genome-wide association study identifies novel loci association with fasting insulin and insulin resistance in African Americans. Hum. Mol. Genet. 2012, 21, 4530–4536. [Google Scholar] [CrossRef] [PubMed]
- Go, M.J.; Hwang, J.Y.; Kim, Y.J.; Hee Oh, J.; Kim, Y.J.; Heon Kwak, S.; Soo Park, K.; Lee, J.; Kim, B.J.; Han, B.G.; et al. New susceptibility loci in myl2, C12orf51 and oas1 associated with 1-h plasma glucose as predisposing risk factors for type 2 diabetes in the Korean population. J. Hum. Genet. 2013, 58, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; Belmont, J.W.; Boerwinkle, E.; Gibbs, R.A. Clan genomics and the complex architecture of human disease. Cell 2011, 147, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Thorleifsson, G.; Sulem, P.; Helgason, H.; Grarup, N.; Sigurdsson, A.; Helgadottir, H.T.; Johannsdottir, H.; Magnusson, O.T.; Gudjonsson, S.A.; et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat. Genet. 2014, 46, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Moltke, I.; Grarup, N.; Jorgensen, M.E.; Bjerregaard, P.; Treebak, J.T.; Fumagalli, M.; Korneliussen, T.S.; Andersen, M.A.; Nielsen, T.S.; Krarup, N.T.; et al. A common greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 2014, 512, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Consortium, S.T.D.; Estrada, K.; Aukrust, I.; Bjorkhaug, L.; Burtt, N.P.; Mercader, J.M.; Garcia-Ortiz, H.; Huerta-Chagoya, A.; Moreno-Macias, H.; Walford, G.; et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 2014, 311, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, J.R.; Jackson, A.U.; Fogarty, M.P.; Buchkovich, M.L.; Stancakova, A.; Stringham, H.M.; Sim, X.; Yang, L.; Fuchsberger, C.; Cederberg, H.; et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat. Genet. 2013, 45, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium; Craddock, N.; Hurles, M.E.; Cardin, N.; Pearson, R.D.; Plagnol, V.; Robson, S.; Vukcevic, D.; Barnes, C.; Conrad, D.F.; et al. Genome-wide association study of cnvs in 16,000 cases of eight common diseases and 3000 shared controls. Nature 2010, 464, 713–720. [Google Scholar]
- Plomin, R.; Haworth, C.M.; Davis, O.S. Common disorders are quantitative traits. Nat. Rev. Genet. 2009, 10, 872–878. [Google Scholar] [CrossRef] [PubMed]
- McClellan, J.; King, M.C. Genetic heterogeneity in human disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J. What is complex about complex disorders? Genome Biol. 2012, 13, e237. [Google Scholar] [CrossRef]
- Agarwala, V.; Flannick, J.; Sunyaev, S.; Go, T.D.C.; Altshuler, D. Evaluating empirical bounds on complex disease genetic architecture. Nat. Genet. 2013, 45, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Nagorny, C.L.; Erdos, M.R.; Wierup, N.; Jonsson, A.; Spegel, P.; Bugliani, M.; Saxena, R.; Fex, M.; Pulizzi, N.; et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat. Genet. 2009, 41, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Bouatia-Naji, N.; Bonnefond, A.; Cavalcanti-Proenca, C.; Sparso, T.; Holmkvist, J.; Marchand, M.; Delplanque, J.; Lobbens, S.; Rocheleau, G.; Durand, E.; et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat. Genet. 2009, 41, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Park, S.Y.; Su, J.; Bailey, K.; Ottosson-Laakso, E.; Shcherbina, L.; Oskolkov, N.; Zhang, E.; Thevenin, T.; Fadista, J.; et al. TCF7L2 is a master regulator of insulin production and processing. Hum. Mol. Genet. 2014, 23, 6419–6431. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Lupi, R.; Marchetti, P.; Del Guerra, S.; Orho-Melander, M.; Almgren, P.; Sjogren, M.; Ling, C.; Eriksson, K.F.; Lethagen, A.L.; et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Investig. 2007, 117, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Eliasson, L.; Kotova, O.; Pilgaard, K.; Wierup, N.; Salehi, A.; Wendt, A.; Jonsson, A.; de Marinis, Y.Z.; Berglund, L.M.; et al. Pleiotropic effects of gip on islet function involve osteopontin. Diabetes 2011, 60, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, A.H.; Braun, M.; Mahdi, T.; Andersson, S.A.; Travers, M.E.; Shigeto, M.; Zhang, E.; Almgren, P.; Ladenvall, C.; Axelsson, A.S.; et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes 2012, 61, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; Langenberg, C.; Hivert, M.F.; Prokopenko, I.; Lyssenko, V.; Dupuis, J.; Magi, R.; Sharp, S.; Jackson, A.U.; Assimes, T.L.; et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic loci regulating glucose and insulin metabolism in humans. Diabetes 2010, 59, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Grarup, N.; Andersen, G.; Krarup, N.T.; Albrechtsen, A.; Schmitz, O.; Jorgensen, T.; Borch-Johnsen, K.; Hansen, T.; Pedersen, O. Association testing of novel type 2 diabetes risk alleles in the JAZF1, CDC123/CAMK1D, TSPAN8, THADA, ADAMTS9, and NOTCH2 loci with insulin release, insulin sensitivity, and obesity in a population-based sample of 4516 glucose-tolerant middle-aged danes. Diabetes 2008, 57, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Axelsson, A.S.; Spegel, P.; Andersson, L.E.; Mulder, H.; Groop, L.C.; Renstrom, E.; Rosengren, A.H. Genotype-based treatment of type 2 diabetes with an alpha2a-adrenergic receptor antagonist. Sci. Transl. Med. 2014, 6, 257ra139. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era—Concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Wray, N.R.; Goddard, M.E.; Visscher, P.M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011, 88, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Yang, J.; Goddard, M.E. A commentary on “common SNPs explain a large proportion of the heritability for human height” by yang et al. (2010). Twin Res. Hum. Genet. 2010, 13, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Forsblom, C.; Lehtovirta, M.; Tuomi, T.; Karanko, S.; Nissen, M.; Ehrnstrom, B.O.; Forsen, B.; Isomaa, B.; Snickars, B.; et al. Metabolic consequences of a family history of niddm (the botnia study): Evidence for sex-specific parental effects. Diabetes 1996, 45, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Li, X.; Sundquist, K.; Sundquist, J. Familial risks for type 2 diabetes in Sweden. Diabetes Care 2010, 33, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Whitelaw, E. Epigenetic germline inheritance. Curr. Opin. Genet. Dev. 2004, 14, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Tollefsbol, T.O. Gene-environment interactions and epigenetic basis of human diseases. Curr. Issues Mol. Biol. 2008, 10, 25–36. [Google Scholar] [PubMed]
- Small, K.S.; Hedman, A.K.; Grundberg, E.; Nica, A.C.; Thorleifsson, G.; Kong, A.; Thorsteindottir, U.; Shin, S.Y.; Richards, H.B.; Soranzo, N.; et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 2011, 43, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.L.; Guo, T.; Muller, Y.L.; Fleming, J.; Knowler, W.C.; Kobes, S.; Bogardus, C.; Baier, L.J. Strong parent-of-origin effects in the association of KCNQ1 variants with type 2 diabetes in American Indians. Diabetes 2013, 62, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. TIG 1991, 7, 45–49. [Google Scholar] [CrossRef]
- Wolf, J.B.; Hager, R. A maternal-offspring coadaptation theory for the evolution of genomic imprinting. PLOS Biol. 2006, 4, e380. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Travers, M.E.; Mackay, D.J.; Dekker Nitert, M.; Morris, A.P.; Lindgren, C.M.; Berry, A.; Johnson, P.R.; Hanley, N.; Groop, L.C.; McCarthy, M.I.; et al. Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets. Diabetes 2013, 62, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Hoggart, C.J.; Venturini, G.; Mangino, M.; Gomez, F.; Ascari, G.; Zhao, J.H.; Teumer, A.; Winkler, T.W.; Tsernikova, N.; Luan, J.; et al. Novel approach identifies SNPs in slc2a10 and kcnk9 with evidence for parent-of-origin effect on body mass index. PLOS Genet. 2014, 10, e1004508. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.C. Epistasis—The essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J.; Wedig, G.C.; Jacobs, K.B.; Elston, R.C. Multilocus linkage tests based on affected relative pairs. Am. J. Hum. Genet. 2000, 66, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.J.; Frigge, M.; Nicolae, D.L.; Concannon, P.; Hanis, C.L.; Bell, G.I.; Kong, A. Loci on chromosomes 2 (niddm1) and 15 interact to increase susceptibility to diabetes in Mexican Americans. Nat. Genet. 1999, 21, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Gayan, J.; Marin, J.; Galan, J.J.; Saez, M.E.; Real, L.M.; Antunez, C.; Ruiz, A. Whole-genome conditional two-locus analysis identifies novel candidate genes for late-onset parkinson’s disease. Neurogenetics 2009, 10, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with il-23 and nf-kappab pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.M.; Marchini, J.; Morris, A.P.; Cardon, L.R. Two-stage two-locus models in genome-wide association. PLOS Genet. 2006, 2, e157. [Google Scholar] [CrossRef] [PubMed]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Diabetic nephropathy—Emerging epigenetic mechanisms. Nat. Rev. Nephrol. 2014, 10, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.L.; McCray, P.B., Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Valverde, S.L.; Taft, R.J.; Mattick, J.S. MicroRNAs in beta-cell biology, insulin resistance, diabetes and its complications. Diabetes 2011, 60, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, M.; Scaria, V.; Brahmachari, S.K. Dbsmr: A novel resource of genome-wide SNPs affecting microRNA mediated regulation. BMC Bioinform. 2009, 10, e108. [Google Scholar] [CrossRef]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the anril locus on chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Eftychi, C.; Howson, J.M.; Barratt, B.J.; Vella, A.; Payne, F.; Smyth, D.J.; Twells, R.C.; Walker, N.M.; Rance, H.E.; Tuomilehto-Wolf, E.; et al. Analysis of the type 2 diabetes-associated single nucleotide polymorphisms in the genes IRS1, KCNJ11, and pparg2 in type 1 diabetes. Diabetes 2004, 53, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.M.; Howson, J.M.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Field, S.F.; Stevens, H.E.; Todd, J.A. No association of multiple type 2 diabetes loci with type 1 diabetes. Diabetologia 2009, 52, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Sterner, M.; Forsen, T.; Karajamaki, A.; Rolandsson, O.; Forsblom, C.; Groop, P.H.; Lahti, K.; Nilsson, P.M.; Groop, L.; et al. Type 2 diabetes susceptibility gene variants predispose to adult-onset autoimmune diabetes. Diabetologia 2014, 57, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Van Hoek, M.; van Herpt, T.W.; Dehghan, A.; Hofman, A.; Lieverse, A.G.; van Duijn, C.M.; Witteman, J.C.; Sijbrands, E.J. Association of an APOC3 promoter variant with type 2 diabetes risk and need for insulin treatment in lean persons. Diabetologia 2011, 54, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The cation efflux transporter znt8 (SLC30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 17040–17045. [Google Scholar] [CrossRef] [PubMed]
- Corona, E.; Dudley, J.T.; Butte, A.J. Extreme evolutionary disparities seen in positive selection across seven complex diseases. PLOS ONE 2010, 5, e12236. [Google Scholar] [CrossRef] [PubMed]
- Frayling, T.M.; Colhoun, H.; Florez, J.C. A genetic link between type 2 diabetes and prostate cancer. Diabetologia 2008, 51, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Shin, S.Y.; Petersen, A.K.; Mohney, R.P.; Meredith, D.; Wagele, B.; Altmaier, E.; CardioGram; Deloukas, P.; Erdmann, J.; et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011, 477, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The kegg resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; Lang, S.; Sharma, A.; Fadista, J.; Zhou, Y.; Ahlqvist, E.; Jonsson, A.; Lyssenko, V.; Vikman, P.; Hansson, O.; et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012, 16, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; Fadista, J.; Ahlqvist, E.; Zhang, M.; Wierup, N.; Renstrom, E.; Groop, L. Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol. Cell. Endocrinol. 2013, 375, 35–42. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, R.B.; Groop, L. Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes 2015, 6, 87-123. https://doi.org/10.3390/genes6010087
Prasad RB, Groop L. Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes. 2015; 6(1):87-123. https://doi.org/10.3390/genes6010087
Chicago/Turabian StylePrasad, Rashmi B., and Leif Groop. 2015. "Genetics of Type 2 Diabetes—Pitfalls and Possibilities" Genes 6, no. 1: 87-123. https://doi.org/10.3390/genes6010087
APA StylePrasad, R. B., & Groop, L. (2015). Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes, 6(1), 87-123. https://doi.org/10.3390/genes6010087